

Abstract
The aim of this study was to demonstrate the value of mechanistic simulations in gaining insight into the behaviors of modified release (MR) formulations in vivo and to use the properly calibrated models for prediction of pharmacokinetics (PK) and pharmacodynamics (PD). GastroPlusTM (Simulations Plus, Inc.) was used to fit mechanistic models for adinazolam and metoprolol that describe the absorption, PK, and PD after intravenous (i.v.) and immediate release (IR) oral (p.o.) administration. The fitted model for adinazolam was then used to predict the PD profile for a MR formulation and to design a new formulation with desired onset and duration of action. The fitted metoprolol model was used to gain insight and to explain the in vivo behaviors of MR formulations. For each drug, a single absorption/PK model was fitted that provided simulated plasma concentration–time profiles closely matching observed in vivo profiles across several different i.v. and p.o doses. Sedation score profiles of adinazolam were fitted with an indirect PD model. For metoprolol, the fitted absorption/PK model for IR p.o. doses was used to select in vitro dissolution conditions that best matched the in vivo release of MR doses. This model also explained differences in exposure after administration of MR formulations with different release rates. Mechanistic absorption/PK models allow for detailed descriptions of all processes affecting the two drugs’ bioavailability, including release/dissolution, absorption, and intestinal and hepatic first pass extraction. The insights gained can be used to design formulations that more effectively overcome identified problems.
Key words: gastrointestinal absorption, mechanistic simulation, modified release, pharmacodynamics, pharmacokinetics
INTRODUCTION
In vitro–in vivo correlations (IVIVCs) have become an integral part of the development of modified release (MR) formulations from the early stages of formulation development where the IVIVC is only assumed to scale up and postapproval changes (1). Several levels of IVIVC are defined in Food and Drug Administration guidance documents, with level A (generally linear correlation with a point-to-point relationship) being the most common method utilized in formulation development (2). A number of approaches for the development of level A correlations (relating the drug appearance in systemic circulation as deconvoluted from the plasma concentration (Cp)–time profile to the in vitro release profile) have been published over the years (3–11) and will not be described in detail here.
The focus of this study is on the mechanistic simulation of absorption, which so far seems to be underutilized in this field. These models can include all major processes relevant to absorption and elimination, including nonlinear dose-dependent absorption, first pass metabolism in gut as well as in liver, limited “absorption window” for the compounds whose absorption is dependent on influx transporters with very narrow regional expression in gut, etc. (12–19). Many pharmaceutical research groups have published their applications of simulation and modeling using the advanced compartmental absorption and transit (ACAT) model in GastroPlus (20–27). By modeling each relevant process explicitly, the in vivo intestinal release profile can be deconvoluted, rather than only a profile of drug appearance in the systemic circulation (i.e., systemic availability as obtained from traditional deconvolution methods). Systemic availability can be affected not only by drug release from the formulation but also by first pass extraction in gut and liver, as well as saturable carrier-mediated transport. The mechanistic approach which incorporates all of these processes and takes into account how they might be affecting each other can provide better insights into a drug’s in vivo behavior. This work describes how several different pieces of information available for the molecule (physicochemical parameters, in vivo data, in vitro dissolution data), when properly linked to the intestinal physiological parameters, can be used to design the formulation for a desired onset and duration of pharmacological effect as well as to avoid intestinal first pass extraction.
MATERIALS AND METHODS
All simulations were performed using GastroPlusTM software (Simulations Plus, Inc.), utilizing several of its features including the ACAT model to describe the drug’s absorption, the PKPlusTM and PBPKPlusTM modules to describe pharmacokinetics, and the PDPlusTM module to describe pharmacodynamics. The ACAT model in GastroPlus simulated the absorption of adinazolam and metoprolol. This model is based on the original compartmental absorption and transit model by Yu et al. (28), which was enhanced in GastroPlus by the incorporation of pH-dependent solubility and its effects on dissolution rate, metabolism in both gut and liver, possible absorption from stomach and colon, and other regional factors in the gastrointestinal tract (12). The default parameters for individual compartments representing a typical adult human physiology (body weight ∼85 kg) in fasted state are listed in Table I.
Table I.
ACAT Model Parameters for Fasted State Adult Human Physiology (Body Weight ∼85 kg)
| Compartment | Volume (mL) | Transit time (h) | pH |
|---|---|---|---|
| Stomach | 50 | 0.25 | 1.3 |
| Duodenum | 48 | 0.26 | 6.0 |
| Jejunum1 | 175 | 0.95 | 6.2 |
| Jejunum2 | 140 | 0.76 | 6.4 |
| Ileum1 | 109 | 0.59 | 6.6 |
| Ileum2 | 79 | 0.43 | 6.9 |
| Ileum3 | 56 | 0.31 | 7.4 |
| Cecum | 53 | 4.50 | 6.4 |
| Ascending colon | 57 | 13.5 | 6.8 |
PKPlus is a module in GastroPlus used to obtain standard compartmental PK parameters by fitting the one-, two-, or three-compartment models to single or multiple Cp–time profiles after intravenous (i.v.) or oral (p.o.) administration. This module was used to fit the PK parameters for adinazolam by fitting a two-compartment model across seven Cp–time profiles after i.v. infusion administration of doses ranging from 5 to 20 mg. PBPKPlus is a module in GastroPlus that allows modeling the drug distribution and clearance using a physiologically based pharmacokinetic (PBPK) model. It includes the Population Estimates for Age-Related Physiology™ module used to create PBPK models with appropriate organ weights, volumes, and blood perfusion rates. All simulations in the current study were performed using a default physiology for a typical adult male. The PDPlus module expands the capability of GastroPlus for fitting direct or indirect PD models to the observed effect data. It was used to fit the PD model to adinazolam sedation score vs. time profiles and to predict changes in the onset and duration of PD response with changes to the adinazolam release rate from the formulation.
Adinazolam
Adinazolam is a triazolobenzodiazepine with antidepressant properties (29). Extensive PK and PD studies have been performed to date using this drug. The in vivo PK data obtained from literature and used in the current simulations include Cp–time profiles after i.v. administration of doses ranging from 5 to 20 mg (30–33), p.o. administration of immediate release (IR) formulations of doses ranging from 15 to 60 mg (31,32,34–39), and p.o. administration of sustained release (SR) formulations of doses ranging from 15 to 60 mg (32,38–40). The in vivo PD data (sedation scores) were reported for p.o. IR formulations of doses ranging from 20 to 60 mg (34,36,38,39), and a 60-mg p.o. SR formulation. All i.v. administrations were constant-rate infusions administered over 10 (5 mg dose) or 30 min (10–20 mg doses).
