

Abstract
Background & Aims
Chronic inflammation is a risk factor for colon cancer (CC). Lysophosphatidic acid (LPA), a naturally produced phospholipid, mediates multiple effects that are vital to disease process, including inflammation and cancer. The expression of LPA receptor 2 (LPA2) is up-regulated in several types of cancer, including ovarian and colon cancer, but the importance of LPA and LPA2 in the development and progression of CC is unclear. In this study, we sought to determine whether LPA and LPA2 regulate the progression of CC in vivo.
Methods
We examined the potential role of LPA in CC progression by administering LPA to ApcMin/+ mice. We determined the loss of LPA2 function in tumorigenesis in the colon by treating mice with genetic deletion of LPA2 (LPA2−/−) with azoxymethane (AOM) and dextran sulfate sodium (DSS).
Results
We found that LPA increased tumor incidence in Apcmin/+ mice. LPA2−/− mice showed reduced mucosal damage and fewer tumors than wild-type (WT) mice. Reduced epithelial cell proliferation and decreases in β-catenin, Krüppel-like factor 5 (KLF5), and cyclooxygenase-2 (COX-2) expression were observed in LPA2−/− mice. Unlike WT mice, induction of monocyte chemoattractant protein-1 (MCP-1) and macrophage migration inhibitory factor (MIF) was significantly attenuated in LPA2−/− mice with reduced infiltration by macrophages.
Conclusion
These results show that LPA is capable of promoting tumorigenesis in the colon. The absence of LPA2 attenuates several effects that contribute to cancer progression in vivo and, hence, the current study identifies LPA2 as an important modulator of CC.
INTRODUCTION
CC is the fourth most common cancer and inflammation is an established risk factor for CC 1. Patients with inflammatory bowel disease are at increased risk for CC and the mortality in patients diagnosed with CC in the setting of IBD is higher than for sporadic colorectal cancer 1.
LPA is an extracellular lipid mediator that evokes multiple growth factor-like effects in almost every cell type 2, 3. LPA mediates its effects primarily by coupling to a family of G protein-coupled receptors, LPA1~LPA5 4. The initial indication that LPA could contribute to tumorigenesis came from studies showing that LPA increases cell proliferation and motility 5. Subsequently, over-expression of LPA2 in ovarian cancer cells has suggested that aberrant LPA-LPA2 signaling axis might have a tumor promoting activity in ovarian cancer 6, 7. Recent evidence shows that deregulation of LPA2 or LPA3 is more commonly found in other types of cancer 8–10.
Mice deficient in individual LPA receptors are available 11–13. Whereas mice with targeted deletion of LPA1 or LPA3 show receptor-specific defects such as craniofacial deformity or delayed implantation of embryos 11, 13, genetic ablation of LPA2 in mice (LPA2−/−) does not cause any obvious phenotypic defect 12. Moreover, transgenic expression of LPA2 in ovaries did not result in ovarian malignancy in mice 14. Although the absence of tumors in LPA2 transgenic mice implies a non-essential or redundant role of LPA2 in tumorigenesis, a body of in vitro experimental evidence tends to suggest the insufficiency of LPA-LPA2 signaling axis to induce malignancy in ovary 14. Therefore, the significance of LPA2 in tumorigenesis remains unclear and specific pathways affected are not known.
AOM, a metabolite of 1,2-dimethylhydrazine, has been used widely to induce the formation of precancerous epithelial lesions, aberrant crypt foci (ACF) 15. Together with AOM, repeated oral administration of DSS is widely employed to induce acute inflammatory reaction and ulceration in the entire colon, and accelerates and increases the incidence of colon carcinogenesis 16. The potential role of LPA in inflammation has extensively been studied 17 and chronic inflammation increases the risk of CC 1. In this study we investigated the loss of LPA2 function in colitis-associated cancer (CAC). Our study revealed that the extent of colon carcinogenesis was markedly decreased in LPA2−/− mice with decreased epithelial cell proliferation and infiltration of inflammatory leukocytes.
METHODS
Mice
LPA2−/− mice 12 were bred into C57BL/6 background for at least 10 generations. LPA2+/− mice were crossbred to derive WT (i.e., LPA2+/+), LPA2+/−, and LPA2−/− littermates and these littermates were used in all studies. Experiments with animals were carried out under approval by the Institutional Animal Care and Use Committee of Emory University.
LPA treatment
Six weeks old male C57BL/6 and ApcMin/+ mice (Jackson Laboratory) were given 1 μg/kg LPA suspended in 0.1% BSA containing PBS (~0.06 μmole per mouse; Avantis) by placing LPA into the stomach using a 22-gauge gavage needle once every three days for one month. Control animal received the same volume of PBS/0.1 % BSA. Animals were killed one month after the last LPA administration. The small intestine was removed, flushed with ice-cold PBS, cut open longitudinally along the main axis, and examined under a dissecting microscope for the presence of adenomas.
AOM and DSS treatment
WT, LPA2+/−, and LPA2−/− littermates of 6 to 8 week-old age were injected with 10 mg/kg of AOM (Midwest Research Institute) intraperitoneally at the beginning of the experiment. After 14 day, mice were given 3% DSS (Sigma-Aldrich) in drinking water for 7 days, followed by a 2 week-period of recovery with normal water. This was followed by another 1-week 3% DSS treatment and 2-weeks of normal water. Body weight of each mouse was measured and recorded daily. Mice were killed on day 36 or day 56. Colon was removed, flushed with ice-cold PBS, cut open longitudinally, and examined under a dissecting microscope for the presence of tumors as previously described 16. Colonic tissues were fixed in 10% buffered formalin overnight for histological analysis. Paraffin embedded sections were stained with hematoxylin and eosin (H&E) for microscopic assessment of colitis.
