

Abstract
For the accurate prediction of in vivo hepatic clearance or drug–drug interaction potential through in vitro microsomal metabolic data, it is essential to evaluate the fraction unbound in hepatic microsomal incubation media. Here, a structure-based in silico predictive model of the nonspecific binding (fumic, fraction unbound in hepatic microsomes) for 86 drugs was successfully developed based on seven selected molecular descriptors. The R2 of the predicted and observed log((1 − fumic)/fumic) for the training set (n = 64) and test set (n = 22) were 0.82 and 0.85, respectively. The average fold error (AFE, calculated by fumic rather than log((1 − fumic)/fumic)) of the in silico model was 1.33 (n = 86). The predictive capability of fumic for neutral drugs compared well to that for basic compounds (R2 = 0.82, AFE = 1.18 and fold error values were all below 2, except for felodipine and progesterone) in our model. This model appears to perform better for neutral compounds when compared to models previously published in the literature. Therefore, this in silico model may be used as an additional tool to estimate fumic and for predicting in vivo hepatic clearance and inhibition potential from in vitro hepatic microsomal studies.
Key words: fraction unbound in hepatic microsomes, in silico prediction, molecular descriptors
INTRODUCTION
Significant progress have been made recently in the prediction of in vivo hepatic clearance (1–3) and drug–drug interaction potential (4–6). The determination of intrinsic clearance (CLint) and inhibition constant (Ki) through in vitro microsomal incubation can provide a basis for these predictions. However, lipophilic drugs tended to bind nonspecifically to microsomal phospholipids, resulting in an underestimation of CLint (7–9) or an overestimation of Ki (10–12). Consequently, in vivo hepatic clearance and the extent of inhibitory drug interactions were often underpredicted.
Investigators have tried to use relative low microsomal protein concentration to avoid the nonspecific binding (13). However, relative high concentrations (1 to 2 mg/mL) were still needed when studying phase II metabolic reactions (14) and in vitro assessment of the time-dependent inhibition potential (15). As such, it is essential to correct the metabolic kinetic parameters (CLint and Ki) by the unbound fraction to microsomes (fumic) in order to ensure accurate pharmacokinetic estimation of potential drug candidates. Unfortunately, currently available experimental methods for measuring the fumic are relatively labor- and time-consuming.
In order to avoid these experimental studies, in silico prediction of fumic has gained great interest recently. Austin et al. (16) reported a linear relationship between log((1 − fumic)/fumic) and logP/D (logP for bases, while logD7.4 for acids and neutrals) of 56 drugs (R2 = 0.82). Hallifax and Houston (17) proposed that the relationship between logP/D and log((1 − fumic)/fumic) was nonlinear. They concluded that the nonlinear empirical equation gave more unbiased predictions of fumic for drugs with low binding affinity (fumic > 0.9) when compared with the model by Austin et al. Later, Gertz et al. addressed the limitations of these empirical predictive tools and their applicability for fumic predictions over a range of lipophilicity and microsomal protein concentrations (18). They concluded that the accuracy of fumic predictions for highly lipophilic drugs was poor by both equations, while the Hallifax equation provided more accurate fumic predictions on average.
Interestingly, Sykes et al. (19) reanalyzed the data reported by Austin et al. and found that the logP values clearly correlated well with the transformed fumic (R2 = 0.90) for bases but less predictive for neutrals (R2 = 0.34) and acids (R2 = 0.10). They obtained good discrimination between drugs classified as strong binders (experimental fumic < 0.50) and those with a lower degree of binding (experimental fumic > 0.50) by molecular modeling approaches.
Recently, Gao et al. (20) developed a quantitative in silico model correlating fumic of 1,223 drug-like molecules with two-dimensional molecular descriptors. These investigators demonstrated that lipophilicity was the most important molecular property contributing to fumic in this high performance model. However, the information of the original dataset was not open to the public. Therefore, a model not only with high prediction accuracy but also with open-sourced dataset would be useful for researchers in assessing quantitative structure vs. fumic relationships.
In this study, quantitative structure–fumic relationship was constructed just based on molecular descriptors for a dataset of 86 drugs covering a large range of molecular properties. Molecular descriptors were calculated using TSARTM software version 3.3 (Accelrys Inc.) (21), preADMET (22), and SciFinder Scholar 2007 (23). Then, the feature selection was performed by stepwise regression, and an in silico model was established with multiple linear regression (MLR) method. The principal objectives of the study were, therefore, (1) to develop a quantitative relationship between the molecular structure descriptors and log((1 − fumic)/fumic); (2) to estimate the predictive accuracy of in silico model, and (3) to understand what structural factors determining fumic.