The PKPlus™ module of GastroPlusTM (Simulations Plus, Inc.) was used to fit the volume of central compartment (Vc), clearance (CL), and the rate constants for drug transfer between central and one peripheral compartment (k12 and k21) for adinazolam simultaneously across seven observed Cp–time profiles after i.v. administration (31,33–35) of doses ranging from 5 to 20 mg. The fitted PK parameters for these i.v. Cp–time profiles were then used for all oral doses (IR and SR formulations). The Cp–time profile from a 15-mg oral solution dose (31) was used to fit the in vivo permeability (Peff) and first pass extraction (FPE). The fitted Peff and FPE values were then used in the simulations of all remaining oral doses. The PDPlus™ module of GastroPlus was then used to fit therapeutic PD response (mean sedation score) with the Cp–time profiles of adinazolam (34). GastroPlus simulations were then used to predict the Cp–time profile and PD response profiles for release profiles with different release rates. Table II summarizes physicochemical, PK, and PD parameters used in adinazolam simulations.
Table II.
Key Physicochemical, Pharmacokinetic, and Pharmacodynamic Parameters of Adinazolam Used in the Simulations
| Property | Value | Reference |
|---|---|---|
| S+LogP | 2.73 | Estimated by ADMET PredictorTM (Simulations Plus, Inc.) |
| Solubility (S+S w) | 0.109 @ pH = 8.75 | Estimated by ADMET PredictorTM (Simulations Plus, Inc.) |
| Passive jejunal permeability (cm/s) | 11 × 10−4 | Fitted using Cp–time profile after 15 mg oral solution dose |
| pKa values | Base = 2.43 | Estimated by ADMET PredictorTM (Simulations Plus, Inc.) |
| Base = 7.02 | ||
| Plasma unbound % | 31 | Fleischaker et al. (31) |
| Blood/plasma concentration ratio | 0.7 | Fleischaker et al. (31) |
| FPE % | 70 | Fitted using Cp–time profile after 15 mg oral solution dose |
| V c (L/kg) | 0.66 | Fitted across Cp–time profiles after i.v. infusion doses 5–20 mg |
| CL (L/h/kg) | 0.355 | |
| k 12 (h−1) | 0.473 | |
| k 21 (h−1) | 0.468 | |
| E 0 | 0 | Set arbitrarily (assumed minimum effect = 0) |
| EC50 (μg/mL) | 0.01 | Approximated by Kd to GABA receptor (41) |
| E max | 1.8 | Fitted to sedation score vs. time profiles after IR p.o. administration of 20 and 40 mg doses |
| γ | 2 | |
| CLe (h−1) | 1 |
PK parameters: FPE first pass extraction, V c volume of central compartment, CL total plasma clearance, k 12 and k 21 rate constants for drug transfer between central and peripheral compartment; PD parameters: E 0 the baseline response, E max the maximum response, EC 50 the concentration at which 50% of the maximum response is observed, γ the Hill parameter, CL e the clearance of drug from plasma to effect compartment
Metoprolol
Metoprolol is a selective β1-receptor antagonist (42) whose absorption and pharmacokinetics have been extensively studied in man. The in vivo PK data obtained from literature and used in the current simulations include Cp–time profiles after i.v. infusion administration (42,43), jejunal and colonic perfusion (44), p.o. administration of IR formulations (45,46), and p.o. administration of MR formulations (46,47).
The distribution and clearance were simulated using the PBPKPlus™ module of GastroPlus with a PBPK model. This model was based on the physiology (height, weight, tissue volumes, and tissue blood flows) of a typical 30-year-old male. The method of Rodgers et al. (48,49) was used to estimate tissue/plasma partition coefficients (Kp). Initial estimates for metabolic clearance parameters (Km, Vmax, and enzyme expressions) in liver and gut were based on in vitro measurements in human liver and intestinal microsomes (50). The in vivo Vmax in milligrams per second was extrapolated from the in vitro Vmax value using an average expression for CYP 2D6 in liver of 13 pmol/mg microsomal protein and an average amount of 38 mg of microsomal protein in human liver (51–56). Due to the high polymorphism in expression of 2D6 in the intestine and liver, the average expressions were modified for certain groups of subjects (the same expression level was used for all formulations from any particular study, while the same ratio of enzyme expression in intestinal compartments and liver was assumed for all groups of subjects). Renal clearance was estimated from glomerular filtration rate (GFR) and unbound fraction of metoprolol in plasma (Fup) as “GFR × Fup” assuming healthy subjects with normal GFR for ∼30-year-old adult. The accuracy of prediction of metoprolol distribution and pharmacokinetics using the PBPK model was verified in simulations of i.v. doses (42,43).
The same PBPK model was then used to simulate jejunal and colonic perfusion studies. In these studies, a solution of metoprolol tartrate (130 mg/L) was perfused directly at the duodenojejunal junction or in the cecum at the flow rate of 4.5 mL/min over 150 min (44). The Cp–time profile after colonic perfusion was used to fit the absorption of metoprolol from the colon. The model with fitted colonic absorption was used in simulation of metoprolol absorption after jejunal perfusion (44) and IR p.o. doses (45,46) without further adjustments to drug absorption from either small intestine or colon.
The parameters fitted by the PBPK model and colonic perfusion data were used to simulate the Cp–time profiles after administration of MR formulations (46,47). The in vitro profiles for drug release from the formulation were used as defined in the same publication which was used as a source for the Cp–time profiles after administration of MR formulations (46,47). Table III summarizes physicochemical and PK parameters used in metoprolol simulations.
Table III.
Key Biopharmaceutical and Pharmacokinetic Properties of Metoprolol Used in the PBPK Simulations
| Property | Value | Reference |
|---|---|---|
| LogD 4 | −1.72 | Wang and Lien (57) |
| Solubility | 171 @ pH = 6.5 | Tubic-Grozdanis et al. (58) |
| Passive jejunal permeability (cm/s) | 1.34 × 10−4 | Kasim et al. (59) |
| pKa values | Base = 9.7 | Regardh et al. (42) |
| Plasma unbound % | 89 | Median of reported values of 88% (58) and 90% (43) |
| Blood/plasma concentration ratio | 1.13 | Average across three plasma concentrations (42) |
| V ss (L/kg) | 3.2 | V ss calculation (48) |
| Renal clearance (L/h/kg) | 0.081 | Set as “GFR × F up” for 30-year-old male |
| 2D6 K m [μM] | 26 | Madani et al. (50) |
| 2D6 V max [pmol/min/mg micros prot] | 423 | Sum of V max for α-hydroxymetoprolol and O-demethylmetoprolol formation (50) |
V ss steady-state volume of distribution
RESULTS AND DISCUSSION
Adinazolam
Cp–time profiles of adinazolam after various levels of i.v. and IR p.o. doses were successfully simulated with a single fitted model. The PK parameters were obtained by fitting to Cp–time profiles after i.v. infusion doses. The resulting correlation coefficients, R2, were in the range 0.8–0.96. The intestinal permeability and first pass extraction were fitted to the Cp–time profile after a 15 mg IR p.o. solution dose (R2 = 0.94) using the PK parameters obtained from fitting the i.v. infusion doses. Without any further adjustments to the parameters, the fitted absorption/PK model was used to predict the 20–40 mg IR p.o. doses. The R2 values for these predicted profiles were in the range 0.52–0.92. The apparent poor predictive performance of the model for some IR p.o. doses is attributed to large variabilities in the observed Cp–time profiles. The R2 values for three Cp–time profiles for the 30 mg IR p.o. dose were 0.52, 0.7, and 0.92. The comparison of simulated and experimental Cp–time profiles is shown in Fig. 1.