AOM treatment
LPA2−/− and WT littermates of 5–6 weeks old age received 4 weekly intraperitoneal injections of AOM at 10 mg/kg body weight. Four weeks after the last AOM injection, all mice were killed, and their colons were removed and fixed in 4% paraformaldehyde. Fixed tissues were stained with methylene blue and ACF were visualized by stereo microscopy. ACF were identified using a published set of criteria 18.
Immunohistochemistry
Myeloperoxidase Activity
Quantitative RT-PCR (qRT-PCR)
Statistic Analysis
Data are expressed as means ± SE. Statistical significance was determined by one-way ANOVA. p-values <0.05 were considered significant.
RESULTS
Effect of LPA on the development of adenomas in ApcMin/+ mice
Previous studies have shown that LPA2 expression is elevated in human CC patients and cell lines 9, 10. qRT-PCR analysis showed that LPA2 mRNA level was elevated 3.5 ± 0.78 fold (n = 6) in intestinal adenomas of ApcMin/+ mice compared to normal intestinal tissue from WT controls (Figure 1A). There was a small statistically significant increase in LPA1 in ApcMin/+ mice (P < 0.05) as well, but LPA3 expression was not different.
Figure 1.

The effect of LPA on tumor incidence in ApcMin/+ mice
(A) The expression of LPA receptors in adenomas of ApcMin/+ mice was determined by quantitative RT-PCR. W: WT, M: ApcMin/+. n = 6. *, P < 0.05. **, P < 0.01. (B) The effect of LPA on colonic tumorigenesis was determined by oral administration of LPA to ApcMin/+ mice. Six weeks old male C57BL/6 and ApcMin/+ mice were given 10 μg/kg LPA suspended in 0.1% BSA containing PBS by gavage once every three days for one month. Control animal received the same volume of PBS+BSA. Animals were killed one month after the last LPA administration and the small intestine was examined under a dissecting microscope for the presence of adenomas. Each data point represents the number of adenomas found in the entire intestine of each mouse. Horizontal bars show the median of each group. n = 9. **, P < 0.001.
LPA plays a vital role in intestinal wound healing, cell proliferation, cell survival, cytokine induction, and regulation of ion transport 9, 10, 19–22. We initially tested whether LPA regulates tumorigenesis in the colon by orally administering LPA to ApcMin/+ mice. Although the stability of orally administered LPA may be a concern 23, a previous study has shown that orally administered LPA is effective in inhibition of fluid secretion in the ileum, demonstrating the bioavailability of orally administered LPA in the intestine 22. Administration of LPA increased the number of adenomas (60 ± 5.2, n = 9, P < 0.001) in ApcMin/+ mice compared to ApcMin/+ mice that received carrier (32 ± 6.7, n = 9) (Figure 1B). This result shows that LPA may potentiate tumorigenesis in the colon, although whether this effect was mediated by LPA2 is unclear.
Reduced tumor incidence in LPA2−/− mice
We next sought to determine the importance of LPA2 in colon tumorigenesis by characterizing the loss of LPA2 function in vivo. To this end, we used a CAC model, which combines the treatment of mice with AOM and DSS (Figure 2A). AOM and DSS induced colitis in WT, LPA2+/−, and LPA2−/− littermates as evidenced by the weight loss (Figure 1B) and appearance of soft stool, but a significant difference in weight loss between WT and LPA2−/− mice was observed. Similarly, the clinical score, which quantifies the appearance of occult blood and diarrhea, was significantly lower in LPA2−/− than WT littermates (Supplementary Figure 1A). Weight loss in LPA2+/− mice was similar to that in WT. Moreover, 3 each of WT and LPA2+/− mice died by day 56 as compared to none among LPA2−/− mice (Figure 2C). WT and LPA2+/− mice developed multiple polypoid tumors in the middle to distal colon, whereas tumors were fewer and smaller in LPA2−/− mice (Figure 2D). The colons of WT and LPA2+/− mice were heavier than that of LPA2−/− mice, indicating occurrence of edema in these mice (Supplementary Figure 1B). There was no difference in macroscopic or microscopic features among untreated mice.
Figure 2.

Tumor formation induced by AOM and DSS treatment
(A) Schematic presentation of AOM and DSS treatment (B) Body weight changes represented as mean ± SE during the course of AOM and DSS treatment. *, P < 0.05 compared with WT. (C) Mortality were followed during the course of AOM and DSS treatment. (D) Representative gross appearance of colon of WT, LPA2+/−, and LPA2−/− littermates.