MATERIALS AND METHODS
Data Collection
The observed fumic values of 86 drugs were obtained from the literatures as described in Table I. These fumic values were measured at the microsomal protein concentration of 1 mg/mL or converted to fumic values at 1 mg/mL based on the equation proposed by Austin et al. (16). The fumic values of each drug were transformed to log((1 − fumic)/fumic). As shown in Fig. 1, the fumic values of 86 drugs did not follow a normal distribution. The transformation of fumic to log((1 − fumic)/fumic) yielded a more desirable distribution and could reduce unequal error variances simultaneously. Therefore, the observed log((1 − fumic)/fumic) was considered as the dependent variable in the model construction.
Table I.
Characteristics of, and Results Obtained for, the 86 Drugs Studied to Establish the Present In Silico Model
| No. | Dataset | Drugs | log((1 − fumic)/fumic) | Chemical class | Observed fumic | Predicted fumic | Fold error | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | Training | Bumetanide | −1.0607 | A | 0.92 | 0.90 | 1.02 | (16) |
| 2 | Training | Cerivastatin | −0.2688 | A | 0.65 | 0.75 | 1.15 | (16) |
| 3 | Training | Cinoxacin | −1.0607 | A | 0.92 | 0.99 | 1.08 | (16) |
| 4 | Training | Diclofenac | −1.5097 | A | 0.97 | 0.81 | 1.20 | (16) |
| 5 | Training | Emodin | 0.6297 | A | 0.19 | 0.12 | 1.57 | (18) |
| 6 | Training | Gemfibrozil | −0.5248 | A | 0.77 | 0.67 | 1.15 | (18) |
| 7 | Test | Glipizide | −1.3802 | A | 0.96 | 0.96 | 1.00 | (16) |
| 8 | Training | Glyburide | −0.6585 | A | 0.82 | 0.85 | 1.04 | (16) |
| 9 | Training | Ibuprofen | −1.0048 | A | 0.91 | 0.85 | 1.07 | (16) |
| 10 | Test | Indomethacin | −0.6297 | A | 0.81 | 0.93 | 1.15 | (16) |
| 11 | Test | Ketoprofen | −1.0607 | A | 0.92 | 0.92 | 1.00 | (16) |
| 12 | Training | Losartan | −0.9542 | A | 0.9 | 0.78 | 1.15 | (16) |
| 13 | Test | Mycophenolic acid | −0.5754 | A | 0.79 | 0.89 | 1.12 | (18) |
| 14 | Training | Oxaprozin | −0.8256 | A | 0.87 | 0.79 | 1.10 | (16) |
| 15 | Test | Piroxicam | −1.0607 | A | 0.92 | 0.88 | 1.04 | (16) |
| 16 | Training | Repaglinide | −0.4320 | A | 0.73 | 0.68 | 1.08 | (18) |
| 17 | Test | Rosiglitazone | −0.4102 | A | 0.72 | 0.87 | 1.21 | (18) |
| 18 | Test | Sulfadoxine | −1.5097 | A | 0.97 | 0.98 | 1.01 | (16) |
| 19 | Training | Sulindac | −0.7884 | A | 0.86 | 0.87 | 1.01 | (16) |
| 20 | Training | Tenidap | −0.1581 | A | 0.59 | 0.86 | 1.45 | (19) |
| 21 | Test | Tenoxicam | −1.5097 | A | 0.97 | 0.85 | 1.14 | (19) |
| 22 | Training | Tolbutamide | −1.5097 | A | 0.97 | 0.97 | 1.00 | (16) |
| 23 | Training | Tolmetin | −1.1950 | A | 0.94 | 0.99 | 1.05 | (16) |
| 24 | Test | Warfarin | −1.1950 | A | 0.94 | 0.68 | 1.38 | (16) |
| 25 | Test | Amiodarone | 2.6981 | B | 0.002 | 0.00 | 1.86 | (16) |
| 26 | Training | Amitriptyline | 1.0548 | B | 0.081 | 0.22 | 2.77 | (16) |
| 27 | Training | Betaxolol | −0.2126 | B | 0.62 | 0.63 | 1.02 | (16) |
| 28 | Training | Buspirone | −0.7533 | B | 0.85 | 0.40 | 2.10 | (18) |
| 29 | Training | Carvedilol | 0.9542 | B | 0.1 | 0.15 | 1.53 | (18) |
| 30 | Test | Chlorpromazine | 0.9080 | B | 0.11 | 0.11 | 1.02 | (16) |
| 31 | Test | Clomipramine | 1.4034 | B | 0.038 | 0.08 | 2.07 | (16) |