Fig. 1.

Simulated and experimental Cp–time profiles of adinazolam modeled with a two-compartmental PK model: a i.v. infusion administration and b IR p.o. administration
For clarity, only the profiles used in PK model fitting (15 mg solution) and in fitting of the PD model (20–40 mg doses) are shown. The fitted parameters for a two-compartment PK model were CL = 0.355 L/h/kg, Vc = 0.663 L/kg, and peripheral compartment distribution rate constants K12 = 0.473 h−1, K21 = 0.468 h−1, where volume of peripheral compartment (V2) = 0.671 L/kg. The fitted FPE was 70%. The fitted PK parameters are in good agreement with previously published values (Table IV).
Table IV.
Comparison of Fitted PK Parameters with Published PK Parameters Obtained by Noncompartmental Analysis of Experimental Cp–Time Profiles
| Current fitted model | Published values (31–33,39) | |
|---|---|---|
| i.v. CL (L/h/kg) | 0.355 | 0.28–0.38 |
| i.v. V ss (L/kg) | 1.334a | 0.98–1.15 |
| p.o. CL/F (L/h/kg) | 1.18b | 0.97–1.2 |
| p.o. F (%) | 30 | 37–40 |
F bioavailability, V ss steady-state volume of distribution, CL clearance
aCalculated as sum of volumes of central and peripheral compartments
bCalculated as ratio of CL obtained by fitting to i.v. data and F obtained by fitting to 15 mg p.o. solution dose
The absorption/PK model fitted to i.v. infusion and IR p.o. doses was applied without further modifications to simulate the absorption and PK of the SR formulations. The in vivo release profile for the SR formulations was obtained by fitting a single release profile to best match the plasma concentration–time profiles for different dose levels (Fig. 2).
Fig. 2.

Optimized in vivo release profile and simulated and experimental Cp–time profiles of adinazolam modeled with two-compartmental PK model after p.o. administration of SR formulations: a complete Cp–time profiles and b initial 5 h of the Cp–time profiles
Simulations with the fitted in vivo release profile match the experimental Cp–time profiles for the 15–45 mg doses with R2 ranging from 0.73 to 0.93. Two experimental Cp–time profiles are available for the 60 mg SR dose. One of the experimental profiles after 60 mg dose is ∼30% lower than the simulated profile using the fitted in vivo release. The second experimental profile is well matched up to ∼9 h with the last data point (12 h) being underpredicted by ∼25%. The fitted in vivo release shows steady release over 12 h with apparent changes in release rates at times when the formulation would be moving between the distinct regions of gastrointestinal tract (stomach, small intestine, colon). The sustained release formulation of adinazolam allows for a gradual release over 12–16 h (60). The in vitro release profile for the SR formulation was not published.
Two IR p.o. doses of adinazolam (20 and 40 mg) were used to fit a PD model to reported sedation score profiles (34). The performance of the model was verified by predicting the 60-mg IR and SR dose (Fig. 3). The model that best described the relationship between Cp–time and PD profiles was an indirect link model with an effect compartment. The use of indirect link model is also supported by the counterclockwise hysteresis in the plot of sedation score vs. adinazolam concentration (34). This model incorporates a delay between the drug entering the plasma compartment and the time of exhibiting the pharmacologic action. The delay is due to redistribution to the effect compartment, the true site of the pharmacologic action. The distribution of drug between plasma and the effect compartment is described by Eq. 1:
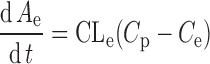 |
1 |
where CLe is the clearance of the drug from plasma to the effect compartment, Cp and Ce are the drug concentrations in plasma and the effect compartment, and Ae is the amount of drug in the effect compartment. The drug concentration in the effect compartment is then linked to the Sigmoid Emax model described by Eq. 2:
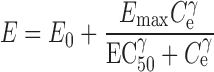 |
2 |
where E0 and Emax are the baseline and the maximum response, respectively, EC50 is the concentration at which 50% of the maximum response is observed, Ce is the instantaneous concentration in the effect compartment, and γ is the Hill parameter. Fitted parameter values were Emax = 1.8; γ = 2; and CLe = 1.0 h−1. E0 was arbitrarily set to 0. Indirect evidence suggests that the pharmacological effects of triazolobenzodiazepines, such as adinazolam, may be due to modulation of gamma-aminobutyric acid (GABA)ergic neurotransmission similar to benzodiazepines (61,62). Using an in vitro Kd = 28 nM for GABA-stimulated CL influx into membrane (41) as an estimate for EC50 resulted in a PD model that described the onset of sedation effect of adinazolam.
Fig. 3.

Pharmacodynamic effect (sedation score) after : a 20 and 40 mg dose of IR p.o. administration and b 60 mg IR and SR p.o. administration. The 20- and 40-mg IR p.o. doses were used to fit the PK/PD model; the 60-mg IR and SR p.o. doses were predicted using the fitted PK/PD model
The PD model fitted to sedation score profiles after 20 and 40 mg IR p.o. doses correctly predicted the onset and duration of action for 60 mg IR and SR p.o. doses (Fig. 3). Administration of a 60-mg IR p.o. formulation which dissolved instantly in the gastrointestinal tract and reached plasma concentration of 0.06 μg/mL in about 20 min (the maximum plasma concentration obtained after administration of 60 mg SR p.o. formulation) results in an observed sedation score of 1 about 40 min after drug administration. As a result of slower release of drug from the SR formulation, the plasma concentration reaches 0.06 μg/mL in about 90 min and results in an observed sedation score of 1 about 130 min after drug administration.
This fitted absorption/PK/PD model combines information about drug properties and pharmacokinetics obtained after i.v. and IR p.o. administration to provide a comprehensive view of the drug’s behavior in vivo. The model was used to predict the PK/PD behavior of SR formulations with different rates of drug release (Fig. 4). This example demonstrates how a comprehensive model can be used to estimate the effect of changing formulation (changing release rate) on both PK and PD response and ultimately guide formulation development for optimum drug performance.
Fig. 4.