The mean number of tumors in the colon of LPA2−/− mice was 0.7±0.2 at day 36 vs. 1.5±0.6 for WT and 1.8±0.5 for LPA2+/− (Figure 3A) (n = 8, P < 0.05). At day 56, an average of 13.8±1.8 and 13.1±1.1 adenomas per animal was found in WT and LPA2+/− mice, respectively (n = 10, P < 0.01). In contrast, LPA2−/− mice developed 8.1±1.6 lesions per animal (Figure 3B). Furthermore, the numbers of tumors larger than 2 mm were significantly less in LPA2−/− mice than in WT or LPA2+/− mice (Figure 3C; 2.8±0.7 for LPA2−/− vs. 6.8 ± 0.8 for WT and 7.3 ± 0.5 for LPA2+/− mice). Histological examination of colonic sections showed no notable difference between untreated WT and LPA2−/− littermates (Figure 3D). By day 36, tubular adenomas were present in WT and LPA2+/− mice, but the numbers and size of lesions were comparatively smaller in LPA2−/− mice. At day 56, the numbers and size of lesions were progressively increased in all mice, but the polyps were flatter and smaller in LPA2−/− mice compared to those in WT or LPA2+/− mice. Together these results show that the absence of LPA2 protected animals from the combination treatment of AOM and DSS and suggest a crucial role of LPA2 in the progression of tumor in the colon. Heterozygosity of LPA2, however, was not sufficient for the protection and we will not display data from LPA2+/− mice hereafter.
Figure 3.

Reduced tumors in LPA2−/− mice
Numbers of tumors at (A) day 36 or (B) day 56 in the colons of WT, LPA2+/−, and LPA2−/− mice were counted under the microscope. Each point represents the number of tumors in the colon of each mouse. Horizontal bars represent the mean. n ≥ 8. *, P < 0.01. (C) The number of tumors larger than 2 mm in diameter at day 56 is shown. Each value represents the mean ± SE. n=10. *, P < 0.01. (D) Representative colon tissues stained with H&E. n = 6. Magnification: x100.
Decreased Cell Proliferation in LPA2−/− mice
The difference in tumor size between WT and LPA2−/− mice suggests that there may be an underlying difference in the rate of cell proliferation. Epithelial cell proliferation was examined by immunohistological staining of Ki67. In untreated mice, Ki67 labeling was confined to the nuclei of epithelial cells located at the base of colonic crypts in WT and LPA2−/− mice and no observable difference was noted between WT and LPA2−/− mice (Figure 4A and D). Following AOM/DSS treatment, WT mice had a large population of proliferating cells in non-tumorous crypts as well as in tumors (Figure 4B and C). Ki67 labeling extended from the base of the crypt to more than half way of the crypt height and elongation of the crypt height compared to untreated animals was evident (Figure 4B). On the contrary, Ki67-positive epithelial cells were largely limited to the base region of the crypt in LPA2−/− mice (Figure 4E and F), suggesting that LPA2 is involved in proliferation of epithelial cells in the crypts following AOM and DSS treatment.
Figure 4.

Reduced cell proliferation in LPA2−/− mice
Paraffin embedded colonic sections from untreated (A and D) and AOM and DSS treated WT (B-C) and LPA2−/− (E-F) mice were deparaffinized and immunolabeled using an antibody against Ki67. n = 5. Magnification at x100.
β-catenin and KLF5 expression in LPA2−/− mice
Previous studies have linked the proliferation of CC cells by LPA to the regulation of β-catenin and KLF5 19, 20. To determine whether differential expression of β-catenin contributes to the cell proliferation, we examined β-catenin expression in WT and LPA2−/− mice. Figure 5A shows immunohistological staining of β-catenin in epithelial cells of colonic section. In treated WT mice, β-catenin labeling was more evident in the cytoplasm and nuclei of epithelial cells in adenomas (Figure 5B–c and b vs. a). In comparison, up-regulation of β-catenin in LPA2−/− mice was substantially lower and less nuclear staining was observed, demonstrating that LPA2 regulates activation of β-catenin in vivo 19, 20.
Figure 5.

Expression of β-catenin, KLF5, and COX-2
(A) Colon tissues of untreated and AOM and DSS-treated WT and LPA2−/− mice were stained with antibodies against β-catenin. WT: a–c; LPA2−/−: d–f. n=5. Magnification: a and d at x630; b–c and e–f at x100 (B) Western blot shows KLF5 protein expression in the colon of WT and LPA2−/− mice, n = 3. (C) Representative immunohistologic staining of colonic tissues for KLF5 are shown (n = 5). WT: a–c; LPA2−/−: e–f. Magnification: a–b and d–e at x100; c and f at x400. (D) COX-2 protein expression in protein extracts from the colons of WT and LPA2−/− mice was determined by Western immunoblotting. n = 3. (B) Immunohistological analysis of COX-2 expression in colon tissues from WT and LPA2−/− mice. n = 5. Magnification; x400.
Figure 5B shows that KLF5 protein expression was significantly up-regulated in treated WT mice. Immunohistological staining of KLF5 shows that all epithelial cells in the crypts of WT and LPA2−/− mice were positive for KLF5 expression (Figure 5C-a and d). In treated WT mice (Figure 5C-b), the staining of KLF5 was more intense in the base of the crypt and KLF5 labeling extended to the epithelial cells near the surface. In comparison, a small increase in KLF5 expression was observed in AOM/DSS-treated LPA2−/− mice (Figure 5B) and the neck of the colon crypts were less populated with KLF5-positive cells in these mice relative to WT mice (Figure 5C–e and f). Together, these results suggest that the differential expression of β-catenin and KLF5 might provide the molecular basis for the decreased cell proliferation in AOM and DSS treated LPA2−/− mice.