| 32 | Training | Clozapine | 0.4543 | B | 0.26 | 0.51 | 1.95 | (16) |
| 33 | Training | Codeine | −1.3802 | B | 0.96 | 0.90 | 1.07 | (18) |
| 34 | Training | Dextromethorphan | −0.4102 | B | 0.72 | 0.38 | 1.90 | (18) |
| 35 | Training | Diltiazerm | −0.7884 | B | 0.86 | 0.33 | 2.59 | (16) |
| 36 | Test | Diphenhydramine | −0.3889 | B | 0.71 | 0.48 | 1.48 | (16) |
| 37 | Training | Ezlopitant | 1.6902 | B | 0.02 | 0.01 | 1.88 | (19) |
| 38 | Training | Fluvoxamine | 0.4320 | B | 0.27 | 0.35 | 1.30 | (28) |
| 39 | Training | Imipramine | 0.7202 | B | 0.16 | 0.26 | 1.61 | (16) |
| 40 | Training | Ketamine | 0.0174 | B | 0.49 | 0.56 | 1.15 | (19) |
| 41 | Test | Levallorphan | −0.7202 | B | 0.84 | 0.37 | 2.29 | (18) |
| 42 | Training | Lorcainide | −0.0348 | B | 0.52 | 0.24 | 2.13 | (19) |
| 43 | Test | Mibefradil | 1.5097 | B | 0.03 | 0.03 | 1.09 | (18) |
| 44 | Training | Miconazole | 1.5097 | B | 0.03 | 0.08 | 2.73 | (28) |
| 45 | Training | Naloxone | −0.8256 | B | 0.87 | 0.73 | 1.19 | (18) |
| 46 | Test | Nicardipine | 1.3802 | B | 0.04 | 0.12 | 3.02 | (19) |
| 47 | Training | Phenytoin | −0.6297 | B | 0.81 | 0.88 | 1.08 | (28) |
| 48 | Training | Promethazine | 0.9080 | B | 0.11 | 0.16 | 1.46 | (16) |
| 49 | Training | Propranolol | 0.1047 | B | 0.44 | 0.58 | 1.31 | (16) |
| 50 | Training | Quinidine | −0.3680 | B | 0.7 | 0.45 | 1.57 | (16) |
| 51 | Training | Quinine | 0.2499 | B | 0.36 | 0.45 | 1.25 | (28) |
| 52 | Training | Tamoxifen | 2.5216 | B | 0.003 | 0.01 | 2.52 | (16) |
| 53 | Training | Terfenadine | 1.6902 | B | 0.02 | 0.03 | 1.65 | (18) |
| 54 | Training | Thioridazine | 2.0418 | B | 0.009 | 0.03 | 3.00 | (16) |
| 55 | Training | Trimeprazine | 1.0548 | B | 0.081 | 0.12 | 1.54 | (16) |
| 56 | Test | Verapamil | 0.2311 | B | 0.37 | 0.17 | 2.24 | (16) |
| 57 | Training | Zolpidem | −0.8653 | B | 0.88 | 0.69 | 1.28 | (19) |
| 58 | Training | 2-Ethoxybenzemide | −1.6902 | N | 0.98 | 0.92 | 1.06 | (16) |
| 59 | Training | Albendazole | −0.1047 | N | 0.56 | 0.68 | 1.22 | (16) |
| 60 | Test | Aldosterone | −0.1761 | N | 0.6 | 0.85 | 1.41 | (28) |
| 61 | Training | Alprazolam | −0.6585 | N | 0.82 | 0.77 | 1.06 | (16) |
| 62 | Training | Amobarbital | −1.5097 | N | 0.97 | 0.94 | 1.03 | (16) |
| 63 | Training | Androstenedione | 0.0174 | N | 0.49 | 0.57 | 1.17 | (28) |
| 64 | Training | Caffeine | −1.9956 | N | 0.99 | 0.99 | 1.00 | (28) |
| 65 | Training | Carbamazepine | −0.8256 | N | 0.87 | 0.84 | 1.04 | (16) |
| 66 | Training | Colchicine | −1.1950 | N | 0.94 | 0.89 | 1.06 | (16) |
| 67 | Test | Diazepam | −0.2881 | N | 0.66 | 0.55 | 1.20 | (16) |
| 68 | Training | Felodipine | 1.1950 | N | 0.06 | 0.16 | 2.72 | (18) |
| 69 | Training | Fluconazole | −1.6902 | N | 0.98 | 0.97 | 1.01 | (28) |
| 70 | Training | Hexobarbital | −1.3802 | N | 0.96 | 0.93 | 1.03 | (16) |
| 71 | Test | Isradipine | 0.2881 | N | 0.34 | 0.38 | 1.12 | (16) |
| 72 | Training | Mebendazole | −0.3680 | N | 0.7 | 0.76 | 1.09 | (16) |
| 73 | Training | Methocarbamol | −0.7202 | N | 0.84 | 0.94 | 1.12 | (16) |
| 74 | Training | Methohexital | −0.7884 | N | 0.86 | 0.89 | 1.04 | (19) |
| 75 | Training | Methoxsalen | −0.9080 | N | 0.89 | 0.82 | 1.09 | (16) |
| 76 | Training | Metyrapone | −1.5097 | N | 0.97 | 0.93 | 1.05 | (16) |