Predicted Cp–time and PD effect-time profiles for two 20-mg adinazolam formulations with different release rates: a drug release over 20 h, b drug release over 10 h
Metoprolol
The GastroPlus default PBPK model combined with in vitro clearance was used to describe metoprolol pharmacokinetics. The tissue distribution was described by Kps calculated using a modified Rodgers’ method (48,49). The modification consisted of combining the two published equations for moderate to strong bases and all other compounds into a single equation. In the new equation, the contribution of the binding of neutral and anionic forms of molecules to tissue albumin and the contribution of the cationic form of molecules to tissue acidic phospholipids are directly related to the molecule’s ionization state in plasma at pH = 7.4. The volume of distribution calculated using the tissue volumes and in silico Kp values was 275 L. The total volumes of distribution fitted to available sets of intravenous data (42–44,63) ranged from 200 to 535 L. The metabolic clearance of metoprolol was predicted by scaling in vitro measured Km and Vmax values to in vivo conditions assuming 38 mg of microsomal protein per gram of liver and average liver weight (∼1,600 g) for a 30-year-old adult male. Variability in the enzyme expression in both intestine and liver was introduced in order to model different sets of data from different studies. This variability was 25% from the published median value of 13 pmol/mg microsomal protein in liver (50). Urinary excretion was estimated from Fup and GFR as “Fup × GFR” using the reported fraction unbound in plasma 89% (43,58) and an average glomerular filtration rate ∼110 mL/min/1.73 m2 for a 30-year-old adult male (64). The simulation resulted in 13% and 6% of metoprolol excreted in urine 70 h after i.v. and p.o. administration, respectively. Reported mean experimental values were 9% and 6% after i.v. and p.o. administration, respectively (42). Simulated Cp–time profiles using this predicted PK model provided a good match to observed Cp–time profiles after i.v. administration (Fig. 5). A comparison of predicted PK parameters with previously published values for metoprolol is shown in Table V.
Fig. 5.

Simulated and experimental Cp–time profiles of metoprolol modeled with PBPK model: a i.v. infusion administration and b colonic perfusion and IR p.o. administration
Table V.
Comparison of PK Parameters Predicted from the GastroPlus PBPK Model Using In Vitro Measurements of Metoprolol Metabolism with Published PK Parameters
| Predicted from PBPK model with in vitro measurements of metoprolol metabolism | Published values (references shown in parentheses) | |
|---|---|---|
| CLm (L/h/kg) | 0.431 | |
| CLr (L/h/kg) | 0.081 | 0.086 (42) |
| CLtotal (L/h/kg) | 0.512 | 0.7–0.97 (43,45,63,65) |
| V ss (L/kg) | 3.2 | 4.4 (43) |
| p.o. F (%) | ∼55 | 40–50 (45,63,65) |
CL m metabolic liver clearance, CL r renal clearance, CL total total systemic clearance, V ss steady-state volume of distribution, F bioavailability, PBPK physiologically based pharmacokinetic
The same PBPK model was then used in simulations of jejunal and colonic perfusion (44). To match the Cp–time profile after colonic perfusion, the absorption of metoprolol from colon was increased 28.5 times from the absorption predicted by the default ACAT model (Fig. 5). No further adjustments to absorption were required to match the Cp–time profile after jejunal perfusion. The final model thus obtained was validated by predicting the Cp–time profiles for IR p.o. formulations (oral solutions). The R2 values for the two predicted profiles were 0.8 and 0.95. The predicted profiles for IR p.o. formulations are shown in Fig. 5.
Simulations of IR p.o. doses showed ∼50–60% bioavailability. Previously published reports showed 40–55% of a metoprolol dose reaching the systemic circulation (42,63). The simulated 40–50% first pass extraction was predominantly due to liver metabolism. The small intestine is predicted to have minimal contribution to the presystemic metabolism of metoprolol (2–3%).
This model with fitted parameters for colonic absorption was then used in simulation of MR formulations with varying in vitro release rates. The simulated MR formulations are hydrophilic matrix extended release formulations using hydroxypropyl methylcellulose as the release-rate-controlling excipient (66). The three tested formulations differ in the rate of drug release from the formulation, with 100% being released in 3, 7, or 10 h in dissolution experiments using Apparatus I operating at 150 rpm and pH = 6.8. The in vivo Cp–time profiles as well as in vitro release profiles obtained from different dissolution experiments (using Apparatus I and II operating at 50–150 rpm and pH = 1.2 or 6.8) for all three formulations were used as reported in literature (46,47). Applying in vitro dissolution profiles from four different in vitro experimental conditions for the fastest releasing formulation (with 100% of drug released in 3 to 8 h in vitro depending on the conditions of dissolution experiment) showed significant differences in the predicted Cp–time profiles (Fig. 6). Among the tested in vitro dissolution conditions, Apparatus I operated at 100 or 150 rpm and pH = 6.8 best represented the in vivo release for this formulation as judged by best match between simulated and experimental Cp–time profiles when using the in vitro release as an estimate of in vivo drug release.
Fig. 6.

Simulated and experimental Cp–time profiles : a with corresponding in vitro release profiles and b used to represent in vivo release from a fast-releasing metoprolol formulation
Applying the dissolution profile from the experiment with Apparatus I operated at 150 rpm and pH = 6.8 for two other formulations with different release rates also provided a reasonable match (R2 = 0.89) for the moderate-release-rate formulation (with 100% released in about 8 h in vitro), but overpredicted the terminal elimination phase for the slow-releasing formulation (with 100% released in about 12 h in vitro; Fig. 7). The area under the plasma concentration–time curve (AUC) for the slow-releasing formulation was overpredicted by 25%.
Fig. 7.

Simulated and experimental Cp–time profiles : a corresponding in vitro release profiles and b measured using Apparatus I at pH 6.8 and 150 rpm for slow- and moderate-releasing formulations
There are several possible explanations for this result:
Higher first pass extraction for the slow-releasing formulation
Faster transit through the gastrointestinal tract
Overestimated colonic absorption/permeability
Incomplete drug release in colon from this MR formulation
In vivo release is not the same as in vitro release
These explanations are discussed below.
Higher First Pass Extraction for the Slow-Releasing Formulation
Differences in exposure (AUC) for formulations with different release rates can often be attributed to saturation of enzymatic or transport processes for fast-releasing formulations. In the case of metoprolol, this could be a saturation of intestinal and/or liver metabolism via CYP2D6, which would result in higher bioavailability for the fast-releasing formulation and lower bioavailability for the slow-releasing formulation. Lower AUC for metoprolol but higher AUC for metabolites would thus be expected with the slow-releasing formulation compared to the faster-releasing formulations. However, this was not the case. As reported in the literature (67), both metoprolol and metabolites show lower AUC with the slowest-releasing formulation. The total AUC for metoprolol and metabolites after administration of the slow-releasing formulation is only ∼83% of the total AUC for metoprolol and metabolites after administration of the fast-releasing formulation.