Reduced COX-2 expression in LPA2−/− mice
An increase in COX-2, which generates prostaglandin E2 (PGE2) from arachidonic acid, has been associated with the progress of CC 24. Hence, we examined the expression of COX-2 proteins by Western blot and immunohistochemical analysis. The expression level of COX-2 protein was slightly lower in untreated LPA2−/− mice than untreated WT mice (Figure 5D). This difference can also be seen in immunohistochemical staining (Figure 5E, a vs. d). AOM/DSS treatment resulted in a significant increase in COX-2 protein expression in WT mice. Epithelial expression of COX-2 in colonic crypt of WT mice was observed (Figure 5E-b), but COX-2 expression was largely found in lamina propria cells (Figure 5E-c). Induction COX-2 expression in LPA2−/− mice was markedly lower compared with WT mice (Figure 5D, 5E-e and f) and clearly fewer COX-2 expressing lamina propria cells were found in LPA2−/− mice.
Cytokine induction in LPA2−/− mice
Previous studies have shown that LPA can induce secretion of cytokines and chemokines from cancer cells, including CC cells 10, 25, 26. We contemplated whether there was a difference in the expression of cytokines and chemokines between WT and LPA2−/− mice. The expression levels of various cytokines and chemokines were determined by qRT-PCR and the results are shown in Figure 6A. There was no apparent difference in TNF-α, INF-γ, IL-1β, and IL-6 expression in untreated WT and LPA2−/− mice, and AOM and DSS markedly stimulated expression of these cytokines in both mice. Although the expression levels of TNF-α, INF-γ, IL-1β, and IL-6 in treated LPA2−/− mice were lower compared to WT mice, they did not reach statistical significance. In contrast, we observed a significant difference in the expression of MCP-1 and MIF between WT and LPA2−/− mice. Whereas the expression levels of MCP-1 and MIF were elevated more than 3-fold in treated WT mice, small increases were observed in LPA2−/− mice. Moreover, the basal expression levels of MCP-1 and MIF were lower in LPA2−/− than WT mice. The induction of MCP-1 and MIF was confirmed in vitro by treating Caco-2 cells with LPA (Supplemental Figure 2). Together, these results suggest LPA2-mediated signaling specifically regulates the expression of pro-inflammatory factors such as MCP-1 and MIF.
Figure 6.

Cytokine gene expression in the colon
(A) Quantitative RT-PCR was performed on total RNAs from the colons of WT and LPA2−/− mice before and after treatment with AOM and DSS. Data are presented as mean ± SE from at least 6 mice from each group. *, P < 0.05; #, P <0.001 compared to untreated WT. +, P < 0.05 compared to untreated LPA2−/−. (C) MPO activity was measured in the colons of WT and LPA2−/− mice. n ≥ 8 mice from each group. (D) immunohiostological analysis of Mac-3-positive macrophage in colon tissues from WT and LPA2−/− mice. n = 5.
Infiltration of inflammatory cells
Early during tumor formation, stromal cells and tumor-infiltrating leukocytes provide signals that regulate cancer cell growth and differentiation. Because MCP-1 mRNA level was markedly decreased in LPA2−/− mice, we next determined whether the lack of LPA2 affected infiltration of inflammatory cells. Histological analysis showed massive infiltration of neutrophils in AOM/DSS-treated mice and quantification of neutrophil infiltration is shown in Figure 6C. Marked increases in MPO activity were observed in WT and LPA2−/− mice, but with no difference between the two groups, implying that infiltration of neutrophils was not affected by the absence of LPA2. On the contrary, immunohistochemical analysis using an anti-Mac-3 antibody showed extensive infiltration of macrophages in WT mice, whereas a reduction in the number of macrophages in the lamina propria of LPA2−/− mice was evident. These results show that a decrease in macrophage infiltration is a part of the protective mechanisms rendered by the absence of LPA2.
AOM-induced neoplasia
The combination treatment of AOM and DSS relies on inflammatory response evoked by DSS intake, which hasten the development of adenocarcinomatous lesions 16. To determine whether enhanced inflammatory response is needed to observe the protective effect of loss of LPA2 function, we subjected WT and LPA2−/− mice with AOM alone. WT and LPA2−/− mice both developed ACF, but significantly smaller numbers of ACF were found in the colons of LPA2−/− mice than in WT control mice (Figure 7). The mean number of ACF in WT mice was 24.3 ± 2.1 vs. 11.6 ± 1.5 for LPA2−/− mice (n = 9, P < 0.01). No ACF was found in saline-treated WT or LPA2−/− mice (data not shown). These results show that the loss of LPA2 function impeded the formation of early neoplastic lesions in the colon without onset of inflammation.
Figure 7.
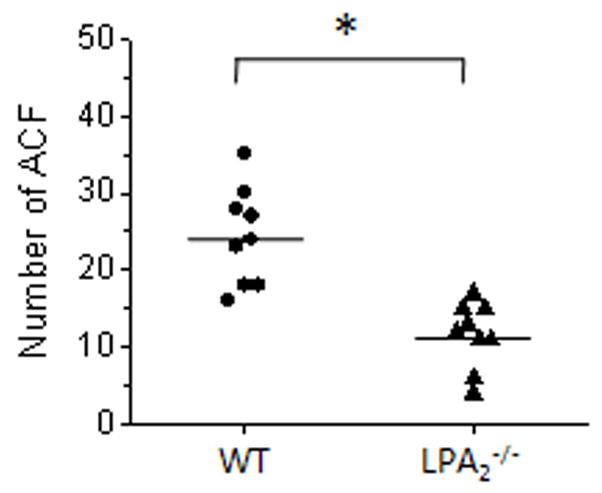
ACF formation by AOM treatment
WT and LPA2−/− mice were given repeated intraperitonial injection of AOM. The number of ACF was determined in methylene blue stained colons. Each data point represents the number of ACF in each mouse. n = 9 per group. *, P < 0.001.