| 77 | Test | Midazolam | −0.0696 | N | 0.54 | 0.47 | 1.16 | (18) |
| 78 | Training | Nifedipine | −0.3680 | N | 0.7 | 0.71 | 1.02 | (18) |
| 79 | Test | Omeprazole | −0.8256 | N | 0.87 | 0.73 | 1.19 | (19) |
| 80 | Training | Oxazepam | −0.4102 | N | 0.72 | 0.66 | 1.08 | (18) |
| 81 | Training | Phensuximide | −0.4771 | N | 0.75 | 0.97 | 1.29 | (16) |
| 82 | Training | Prednisone | −0.1047 | N | 0.56 | 0.64 | 1.15 | (16) |
| 83 | Training | Progesterone | 0.7884 | N | 0.14 | 0.30 | 2.15 | (28) |
| 84 | Training | Simvastatin | 1.1950 | N | 0.06 | 0.09 | 1.49 | (18) |
| 85 | Training | Testosterone | 0.0174 | N | 0.49 | 0.42 | 1.17 | (28) |
| 86 | Training | Triazolam | −0.5497 | N | 0.78 | 0.75 | 1.04 | (16) |
Fig. 1.

Distribution of fumic and log((1 − fumic)/fumic) for 86 drugs
Calculation of Molecular Descriptors
The 2D structures of 86 drugs were searched in SciFinder Scholar 2007 and the mol files were saved for further calculation. Then, the molecular descriptors that were known to influence almost all pharmacokinetic properties were selected as original independent variables. A set of 32 descriptors was obtained from TSAR 3.3, preADMET online, and SciFinder Scholar 2007, including: molecular refractivity, cosmic torsional/electrostatic/total energy, number of atoms/halogen atoms/heteroatoms, heat of formation, energy of the lowest unoccupied molecular orbital (LUMO), energy of the highest occupied molecular orbital (HOMO), ΔE (LUMO–HOMO), number of primary/secondary/tertiary amine groups, number of carboxylic acid groups, number of single/double/aromatic bonds, total absolute atomic charge, total/aromatic rings, number of negatively/positively charged groups, rigid/rotable bonds, number of hydrogen bond acceptors/donors, logD7, logP, molecular weight, mean net charge per molecule of the compounds (fi) (24), and polar surface area.
Feature Selection
As expected, only some of the 32 descriptors are significantly correlated with log((1 − fumic)/fumic). Furthermore, many of the descriptors are intercorrelated, which has a negative effect on the accuracy and interpretability of the final quantitative model. Therefore, stepwise regression method was employed to perform the feature selection process in this study.
Model Development and Evaluation
MLR analysis was applied to develop the in silico model. In order to examine the predictive power and robustness of our model, the entire dataset should be subdivided into training and test set. In general, there are three methods for the selection of training and test set: (1) selection based on a random manner; (2) selection based on clusters of the dependent variable; (3) selection based on clusters of factor scores of the descriptor space along with or without the biological activity values. Due to the skew distribution of fumic, the entire dataset was categorized into training set (n = 64) and test set (n = 22) by the cluster analysis of log((1 − fumic)/fumic). The whole range of log((1 − fumic)/fumic) was divided into bins, and compounds belonging to each bin were randomly assigned to the training or test set. Meanwhile, leave-one-out (LOO) cross-validation was performed. Then, R2 and Q2 resulted from LOO (Q2LOO) were calculated to evaluate the model predictability.