Faster Transit Through Gastrointestinal Tract
Both small intestine and colon transit times were adjusted to match the times previously published for a nondisintegrating tablet (68). The resulting total transit time for small intestine was 2 h and total transit time for colon was 15 h; however, significant overprediction of metoprolol concentration in plasma after ∼10 h remained. The total transit time through small intestine and colon would need to be decreased to 9.2 h (∼40% of typical intestinal transit time and ∼53% of intestinal transit time reported by Abrahamsson et al. (68)) to obtain a good match to the experimental Cp–time profile for the slow-releasing formulation.
Overestimated Colonic Absorption/Permeability
The absorption/permeability in cecum and colon was calibrated against direct colonic perfusion data (44) which showed that there is significant metoprolol absorption if the drug is present in solution. The colonic perfusion was administered by positioning the end of a double-lumen tube in cecum. If the absorption rate from the cecum was much faster than the transit time into the ascending colon, all the perfused drug might have been absorbed directly from cecum without any significant absorption from colon. Refitting the absorption from cecum and colon under this assumption resulted in an acceptable match to the Cp–time profile after colonic perfusion (R2 = 0.85) with cecum/colon absorption ratio after colonic perfusion 80:20. The match with the Cp–time profile after colonic perfusion was therefore slightly worse than the original optimization of drug absorption from cecum and colon with unrestricted colon absorption (R2 = 0.95) where the cecum/colon absorption ratio was 60:40. However, the new absorption model resulted in a slight improvement in the prediction of the Cp–time profile from the slow-releasing formulation with only 20% overprediction in AUC (Fig. 8). The simulation results for the IR p.o. and the two-faster releasing MR p.o. formulations were not significantly affected by the change in cecum/colon absorption ratio.
Fig. 8.

Simulated and experimental Cp–time profiles from colonic perfusion : a slow-releasing MR formulation and b simulated using the incomplete release hypothesis and low colon absorption hypothesis
Incomplete Drug Release in Colon
By the time this slow-releasing formulation reaches the colon, there is only about 10% of the drug left in the core of the tablet and the low volume of fluid available in colon may not be sufficient to penetrate into the formulation and dissolve the remaining amount of drug. This option should not be understood as the inability to design a MR formulation to release drug in colon, rather, only as a possibility that this particular formulation might not be releasing the drug completely. Simulation with incomplete release of drug from slow-releasing formulation resulted in improved prediction of the Cp–time profile with AUC overpredicted by only 14% (Fig. 8).
In Vitro Release is not the Same as In Vivo Release
Based on the in vitro release and Cp–time profiles for the fast-releasing formulation, it appears that Apparatus I operated at 150 rpm and pH = 6.8 sufficiently reproduces in vivo conditions. However, it is possible that assuming that in vitro release = in vivo release does not extrapolate well to formulations with slower release. A separate IVIVC was constructed by fitting an in vivo release profile to obtain the best possible match to the Cp–time profile for the slow-releasing formulation, followed by finding the correlation between the fitted in vivo release and in vitro release obtained using Apparatus I operated at 150 rpm and pH = 6.8. The final correlation function was 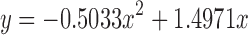 , where y represents in vivo release and x is in vitro release. Using the fitted in vivo release profile improved the shape of the simulated Cp–time profile, but the total AUC was still overpredicted by ∼25%.
, where y represents in vivo release and x is in vitro release. Using the fitted in vivo release profile improved the shape of the simulated Cp–time profile, but the total AUC was still overpredicted by ∼25%.
To summarize these options, the most likely explanations are either the incomplete release of drug from the slow-releasing formulation or a very low absorption rate of metoprolol from colon (assuming that most of the drug after colonic perfusion was being absorbed from cecum). Either one of these hypotheses is partially supported by the lower AUC for the slow-releasing formulation compared to the faster releasing formulations, as well as by direct comparison of the experimental Cp–time profiles from moderate- and slow-releasing formulations (Fig. 7). The terminal elimination phases (after ∼10 h) in vivo overlap for the two formulations, but at the early times, the slow-releasing formulation results in significantly lower plasma concentrations. The lower plasma concentration in the initial phase is a result of the smaller amount of drug that is released in the first few hours. However, if the slow-releasing formulation continued releasing the entire amount at the rate observed in vitro and the drug was getting absorbed from colon, it should produce plasma concentrations higher than the moderate formulation in the later phases (after ∼10 h). The match between experimental and simulated Cp–time profiles after colonic perfusion and MR p.o. administration of slow-releasing formulation as well as comparison of experimental and simulated AUCs from slow-releasing formulation slightly favors the incomplete release hypothesis (colonic perfusion R2 = 0.95, slow-releasing formulation R2 = 0.96, AUC overpredicted by 14%) over the low colon absorption hypothesis (colonic perfusion R2 = 0.85, slow-releasing formulation R2 = 0.94, AUC overpredicted by 20%). The results of both simulations are shown in Fig. 8.
CONCLUSIONS
Mechanistic absorption/PK models were constructed for adinazolam and metoprolol. Both models have shown applicability across a wide range of i.v. and IR p.o. doses. The adinazolam model was linked to a PD model for prediction of PD effect for different MR formulations and to design a release rate to produce a desired onset and duration of PD effect. The metoprolol model provided insights into the possible behavior of MR formulations in colon.
References
- 1.Emami J. In vitro–in vivo correlation: from theory to applications. J Pharm Pharmaceut Sci. 2006;9:31–51. [PubMed] [Google Scholar]
- 2.FDA CDER . Guidance for industry: extended release oral dosage forms: development, evaluation, and application of in vitro/in vivo correlations. Silver Spring, MD: FDA CDER; 1997. [DOI] [PubMed] [Google Scholar]
- 3.Wagnerand JG, Ayres JW. Bioavailability assessment: methods to estimate total area (AUC 0-inf) and total amount excreted and importance of blood and urine sampling scheme with application to digoxin. J Pharmacokinet Biopharm. 1977;5:533–557. doi: 10.1007/BF01061733. [DOI] [PubMed] [Google Scholar]
- 4.Wagner JG. Pharmacokinetic absorption plots from oral data alone or oral/intravenous data and an exact Loo–Riegelman equation. J Pharm Sci. 1983;72:838–842. doi: 10.1002/jps.2600720738. [DOI] [PubMed] [Google Scholar]
- 5.Proost JH. Wagner's exact Loo–Riegelman equation: the need for a criterion to choose between the linear and logarithmic trapezoidal rule. J Pharm Sci. 1985;74:793–794. doi: 10.1002/jps.2600740724. [DOI] [PubMed] [Google Scholar]
- 6.Polli JE, Crison JR, Amidon GL. Novel approach to the analysis of in vitro–in vivo relationships. J Pharm Sci. 1996;85:753–760. doi: 10.1021/js9503587. [DOI] [PubMed] [Google Scholar]
- 7.Lobenberg R, Kramer J, Shah VP, Amidon GL, Dressman JB. Dissolution testing as a prognostic tool for oral drug absorption: dissolution behavior of Glibenclamide. Pharm Res. 2000;17:439–444. doi: 10.1023/A:1007529020774. [DOI] [PubMed] [Google Scholar]
- 8.Langenbucher F. Handling of computational in vitro/in vivo correlation problems by Microsoft Excel: I. principles and some general algorithms. Eur J Pharm Biopharm. 2002;53:1–7. doi: 10.1016/S0939-6411(01)00214-4. [DOI] [PubMed] [Google Scholar]
- 9.Nielsen FS, Jacobsen LO, Magnussen T, Schou HM, Kristensen K, Lue BM, Mullertz A. Relating dissolution in biorelevant media with bioavailability for a poorly soluble compound. AAPS Annual Meeting. 2005.