DISCUSSION
The prominent production of LPA and the effects of LPA in most ovarian and prostate cancer cells along with over-expression of LPA2 have suggested the autocrine system of LPA-LPA2 axis promotes growth and invasion of such cancers 3, 6, 7. More recently, over-expression of LPA2 in colon cancer patients has been shown and suggests a potential role of LPA2 in colon cancer 9, 10. However, the absence of tumor in ovaries of LPA2 transgenic mice has left the role of LPA2 in tumorigenesis uncertain 14. In this study, we determined the importance of LPA2 in CC by characterizing the loss of LPA2 function in mice. By challenging LPA2−/− mice with agents that induce dysplasia in the colon, we were able to reveal the pathological importance of LPA2 in CC. Our data herein demonstrate that the absence of LPA2 results in a marked decrease in tumor incidence and progression of colon adenocarcinomas. The amelioration of CC in LPA2−/− mice was accompanied by a decrease in both cell proliferation and chemokine expression, providing insights into the protection against CC in LPA2−/− mice. Although our study was focused on the characterization of a CAC model, the propensity to protect animals was consistent in the study using AOM alone. To our knowledge, the current study is the first direct evidence demonstrating the importance of LPA2 in colon carcinogenesis in vivo and our data suggest LPA2 as a potential target for preventive strategies for CC.
The absence of tumors in the transgenic LPA2 ovaries suggests incomplete capacity of the LPA-LPA2 signaling axis to initiate cancer and it has been suggested that LPA2 might increase the susceptibility to develop cancer or hasten cancer progress when other genetic factors are present 14. This is in part supported by our current study in which LPA increased the number and size of adenomas in ApcMin/+ mice that are predisposed to forming adenomas in the small intestine 27. However, our study does not specifically address whether the increase in adenomas in Apcmin/+ mice by LPA resulted from signaling by LPA2 despite upregulation of LPA2 expression in these mice. A further study based on deletion of LPA2 in ApcMin/+ background is needed to fully recapitulate this aspect.
The Wnt/β-catenin cascade plays a single most dominant role in controlling cell fate along the crypt-villus axis. Once the Wnt/β-cascade is mutationally activated, the adenoma cells maintain their progenitor status populating entire villi 28. LPA had been shown to inactivate glycogen synthase kinase-3 in NIH3T3 and HEK293 cells although nuclear accumulation of β-catenin by LPA was not reported in these cells 29. On the other hand, Yang et al. 19 have shown that LPA activates the β-catenin pathway in HCT116 and LS174T human CC cells, resulting in an accumulation of β-catenin in the nucleus. However, we previously reasoned that LPA might be able to influence proliferation of CC cells independent of the Wnt/β-catenin cascade as the Wnt/β-catenin cascade is often mutated in CC cells 20. This led us to identify KLF5 as an additional target of LPA-elicited signaling that regulates proliferation of CC cells 20. KLF5, a member of a family of zinc finger-containing transcription factors, is predominantly expressed in the crypt compartment of intestine where cells rapidly replicate 30. Our data show a number of features demonstrating LPA2-dependent proliferation of epithelial cells in the crypts. AOM and DSS induced hyperplasia, as evidenced by the expansion of Ki67-positive cells and concurrent up-regulation of β-catenin and KLF5 expression in WT mice. In contrast, the crypt height of LPA2−/− mice was not only shorter but proliferating cells were largely confined to the crypt base along with significantly lower presence of nuclear β-catenin and KLF5. β-catenin and KLF5 are transcriptional factors, which can activate targets such as c-Myc, cyclin D, cyclin B1, and Cdc2 28, 30. In addition, LPA has previously been shown to induce the generation of reactive oxygen species and subsequent activation of downstream genes 31. Hence, this ability of LPA to regulate oncogenes and tumor suppressor genes suggests a potential role of LPA as a mutagen.
Whereas our study demonstrates the pro-proliferative effect of LPA2-mediated signaling on intestinal epithelial cells, increased epithelial cell proliferation alone is insufficient to cause cancer. LPA, however, is capable of inducing effects that potentially contribute to cancer progress. Herein we showed that COX-2 induction following AOM and DSS administration was markedly reduced in LPA2−/− mice. The link between COX-2 and CC is supported by a variety of studies 24, 32. Studies have shown that COX-2 expression is elevated in human CC patient and ApcMin/+ mice and epidemiological studies have demonstrated the efficacy of COX-2 inhibitors in reducing the tumor incidence in CC patients 24, 32. In AOM and DSS-treated LPA2−/− mice, significantly fewer COX-2 positive cells was detected compared with WT mice. Moreover, the basal COX-2 expression in LPA2−/− mice was lower in untreated LPA2−/− mice than WT, suggesting a role of LPA2 in maintenance of basal COX-2 expression. These findings are consistent with previous studies that LPA is an inducer of COX-2 expression in ovarian cancer cells 33, 34 and that LPA3−/− mice have reduced levels of COX-2 and PGE2/PGI2 that result in a defect in implantation 13.