Two other commonly employed accuracy test criteria, the fold error and the average fold error (AFE), were used to evaluate the predictive accuracy, as represented by Eqs. 1 and 2, respectively. The percentages of drugs with the fold error more than two (E2-fold) and three (E3-fold) were calculated to estimate the accuracy of the model in our study, respectively. A prediction is usually thought to be successful if the value of AFE is less than two (25).
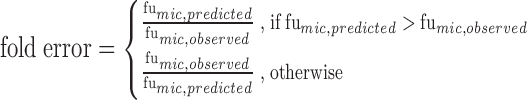 |
1 |
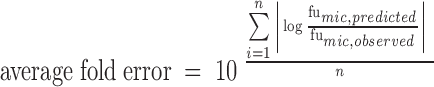 |
2 |
RESULTS AND DISCUSSION
The Predictive Performance and Accuracy of the Model
Seven descriptors were chosen via the feature selection to construct the in silico model. Then, the model for the training set was built with 64 drugs, as represented by Eq. 3.
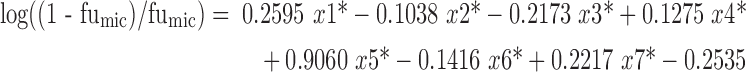 |
3 |
where x1 is the cosmic electrostatic energy; x2 is the number of aromatic bonds; x3 is the number of negatively charged groups; x4 is the number of positively charged groups; x5 is logP; x6 is the mean net charge per molecule of the compounds; x7 is polar surface area (PSA). All of the selected descriptors, the values of which can be obtained directly from the authors, are standardized to ensure that all descriptors had equally determinant strength affecting log((1 − fumic)/fumic). The standardized values with a mean value of zero and a variance of unity are represented as “x*” in Eq. 3.
The correlation between predicted log((1 − fumic)/fumic) and observed log((1 − fumic)/fumic) from in silico model is shown in Fig. 2. It is seen that the in silico model exhibits high predictive performance: for the training set, n = 64, R2 = 0.82 (R2 = 0.85 for test set), Q2LOO = 0.75, RMSE = 0.45, F = 36.31, p < 0.0001, the slope equal to unity and the intercept to zero (the slope is 0.94 and the intercept is −0.08 for test set).
Fig. 2.

Correlation between the predicted and observed log(1 − fumic)/ fumic) for acidic, basic, and neutral drugs (empty circles acids, n = 24, y = 0.78x − 0.19, R 2 = 0.43; multiplication symbols bases, n = 33, y = 0.73x + 0.20, R 2 = 0.82; empty triangles neutrals, n = 29, y = 0.76x − 0.14, R 2 = 0.82; training set (black symbols): n = 64, y = 1x, R 2 = 0.82; test set (red symbols): n = 22, y = 0.94x − 0.08, R 2 = 0.85))
The observed and predicted values of fumic, and the fold error values of 86 drugs are shown in Table I. As can be seen, 75% of drugs are found with fold error < 2 and only 2% of drugs with fold error > 3 and AFE = 1.33 (Table II) in our model. For training set, 82% of drugs are found with fold error < 2 and only 5% of drugs (one drug) with fold error > 3 and AFE = 1.34. And for test set, 86% of drugs are found with fold error < 2 and only 0% of drugs with fold error > 3 and AFE = 1.33. Therefore, the fumic can be predicted accurately by our model.
Table II.
The Predictive Accuracy of the Present In Silico Model vs. Literature Models
| Chemical class | No. | Present study | Austin et al. model | Hallifax-Houston model | |||||
|---|---|---|---|---|---|---|---|---|---|
| E 2-fold (%) | E 3-fold (%) | AFE | R 2 | AFE | R 2 | AFE | R 2 | ||
| Total | 86 | 15 | 2 | 1.33 | 0.85 | 1.47 | 0.75 | 1.33 | 0.79 |
| A | 24 | 0 | 0 | 1.12 | 0.43 | 1.16 | 0.49 | 1.12 | 0.56 |
| B | 33 | 33 | 6 | 1.68 | 0.82 | 1.92 | 0.79 | 1.68 | 0.76 |
| N | 29 | 7 | 0 | 1.18 | 0.82 | 1.34 | 0.61 | 1.18 | 0.66 |
Predictive Differences among Acids, Bases, and Neutrals
Figure 2 also describes the respective correlations between predicted and observed values of log((1 − fumic)/fumic) for acids, bases, and neutral compounds. As stated earlier, the good prediction of bases is more easily achieved than neutral compounds and acids. Our model predicted fumic for bases well (R2 = 0.82, AFE = 1.68). Furthermore, prediction of fumic for neutral drugs was comparable (R2 = 0.82, AFE = 1.18), which might be a positive feature of this model. Unfortunately, for acids, the correlation between log((1 − fumic)/fumic) and the seven descriptors was still poor (as shown in Fig. 2; R2 = 0.43). However, the fold error values of acids were all below 2 (Table II), indicating that the predictive accuracy of acids in our model might still be useful in some circumstances. The slope of the fitted line (Fig. 2; 0.78) for acids was similar to bases (0.73) and neutral compounds (0.76). These findings indicate that the prediction of fumic for acids in our model is still reasonable. In fact, the poor correlation of acids was probably due to the relative narrow distribution of observed fumic values (or the log((1 − fumic)/fumic) values). As can be seen in Table I, except for emodin, the range of the log((1 − fumic)/fumic) for acids is from −1.5097 to −0.1581, with a log unit span of 1.4, (most of the fumic values are within the range of 0.6–0.9). In contrast, for bases and neutral compounds, the log unit spans of the log((1 − fumic)/fumic) are 4.0 and 3.2, respectively. The relative low nonspecific binding of acids to hepatic microsomes likely results in the skew distribution of the fumic and the poor correlation between log((1 − fumic)/fumic) and the selected seven descriptors.