- 10.Zhou R, Moench P, Heran C, Lu X, Mathias N, Faria TN, Wall DA, Hussain MA, Smith RL, Sun D. pH-dependent dissolution in vitro and absorption in vivo of weakly basic drugs: development of a canine model. Pharm Res. 2005;22:188–192. doi: 10.1007/s11095-004-1185-3. [DOI] [PubMed] [Google Scholar]
- 11.Gaynor C, Dunne A, Davis J. A comparison of the prediction accuracy of two IVIVC modeling techniques. J Pharm Sci. 2008;97:3422–3432. doi: 10.1002/jps.21220. [DOI] [PubMed] [Google Scholar]
- 12.Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev. 2001;50:S41–S67. doi: 10.1016/S0169-409X(01)00179-X. [DOI] [PubMed] [Google Scholar]
- 13.Bolger MB, Gilman TM, Fraczkiewicz R, Steere B, Woltosz WS. Predicting drug absorption by computational methods. In: Lehr CM, editor. Cell culture models of biological barriers: in-vitro test systems for drug absorption and delivery. Saarbrucken: Taylor & Francis; 2002. [Google Scholar]
- 14.Hendriksen BA, Felix MV, Bolger MB. The composite solubility versus pH profile and its role in intestinal absorption prediction. AAPS Pharm Sci. 2003;5:E4. doi: 10.1208/ps050104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolger MB, Agoram B, Fraczkiewicz R, Steere B. Simulation of absorption, metabolism, and bioavailability. In: Waterbeemd H, Lennernas H, van de Artursson P, editors. Drug bioavailability: estimation of solubility, permeability and bioavailability. New York: Wiley; 2003. [Google Scholar]
- 16.Bolger MB, Woltosz WS, Chittenden J, Fraczkiewicz G. Accurate simulation of the non-linear dose dependence for absorption of valacyclovir and amoxicillin: influence of PepT1 and HPT1 intestinal distribution. AAPS Drug Transport Meeting, Peachtree City, GA; 2003.
- 17.Tubic M, Wagner D, Spahn-Langguth H, Bolger MB, Langguth P. In silico modeling of non-linear drug absorption for the P-gp substrate talinolol and of consequences for the resulting pharmacodynamic effect. Pharm Res. 2006;23:1712–1720. doi: 10.1007/s11095-006-9020-7. [DOI] [PubMed] [Google Scholar]
- 18.Bolger MB, Fraczkiewicz R, Entzeroth M, Steere B. Concepts for in vitro profiling: drug activity, selectivity, and liability. In: Entzeroth M, editor. Exploiting chemical diversity for drug discovery. London: Royal Society of Chemistry; 2006. pp. 336–362. [Google Scholar]
- 19.Bolger MB, Fraczkiewicz R, Steere B. In silico surrogates for vivo properties: profiling for ADME and toxicological behavior. In: Entzeroth M, editor. Exploiting chemical diversity for drug discovery. London: Royal Society for Chemistry; 2006. pp. 364–381. [Google Scholar]
- 20.Dannenfelser RM, He H, Joshi Y, Bateman S, Serajuddin AT. Development of clinical dosage forms for a poorly water soluble drug I: application of polyethylene glycol-polysorbate 80 solid dispersion carrier system. J Pharm Sci. 2004;93:1165–1175. doi: 10.1002/jps.20044. [DOI] [PubMed] [Google Scholar]
- 21.De Buck SS, Sinha VK, Fenu LA, Gilissen RA, Mackie CE, Nijsen MJ. The prediction of drug metabolism, tissue distribution, and bioavailability of 50 structurally diverse compounds in rat using mechanism-based absorption, distribution, and metabolism prediction tools. Drug Metab Dispos. 2007;35:649–659. doi: 10.1124/dmd.106.014027. [DOI] [PubMed] [Google Scholar]
- 22.De Buck SS, Sinha VK, Fenu LA, Nijsen MJ, Mackie CE, Gilissen RA. Prediction of human pharmacokinetics using physiologically based modeling: a retrospective analysis of 26 clinically tested drugs. Drug Metab Dispos. 2007;35:1766–1780. doi: 10.1124/dmd.107.015644. [DOI] [PubMed] [Google Scholar]
- 23.Jones HM, Parrott N, Ohlenbusch G, Lave T. Predicting pharmacokinetic food effects using biorelevant solubility media and physiologically based modelling. Clin Pharmacokinet. 2006;54:1213–1226. doi: 10.2165/00003088-200645120-00006. [DOI] [PubMed] [Google Scholar]
- 24.Kuentz M, Nick S, Parrott N, Rothlisberger D. A strategy for preclinical formulation development using GastroPlus as pharmacokinetic simulation tool and a statistical screening design applied to a dog study. Eur J Pharm Sci. 2006;27:91–99. doi: 10.1016/j.ejps.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 25.Lindahl A, Sjoberg A, Bredberg U, Toreson H, Ungell AL, Lennernas H. Regional intestinal absorption and biliary excretion of fluvastatin in the rat: possible involvement of mrp2. Mol Pharmacol. 2004;1:347–356. doi: 10.1021/mp0499297. [DOI] [PubMed] [Google Scholar]
- 26.Parrott N, Lave T. Prediction of intestinal absorption: comparative assessment of GastroPlus and IDEA. Eur J Pharm Sci. 2002;17:51–61. doi: 10.1016/S0928-0987(02)00132-X. [DOI] [PubMed] [Google Scholar]
- 27.Brandl M, Wu X, Holper M, Hong L, Jia Z, Birudaraj R, Reddy M, Alfredson T, Tran T, Larrabee S, Hadig X, Sarma K, Washington C, Hill G, Smith DB. Physicochemical properties of the nucleoside prodrug R1626 leading to high oral bioavailability. Drug Dev Ind Pharm. 2008;34:683–691. doi: 10.1080/03639040701836636. [DOI] [PubMed] [Google Scholar]
- 28.Yu LX, Lipka E, Crison JR, Amidon GL. Transport approaches to the biopharmaceutical design of oral drug delivery system: prediction of intestinal absorption. Adv Drug Deliv Rev. 1996;19:359–376. doi: 10.1016/0169-409X(96)00009-9. [DOI] [PubMed] [Google Scholar]