LPA is a potent inducer of cytokines and angiogenic factors 10, 25, 35. LPA2 transgenic mice secreted increased amounts of vascular endothelial growth factor and urokinase-type plasminogen activator 14. The down-regulation of MCP-1 and MIF in LPA2−/− mice is especially significant as this represents one potential mechanism where the absence of LPA2 ameliorates CC progress. MCP-1 is a chemoattractant for monocytes, endothelial cells, and a subset of T lymphocytes, and its expression correlates with macrophage infiltration and tumor vessel density in human colorectal cancer 36. The reduced MCP-1 expression is in part thought to be responsible for the decrease in the number of infiltrating macrophages in LPA2−/− mice. Macrophages are one of the predominant stromal cells that are found in cancers and studies have correlated the macrophage infiltration and the prognosis of cancers 37. Macrophages contribute to tumor progression by producing matrix metalloproteinases as well as COX-2 and PGE2 37, 38. Previously, F4/80-positive macrophages and to a lesser extent neutrophils were shown to be the major COX-2 producing leukocytes in the colon of AOM and DSS treated mice 39. Hence, our finding of reduced MCP-1 expression in LPA2−/− mice parallels the decreased infiltration of macrophages and COX-2 expression. Our in vitro study showed that LPA can stimulate MCP-1 secretion from human CC cells and, hence, we suggest that LPA-LPA2 signaling axis is a major contributor of MCP-1 in the intestine and that under the pathological conditions, such as cancer, MCP-1 secreted by the epithelial cells induces transepithelial migration of macrophages, which adds to tumor progression.
Another effect observed in LPA2−/− mice was the induction of MIF. Over-expression of MIF has been shown in several neoplasms, including prostate and colorectal cancer 40, 41. In addition, increased expression of MIF in human and Apcmin/+ mouse adenomas has been reported and homozygous deletion of mif gene in Apcmin/+ mice decreased the number and size of adenomas, providing direct evidence for its function in colon carcinogenesis 42. Multiple effects have been ascribed to MIF, including tumor invasion, angiogenesis, and down-regulation of the tumor suppressor p53, suggesting that increased expression of MIF might exacerbate tumor progression by suppressing p53-mediated growth arrest and apoptosis 42–44. MIF is abundantly expressed in intestinal epithelial cells, but during inflammatory processes as well as cancer, macrophages are thought to be a major producer of MIF 45. The mechanisms underlying the up-regulation of MIF expression during the early stage of intestinal tumorigenesis is unclear, but our data suggest that the LPA-LPA2 signaling axis is a significant contributor to this regulation.
In summary, we demonstrated that the loss of LPA2 significantly attenuated the progression of CC. During the recent years, several inhibitors targeting LPA receptors have been synthesized 46–48, but these inhibitors target LPA1 or LPA3, but not LPA2. Recently, LPA2-specific compound was reported, but this compound was not available to examine the efficacy of LPA2-specific inhibition as a therapeutic strategy for CC prevention. However, our current study show that inhibiting LPA2-mediated signaling has potential to restrict tumor progression and targeting LPA2 might be a new strategy for CC prevention and treatment.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Asma Nusrat for valuable comments and Dr. Mauricio Rojas for the use of a microtome.
Funding sources: This work was supported by grants from the National Institutes of Health (DK071597 (C.C.Y.), DK052230 (V.W.Y.), MH51699, HD050685, and NS048478 (J.C.). S.L. is supported by a Research Fellowship Award from the Crohn’s and Colitis Foundation of America. We acknowledge the Emory Digestive Disease Research Development Center (supported by DK064399) for the use of light microscope.
Abbreviations
- LPA
lysophosphatidic acid
- AOM
azoxymethane
- DSS
dextran sulfate sodium
- CAC
colitis-associated cancer
- Apc
adenomatous polyposis coli
- ACF
aberrant crypt foci
- COX-2
cyclooxygenase-2
- PG
prostaglandin
- MCP-1
monocyte chemoattractant protein 1
- MIF
Macrophage migration inhibitory factor
Footnotes
No conflict of interest exist
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Itzkowitz SH, Yio X. Inflammation and Cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol. 2004;287:G7–17. doi: 10.1152/ajpgi.00079.2004. [DOI] [PubMed] [Google Scholar]
- 2.Ye X, Ishii I, Kingsbury MA, et al. Lysophosphatidic acid as a novel cell survival/apoptotic factor. Biochim Biophys Acta. 2002;1585:108–13. doi: 10.1016/s1388-1981(02)00330-x. [DOI] [PubMed] [Google Scholar]