Identification of Important Molecular Descriptors Affecting Fumic
Our results shown in Eq. 3 suggest that the descriptors chosen strongly correlate with fumic, thus, allowing some mechanistic interpretations of the model. In general, these molecular descriptors relate to molecular lipophilicity, charge state, flexibility, polarity, and extent of ionization at pH 7.4, as shown in Fig. 3.
Fig. 3.

Coefficients plot of the seven selected molecular descriptors
The molecular mechanism of nonspecific binding is presently unclear, but it is believed to depend on the lipophilicity and the electronic charge. The main binding contributors can be divided into non-electrostatic and electrostatic terms, wherein the non-electrostatic contributions include lipophilic interactions, van der Waals interactions, and translational, rotational, and configurational entropies (26).
As can be expected, the extent of microsomal binding generally increases with increasing lipophilicity of the drug. Especially, as the main structural contributor, logP is positively correlated with fumic (Eq. 3), consistent with the above analysis. The cosmic electrostatic energy, parameter x6 (fi), the number of positively charged groups and the number of negatively charged groups are descriptors representing the above electrostatic term contributing to the nonspecific binding. As shown in Eq. 3 and Fig. 3, the cosmic electrostatic energy is the second important descriptor in our model. It is energy descriptor accounting for the noncovalent interaction potential energy, which determines the binding affinity of a molecule to the pertinent receptor(s). The parameter x6 (fi) is calculated from the pKa and pH7.4, and its value is equal to the ionization fraction for compounds at pH7.4 (24). Thus, it denotes the contribution of electrostatic interaction to the nonspecific binding based on the ionization of the compounds. However, the basic compounds clearly exhibit enhanced binding over neutral or acidic compounds with similar lipophilicity. This enhanced phospholipid binding of bases is thought to be due to a favorable electrostatic interaction between the protonated base and phosphate groups of the phospholipids (27). The negative charges for acidic drugs at pH 7.4 would likely limit their nonspecific binding. This conclusion can be used to explain the positive effect of the x4 (the number of positively charged groups) on fumic, and the negative effect of the x3 (the number of negatively charged groups) on fumic (Eq. 3). The PSA and the number of aromatic bonds are two other contributors in our model.
Comparison between In Silico and Published Models
The performance of the present model vs. the models published by Austin et al. and Hallifax-Houston were compared (Table II).
In general, our model compared favorably to these models for basic and neutral compounds but fared equally inadequately for acidic compounds. The present model differs, however, in its approach in that it utilizes more structural specific parameters such as the number of positively or negatively charged groups, the cosmic electrostatic energy and PSA, etc., in addition to log P and log D. The involvement of these parameters provided additional insights into the molecular mechanisms of nonspecific binding of drugs to hepatic microsomes, especially for the electrostatic interaction.
CONCLUSIONS
A structure-based in silico model was developed successfully for the prediction of the nonspecific binding of drugs to hepatic microsomes. Especially, the prediction of fumic for neutral drugs demonstrated similar capability to that for basic drugs (R2 = 0.82, AFE = 1.18 and fold error values were all below 2, except for felodipine and progesterone). The lipophilicity, charge state, and the extent of ionization at pH 7.4 were identified as important properties affecting fumic. One obvious weakness of the present model is the skew distribution of fumic in the entire dataset (most of the compounds were in the range of fumic > 0.7, especially for the acids). A larger dataset, composed of drugs with uniform distribution of fumic values, is necessary for accurate fumic prediction and for further reliable evaluation of the free clearance and drug–drug interaction.