- 29.Dunner D, Myers J, Khan A, Avery D, Ishiki D, Pyke R. Adinazolam—a new antidepressant: findings of a placebo-controlled, double-blind study in outpatients with major depression. J Clin Psychopharmacol. 1987;7:170–172. doi: 10.1097/00004714-198706000-00010. [DOI] [PubMed] [Google Scholar]
- 30.Venkatakrishnan K, Culm KE, Ehrenberg BL, Harmatz JS, Corbett KE, Fleishaker JC, Greenblatt DJ. Kinetics and dynamics of intravenous adinazolam, N-desmethyl adinazolam, and alprazolam in healthy volunteers. J Clin Pharmacol. 2005;45:529–537. doi: 10.1177/0091270004269105. [DOI] [PubMed] [Google Scholar]
- 31.Fleishaker JC, Friedman H, Pollock SR. Extent and variability of the first-pass elimination of adinazolam mesylate in healthy male volunteers. Pharm Res. 1991;8:162–167. doi: 10.1023/A:1015875516834. [DOI] [PubMed] [Google Scholar]
- 32.Fleishaker JC, Hulst LK, Ekernas SA, Grahnen A. Pharmacokinetics and pharmacodynamics of adinazolam and N-desmethyladinazolam after oral and intravenous dosing in healthy young and elderly volunteers. J Clin Psychopharmacol. 1992;12:403–414. doi: 10.1097/00004714-199212000-00006. [DOI] [PubMed] [Google Scholar]
- 33.Fleishaker JC, Hulst LK, Smith TC, Friedman H. Clinical pharmacology of adinazolam and N-desmethyladinazolam mesylate following single intravenous infusions of each compound in health volunteers. Eur J Clin Pharmacol. 1992;42:287–294. doi: 10.1007/BF00266350. [DOI] [PubMed] [Google Scholar]
- 34.Fleishaker JC, Phillips JP. Adinazolam pharmacokinetics and behavioral effects following administration of 20–60 mg oral doses of its mesylate salt in healthy volunteers. Psychopharmacology. 1989;99:34–39. doi: 10.1007/BF00634449. [DOI] [PubMed] [Google Scholar]
- 35.Linnoila M, Stapleton JM, Lister R, Moss H, Lane E, Granger A, Greenblatt DJ, Eckardt MJ. Effects of adinazolam and diazepam, alone and in combination with ethanol, on psychomotor and cognitive performance and on autonomic nervous system reactivity in healthy volunteers. Eur J Clin Pharmacol. 1990;38:371–377. doi: 10.1007/BF00315578. [DOI] [PubMed] [Google Scholar]
- 36.Kroboth PD, Maxwell RA, Fleishaker JC, Van Thiel DH, Smith RB. Comparison of adinazolam pharmacokinetics and effects in healthy and cirrhotic subjects. J Clin Pharmacol. 1991;31:580–586. doi: 10.1002/j.1552-4604.1991.tb03741.x. [DOI] [PubMed] [Google Scholar]
- 37.Suttle AB, Songer SS, Dukes GE, Hak LJ, Koruda M, Fleishaker JC, Brouwer KL. Ranitidine does not alter adinazolam pharmacokinetics or pharmacodynamics. J Clin Psychopharmacol. 1992;12:282–287. doi: 10.1097/00004714-199208000-00012. [DOI] [PubMed] [Google Scholar]
- 38.Fleishaker JC, Sisson TA, Sramek JJ, Conrad J, Veroff AE, Cutler NR. Psychomotor and memory effects of two adinazolam formulations assessed by a computerized neuropsychological test battery. J Clin Pharmacol. 1993;33:463–469. doi: 10.1002/j.1552-4604.1993.tb04689.x. [DOI] [PubMed] [Google Scholar]
- 39.Fleishaker JC, Wright CE. Pharmacokinetic and pharmacodynamic comparison of immediate-release and sustained-release adinazolam mesylate tablets after single- and multiple-dose administration. Pharm Res. 1992;9:457–463. doi: 10.1023/A:1015875910222. [DOI] [PubMed] [Google Scholar]
- 40.Fleishaker JC, Garzone PD, Chambers JH, Sirocco K, Weingartner H. Comparison of the spectrum of cognitive effects of alprazolam and adinazolam after single doses in healthy subjects. Psychopharmacology. 1995;120:169–176. doi: 10.1007/BF02246190. [DOI] [PubMed] [Google Scholar]
- 41.Obata T, Yamamura HI. Modulation of GABA-stimulated chloride influx into membrane vesicles from rat cerebral cortex by triazolobenzodiazepines. Life Sci. 1988;42:659–665. doi: 10.1016/0024-3205(88)90457-2. [DOI] [PubMed] [Google Scholar]
- 42.Regardh CG, Borg KO, Johansson R, Johnsson G, Palmer L. Pharmacokinetic studies on the selective α1-receptor antagonist metoprolol in man. J Pharmacokinet Biopharm. 1974;2:347–364. doi: 10.1007/BF01061407. [DOI] [PubMed] [Google Scholar]
- 43.Regardh CG, Johnsson G, Jordo L, Lundborg P, Persson B-A, Ronn O. Plasma concentrations and beta-blocking effects in normal volunteers after intravenous doses of metoprolol and propranolol. J Cardiovasc Pharmacol. 1980;2:715–723. doi: 10.1097/00005344-198011000-00002. [DOI] [PubMed] [Google Scholar]
- 44.Godbillon J, Evard D, Vidon N, Duval M, Schoeller JP, Bernier JJ, Hirtz J. Investigation of drug absorption from the gastrointestinal tract of man. III. metoprolol in the colon. Br J Clin Pharmacol. 1985;19:113S–118S. doi: 10.1111/j.1365-2125.1985.tb02751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sandberg A, Abrahamsson B, Sjogren J. Influence of dissolution rate on the extent and rate of bioavailability of metoprolol. Int J Pharm. 1991;68:167–177. doi: 10.1016/0378-5173(91)90139-F. [DOI] [Google Scholar]
- 46.Eddington ND, Marroum P, Uppoor R, Hussain A, Augsburger L. Development and internal validation of an in vitro–in vivo correlation for a hydrophilic metoprolol tartrate extended release tablet formulation. Pharm Res. 1998;15:466–473. doi: 10.1023/A:1011988601696. [DOI] [PubMed] [Google Scholar]
- 47.Eddington ND, Marroum P, Uppoor R, Hussain A, Augsburger L. Development and internal validation of an in vitro–in vivo correlation for a hydrophilic metoprolol tartrate extended release tablet formulation—erratum. Pharm Res. 1998;15:1320. doi: 10.1023/A:1011988601696. [DOI] [PubMed] [Google Scholar]