- 3.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582–91. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 4.van Meeteren LA, Moolenaar WH. Regulation and biological activities of the autotaxin-LPA axis. Prog Lipid Res. 2007;46:145–160. doi: 10.1016/j.plipres.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 5.van Corven EJ, Groenink A, Jalink K, et al. Lysophosphatidate-induced cell proliferation: Identification and dissection of signaling pathways mediated by G proteins. Cell. 1989;59:45–54. doi: 10.1016/0092-8674(89)90868-4. [DOI] [PubMed] [Google Scholar]
- 6.Xu Y, Shen Z, Wiper DW, et al. Lysophosphatidic acid as a potential biomarker for ovarian and other gynecologic cancers. JAMA. 1998;280:719–23. doi: 10.1001/jama.280.8.719. [DOI] [PubMed] [Google Scholar]
- 7.Goetzl EJ, Dolezalova H, Kong Y, et al. Distinctive Expression and Functions of the Type 4 Endothelial Differentiation Gene-encoded G Protein-coupled Receptor for Lysophosphatidic Acid in Ovarian Cancer. Cancer Res. 1999;59:5370–5375. [PubMed] [Google Scholar]
- 8.Nakamoto T, Yasuda K, Yasuhara M, et al. Expression of the endothelial cell differentiation gene 7, a lysophosphatidic acid receptor, in ovarian tumor. J Obst Gyn Res. 2005;31:344–351. doi: 10.1111/j.1447-0756.2005.00299.x. [DOI] [PubMed] [Google Scholar]
- 9.Shida D, Watanabe T, Aoki J, et al. Aberrant expression of lysophosphatidic acid (LPA) receptors in human colorectal cancer. Lab Invest. 2004;84:1352–62. doi: 10.1038/labinvest.3700146. [DOI] [PubMed] [Google Scholar]
- 10.Yun CC, Sun H, Wang D, et al. LPA2 receptor mediates mitogenic signals in human colon cancer cells. Am J Physiol. 2005;289:C2–11. doi: 10.1152/ajpcell.00610.2004. [DOI] [PubMed] [Google Scholar]
- 11.Contos JJA, Fukushima N, Weiner JA, et al. Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc Natl Acad Sci USA. 2000;97:13384–13389. doi: 10.1073/pnas.97.24.13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Contos JJA, Ishii I, Fukushima N, et al. Characterization of lpa2 (Edg4) and lpa1/lpa2 (Edg2/Edg4) Lysophosphatidic Acid Receptor Knockout Mice: Signaling Deficits without Obvious Phenotypic Abnormality Attributable to lpa2. Mol. Cell. Biol. 2002;22:6921–6929. doi: 10.1128/MCB.22.19.6921-6929.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye X, Hama K, Contos JJA, et al. LPA3-mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature. 2005;435:104–108. doi: 10.1038/nature03505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang MC, Lee HY, Yeh CC, et al. Induction of protein growth factor systems in the ovaries of transgenic mice overexpressing human type 2 lysophosphatidic acid G protein-coupled receptor (LPA2) Oncogene. 2004;23:122–9. doi: 10.1038/sj.onc.1206986. [DOI] [PubMed] [Google Scholar]
- 15.Pretlow TP, Barrow BJ, Ashton WS, et al. Aberrant crypts: putative preneoplastic foci in human colonic mucosa. Cancer Res. 1991;51:1564–7. [PubMed] [Google Scholar]
- 16.Suzuki R, Kohno H, Sugie S, et al. Sequential observations on the occurrence of preneoplastic and neoplastic lesions in mouse colon treated with azoxymethane and dextran sodium sulfate. Cancer Sci. 2004;95:721–7. doi: 10.1111/j.1349-7006.2004.tb03252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang MC, Graeler M, Shankar G, et al. Lysophospholipid mediators of immunity and neoplasia. Biochim Biophys Acta. 2002;1582:161–7. doi: 10.1016/s1388-1981(02)00151-8. [DOI] [PubMed] [Google Scholar]
- 18.Papanikolaou A, Wang QS, Papanikolaou D, et al. Sequential and morphological analyses of aberrant crypt foci formation in mice of differing susceptibility to azoxymethane-induced colon carcinogenesis. Carcinogenesis. 2000;21:1567–72. [PubMed] [Google Scholar]
- 19.Yang M, Zhong WW, Srivastava N, et al. G protein-coupled lysophosphatidic acid receptors stimulate proliferation of colon cancer cells through the {beta}-catenin pathway. Proc Natl Acad Sci USA. 2005;102:6027–32. doi: 10.1073/pnas.0501535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H, Bialkowska A, Rusovici R, et al. Lysophosphatidic acid facilitates proliferation of colon cancer cells via induction of Kruppel-like factor 5. J Biol Chem. 2007;282:15541–15549. doi: 10.1074/jbc.M700702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mori K, Kitayama J, Shida D, et al. Lysophosphatidic Acid-Induced Effects in Human Colon Carcinoma DLD1 Cells Are Partially Dependent on Transactivation of Epidermal Growth Factor Receptor. J Surgical Res. 2006;132:56–61. doi: 10.1016/j.jss.2005.07.040. [DOI] [PubMed] [Google Scholar]
- 22.Li C, Dandridge KS, Di A, et al. Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J Exp Med. 2005;202:975–86. doi: 10.1084/jem.20050421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deng, Tsukahara, Valentine, et al. The Lysophosphatidic Acid Type 2 Receptor Is Required for Protection Against Radiation-Induced Intestinal Injury. Gastroenterology. 2007;132:1834–1851. doi: 10.1053/j.gastro.2007.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turini ME, DuBois RN. Cyclooxygenase-2: a therapeutic target. Annu Rev Med. 2002;53:35–57. doi: 10.1146/annurev.med.53.082901.103952. [DOI] [PubMed] [Google Scholar]
- 25.Fang X, Yu S, Bast RC, et al. Mechanisms for lysophosphatidic acid-induced cytokine production in ovarian caner cells. J Biol Chem. 2004;279:9653–61. doi: 10.1074/jbc.M306662200. [DOI] [PubMed] [Google Scholar]