Acknowledgements
We are thankful to Accelrys Inc. for providing 1-month free evaluation of TSAR software in 2007.
Contributor Information
Jin Sun, Phone: +86-24-23986321, FAX: +86-24-23986321, Email: sunjin66@21cn.com.
Zhonggui He, Phone: +86-24-23986321, FAX: +86-24-23986321, Email: hezhonggui@gmail.com, Email: hezhonggui@gmail.com.
References
- 1.Ito K, Houston JB. Prediction of human drug clearance from in vitro and preclinical data using physiologically based and empirical approaches. Pharm Res. 2005;22:103–12. doi: 10.1007/s11095-004-9015-1. [DOI] [PubMed] [Google Scholar]
- 2.Rostami-Hodjegan A, Tucker GT. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat Rev Drug Discov. 2007;6:140–8. doi: 10.1038/nrd2173. [DOI] [PubMed] [Google Scholar]
- 3.Miners JO, Smith PA, Sorich MJ, McKinnon RA, Mackenzie PI. Predicting human drug glucuronidation parameters: application of in vitro and in silico modeling approaches. Annu Rev Pharmacol Toxicol. 2004;44:1–25. doi: 10.1146/annurev.pharmtox.44.101802.121546. [DOI] [PubMed] [Google Scholar]
- 4.Ito K, Brown HS, Houston JB. Database analyses for the prediction of in vivo drug–drug interactions from in vitro data. Br J Clin Pharmacol. 2004;57:473–86. doi: 10.1111/j.1365-2125.2003.02041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Polasek TM, Miners JO. Quantitative prediction of macrolide drug–drug interaction potential from in vitro studies using testosterone as the human cytochrome P4503A substrate. Eur J Clin Pharmacol. 2006;62:203–8. doi: 10.1007/s00228-005-0091-x. [DOI] [PubMed] [Google Scholar]
- 6.Uchaipichat V, Winner LK, Mackenzie PI, Elliot DJ, Williams JA, Miners JO. Quantitative prediction of in vivo inhibitory interactions involving glucuronidated drugs from in vitro data: the effect of fluconazole on zidovudine glucuronidation. Br J Clin Pharmacol. 2006;61:427–39. doi: 10.1111/j.1365-2125.2006.02588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grime K, Riley RJ. The impact of in vitro binding on in vitro-in vivo extrapolations, projections of metabolic clearance and clinical drug–drug interactions. Curr Drug Metab. 2006;7:251–64. doi: 10.2174/138920006776359266. [DOI] [PubMed] [Google Scholar]
- 8.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27:1350–9. [PubMed] [Google Scholar]
- 9.Riley RJ, McGinnity DF, Austin RP. A unified model for predicting human hepatic, metabolic clearance from in vitro intrinsic clearance data in hepatocytes and microsomes. Drug Metab Dispos. 2005;33:1304–11. doi: 10.1124/dmd.105.004259. [DOI] [PubMed] [Google Scholar]
- 10.Margolis JM, Obach RS. Impact of nonspecific binding to microsomes and phospholipid on the inhibition of cytochrome P4502D6: implications for relating in vitro inhibition data to in vivo drug interactions. Drug Metab Dispos. 2003;31:606–11. doi: 10.1124/dmd.31.5.606. [DOI] [PubMed] [Google Scholar]
- 11.Brown HS, Galetin A, Hallifax D, Houston JB. Prediction of in vivo drug–drug interactions from in vitro data : factors affecting prototypic drug–drug interactions involving CYP2C9, CYP2D6 and CYP3A4. Clin Pharmacokinet. 2006;45:1035–50. doi: 10.2165/00003088-200645100-00006. [DOI] [PubMed] [Google Scholar]
- 12.Tran TH, Von Moltke LL, Venkatakrishnan K, Granda BW, Gibbs MA, Obach RS, Harmatz JS, Greenblatt DJ. Microsomal protein concentration modifies the apparent inhibitory potency of CYP3A inhibitors. Drug Metab Dispos. 2002;30:1441–5. doi: 10.1124/dmd.30.12.1441. [DOI] [PubMed] [Google Scholar]
- 13.Jones HM, Houston JB. Substrate depletion approach for determining in vitro metabolic clearance: time dependencies in hepatocyte and microsomal incubations. Drug Metab Dispos. 2004;32:973–82. doi: 10.1124/dmd.104.000125. [DOI] [PubMed] [Google Scholar]