- 48.Rodgers T, Leahy D, Rowland M, Rodgers T, Leahy D, Rowland M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci. 2005;94:1259–1276. doi: 10.1002/jps.20322. [DOI] [PubMed] [Google Scholar]
- 49.Rodgers T, Rowland M, Rodgers T, Rowland M. Physiologically-based pharmacokinetic modeling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95:1238–1257. doi: 10.1002/jps.20502. [DOI] [PubMed] [Google Scholar]
- 50.Madani S, Paine MF, Lewis L, Thummel KE, Shen DD. Comparison of CYP2D6 content and metoprolol oxidation between microsomes isolated from human lives and small intestines. Pharm Res. 1999;16:1199–1205. doi: 10.1023/A:1018989211864. [DOI] [PubMed] [Google Scholar]
- 51.Ito K, Houston JB. Prediction of human drug clearance from in vitro and preclinical data using physiologically based and empirical approaches. Pharm Res. 2005;22:103–112. doi: 10.1007/s11095-004-9015-1. [DOI] [PubMed] [Google Scholar]
- 52.Schoene B, Fleischmann RA, Remmer H, von Oldershausen HF. Determination of drug metabolizing enzymes in needle biopsies of human liver. Eur J Clin Pharmacol. 1972;4:65–73. doi: 10.1007/BF00562499. [DOI] [PubMed] [Google Scholar]
- 53.Knaak JB, al-Bayati MA, Raabe OG, Blancato JN. Development of in vitro Vmax and Km values for the metabolism of isofenphos by P-450 liver enzymes in animals and human. Toxicol Appl Pharmacol. 1993;120:106–113. doi: 10.1006/taap.1993.1092. [DOI] [PubMed] [Google Scholar]
- 54.Iwatsubo T, Hirota N, Ooie T, Suzuki H, Shimada N, Chiba K, Ishizaki T, Green CE, Tyson CA, Sugiyama Y. Prediction of in vivo drug metabolism in the human liver from in vitro metabolism data. Pharmacol Ther. 1997;73:147–171. doi: 10.1016/S0163-7258(96)00184-2. [DOI] [PubMed] [Google Scholar]
- 55.Lipscomb JC, Fisher JW, Confer PD, Byczkowski JZ. In vitro to in vivo extrapolation for trichloroethylene metabolism in humans. Toxicol Appl Pharmacol. 1998;152:376–387. doi: 10.1006/taap.1998.8485. [DOI] [PubMed] [Google Scholar]
- 56.Wilson ZE, Rostami-Hodjegan A, Burn JL, Tooley A, Boyle J, Ellis SW, Tucker GT. Inter-individual variability in levels of human microsomal protein and hepatocellularity per gram of liver. Br J Clin Pharmacol. 2003;56:433–440. doi: 10.1046/j.1365-2125.2003.01881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang PH, Lien EJ. Effects of different buffer species on partition coefficients of drugs used in quantitative structure–activity relationships. J Pharm Sci. 1980;69:662–668. doi: 10.1002/jps.2600690614. [DOI] [PubMed] [Google Scholar]
- 58.Tubic-Grozdanis M, Bolger MB, Langguth P. Application of gastrointestinal simulation for extensions for biowaivers of highly permeable compounds. AAPS J. 2008;10:213–226. doi: 10.1208/s12248-008-9023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kasim NA, Whitehouse M, Ramachandran C, Bermejo M, Lennernas H, Hussain AS, Junginger HE, Stavchansky SA, Midha KK, Shah VP, Amidon GL. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol Pharm. 2004;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 60.Carter CS, Fawcett J, Hertzman M, Papp LA, Jones W, Patterson WM, Swinson RP, Weise CC, Maddock RJ, Denahan AQ. Adinazolam-SR in panic disorder with agoraphobia: relationship of daily dose to efficacy. J Clin Psychiatry. 1995;56:202–210. [PubMed] [Google Scholar]
- 61.Haefely W, Kulcsar A, Mohler H, Pieri L, Polc P, Schaffner R. Possible involvement of GABA in the central actions of benzodiazepines. Adv Biochem Psychopharmacol. 1975;14:131–151. [PubMed] [Google Scholar]
- 62.Lahti RA, Sethy VH, Barsuhn C, Hester JB. Pharmacological profile of the antidepressant adinazolam, a triazolobenzodiazepine. Neuropharmacology. 1983;22:1277–1282. doi: 10.1016/0028-3908(83)90200-9. [DOI] [PubMed] [Google Scholar]
- 63.Regardh CG, Landahl S, Larsson M, Lundborg P, Steen B, Hoffmann K-J, Lagerstrom P-O. Pharmacokinetics of metoprolol and its metabolite α-OH-metoprolol in healthy, non-smoking, elderly individuals. Eur J Clin Pharmacol. 1983;24:221–226. doi: 10.1007/BF00613821. [DOI] [PubMed] [Google Scholar]
- 64.Stevens LA, Levey AS. Frequently asked questions about GFR estimates, national kidney foundation. New York, NY: National Kidney Foundation; 2007. pp. 1–17. [Google Scholar]
- 65.Larsson M, Landahl S, Lundborg P, Regardh CG. Pharmacokinetics of metoprolol in healthy, elderly, non-smoking individuals after a single dose and two weeks of treatment. Eur J Clin Pharmacol. 1984;27:217–222. doi: 10.1007/BF00544048. [DOI] [PubMed] [Google Scholar]
- 66.Nellore RV, Rekhi GS, Hussain A, Tillman LG, Augsburger L. Development of metoprolol tartrate extended-release matrix tablet formulations for regulatory policy consideration. J Contr Rel. 1998;50:247–256. doi: 10.1016/S0168-3659(97)00141-7. [DOI] [PubMed] [Google Scholar]
- 67.Sirisuth N, Eddington ND. The influence of first pass metabolism on the development and validation of an IVIVC for metoprolol extended release tablets. Eur J Pharm Biopharm. 2002;53:301–309. doi: 10.1016/S0939-6411(01)00248-X. [DOI] [PubMed] [Google Scholar]
- 68.Abrahamsson B, Alpsten M, Jonsson UE, Lundberg PJ, Sandberg A, Sundgren M, Svenheden A, Tolli J. Gastro-intestinal transit of a multiple-unit formulation (metoprolol CR/ZOK) and a non-disintegrating tablet with the emphasis on colon. Int J Pharm. 1996;140:229–235. doi: 10.1016/0378-5173(96)04604-2. [DOI] [Google Scholar]