- 14.Soars MG, Burchell B, Riley RJ. in vitro analysis of human drug glucuronidation and prediction of in vivo metabolic clearance. J Pharmacol Exp Ther. 2002;301:382–90. doi: 10.1124/jpet.301.1.382. [DOI] [PubMed] [Google Scholar]
- 15.Ghanbari F, Rowland-Yeo K, Bloomer JC, Clarke SE, Lennard MS, Tucker GT, Rostami-Hodjegan A. A critical evaluation of the experimental design of studies of mechanism based enzyme inhibition, with implications for in vitro-in vivo extrapolation. Curr Drug Metab. 2006;7:315–34. doi: 10.2174/138920006776359293. [DOI] [PubMed] [Google Scholar]
- 16.Austin RP, Barton P, Cockroft SL, Wenlock MC, Riley RJ. The influence of nonspecific microsomal binding on apparent intrinsic clearance, and its prediction from physicochemical properties. Drug Metab Dispos. 2002;30:1497–503. doi: 10.1124/dmd.30.12.1497. [DOI] [PubMed] [Google Scholar]
- 17.Hallifax D, Houston JB. Binding of drugs to hepatic microsomes: comment and assessment of current prediction methodology with recommendation for improvement. Drug Metab Dispos. 2006;34:724–6. doi: 10.1124/dmd.105.007658. [DOI] [PubMed] [Google Scholar]
- 18.Gertz M, Kilford PJ, Houston JB, Galetin A. Drug lipophilicity and microsomal protein concentration as determinants in the prediction of the fraction unbound in microsomal incubations. Drug Metab Dispos. 2008;36:535–42. doi: 10.1124/dmd.107.018713. [DOI] [PubMed] [Google Scholar]
- 19.Sykes MJ, Sorich MJ, Miners JO. Molecular modeling approaches for the prediction of the nonspecific binding of drugs to hepatic microsomes. J Chem Inf Model. 2006;46:2661–73. doi: 10.1021/ci600221h. [DOI] [PubMed] [Google Scholar]
- 20.Gao H, Yao L, Mathieu HW, Zhang Y, Maurer TS, Troutman MD, Scott DO, Ruggeri RB, Lin J. In silico modeling of non-specific binding to human liver microsomes. Drug Metab Dispos. 2008;36(10):2130–5. doi: 10.1124/dmd.107.020131. [DOI] [PubMed] [Google Scholar]
- 21.TSAR. 3.3 Reference Guide, Oxford Molecular Limited, 2000.
- 22.PreADME. http://preadmet.bmdrc.org/preadmet/index.php
- 23.O’Reilly SA, Wilson AM, Howes B. Utilization of SciFinder Scholar at an Undergraduate Institution. J Chem Educ. 2002;79:524–6. doi: 10.1021/ed079p524. [DOI] [Google Scholar]
- 24.Li J, Sun J, Cui S, He Z. Quantitative structure-retention relationship studies using immobilized artificial membrane chromatography I: amended linear solvation energy relationships with the introduction of a molecular electronic factor. J Chromatogr A. 2006;1132:174–82. doi: 10.1016/j.chroma.2006.07.073. [DOI] [PubMed] [Google Scholar]
- 25.Houston JB, Carlile DJ. Prediction of hepatic clearance from microsomes, hepatocytes, and liver slices. Drug Metab Rev. 1997;29:891–922. doi: 10.3109/03602539709002237. [DOI] [PubMed] [Google Scholar]
- 26.Honig B, Sharp K, Yang A. Macroscopic models of aqueous solutions: biological and chemical application. J Phys Chem. 1993;97:1101–9. doi: 10.1021/j100108a002. [DOI] [Google Scholar]
- 27.Krämer SD, Braun A, Jakits-Deiser C, Wunderli-Allenspach H. Towards the predictability of drug-lipid membrane interactions: the pH-dependent affinity of propanolol to phosphatidylinositol containing liposomes. Pharm Res. 1998;15:739–44. doi: 10.1023/A:1011923103938. [DOI] [PubMed] [Google Scholar]
- 28.Kilford PJ, Gertz M, Houston JB, Galetin A. Hepatocellular binding of drugs: correction for unbound fraction in hepatocyte incubations using microsomal binding or drug lipophilicity data. Drug Metab Dispos. 2008;36:1194–7. doi: 10.1124/dmd.108.020834. [DOI] [PubMed] [Google Scholar]