- 26.Zhao Y, He D, Saatian B, et al. Regulation of lysophosphatidic acid- induced epidermal growth factor receptor transactivation and interleukin-8 secretion in human bronchial epithelial cells by protein kinase C delta, Lyn kinase and matrix metalloproteinases. J Biol Chem. 2006;281:19501–19511. doi: 10.1074/jbc.M511224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moser AR, Dove WF, Roth KA, et al. The Min (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. J Cell Biol. 1992;116:1517–26. doi: 10.1083/jcb.116.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–50. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 29.Fang X, Yu S, Tanyi JL, et al. Convergence of multiple signaling cascades at glycogen synthase kinase 3: Edg receptor-mediated phosphorylation and inactivation by lysophosphatidic acid through a protein kinase C-dependent intracellular pathway. Mol Cell Biol. 2002;22:2099–110. doi: 10.1128/MCB.22.7.2099-2110.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghaleb AM, Nandan MO, Chanchevalap S, et al. Kruppel-like factors 4 and 5: the yin and yang regulators of cellular proliferation. Cell Res. 2005;15:92–6. doi: 10.1038/sj.cr.7290271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Q, Olashaw N, Wu J. Participation of reactive oxygen species in the lysophosphatidic acid-stimulated mitogen-activated protein kinase kinase activation pathway. J Biol Chem. 1995;270:28499–502. doi: 10.1074/jbc.270.48.28499. [DOI] [PubMed] [Google Scholar]
- 32.Eberhart CE, Coffey RJ, Radhika A, et al. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–8. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 33.Symowicz J, Adley BP, Woo MMM, et al. Cyclooxygenase-2 Functions as a Downstream Mediator of Lysophosphatidic Acid to Promote Aggressive Behavior in Ovarian Carcinoma Cells. Cancer Res. 2005;65:2234–2242. doi: 10.1158/0008.5472.CAN-04-2781. [DOI] [PubMed] [Google Scholar]
- 34.Oyesanya RA, Lee ZP, Wu J, et al. Transcriptional and post-transcriptional mechanisms for lysophosphatidic acid-induced cyclooxygenase-2 expression in ovarian cancer cells. FASEB J. 2008 doi: 10.1096/fj.07-101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shida D, Kitayama J, Yamaguchi H, et al. Lysophosphatidic acid (LPA) enhances the metastatic potential of human colon carcinoma DLD1 cells through LPA1. Cancer Res. 2003;63:1706–11. [PubMed] [Google Scholar]
- 36.Salcedo R, Ponce ML, Young HA, et al. Human endothelial cells express CCR2 and respond to MCP-1: direct role of MCP-1 in angiogenesis and tumor progression. Blood. 2000;96:34–40. [PubMed] [Google Scholar]
- 37.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–50. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka S, Tatsuguchi A, Futagami S, et al. Monocyte chemoattractant protein 1 and macrophage cyclooxygenase 2 expression in colonic adenoma. Gut. 2006;55:54–61. doi: 10.1136/gut.2004.059824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Popivanova BK, Kitamura K, Wu Y, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008;118:560–70. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyer-Siegler KL, Vera PL, Iczkowski KA, et al. Macrophage migration inhibitory factor (MIF) gene polymorphisms are associated with increased prostate cancer incidence. Genes Immun. 2007;8:646–652. doi: 10.1038/sj.gene.6364427. [DOI] [PubMed] [Google Scholar]
- 41.Legendre H, Decaestecker C, Nagy N, et al. Prognostic Values of Galectin-3 and the Macrophage Migration Inhibitory Factor (MIF) in Human Colorectal Cancers. Mod Pathol. 2003;16:491–504. doi: 10.1097/01.MP.0000068235.45178.C1. [DOI] [PubMed] [Google Scholar]
- 42.Wilson JM, Coletta PL, Cuthbert RJ, et al. Macrophage Migration Inhibitory Factor Promotes Intestinal Tumorigenesis. Gastroenterology. 2005;129:1485–1503. doi: 10.1053/j.gastro.2005.07.061. [DOI] [PubMed] [Google Scholar]
- 43.Sun B, Nishihira J, Yoshiki T, et al. Macrophage Migration Inhibitory Factor Promotes Tumor Invasion and Metastasis via the Rho-Dependent Pathway. Clin Cancer Res. 2005;11:1050–1058. [PubMed] [Google Scholar]
- 44.Hudson JD, Shoaibi MA, Maestro R, et al. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. 1999;190:1375–82. doi: 10.1084/jem.190.10.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maaser C, Eckmann L, Paesold G, et al. Ubiquitous production of macrophage migration inhibitory factor by human gastric and intestinal epithelium. Gastroenterology. 2002;122:667–680. doi: 10.1053/gast.2002.31891. [DOI] [PubMed] [Google Scholar]
- 46.Hasegawa Y, Erickson JR, Goddard GJ, et al. Identification of a phosphothionate analogue of lysophosphatidic acid (LPA) as a selective agonist of the LPA3 receptor. J Biol Chem. 2003;278:11962–9. doi: 10.1074/jbc.M209168200. [DOI] [PubMed] [Google Scholar]
- 47.Virag T, Elrod DB, Liliom K, et al. Fatty alcohol phosphates are subtype-selective agonists and antagonists of lysophosphatidic acid receptors. Mol Pharmacol. 2003;63:1032–42. doi: 10.1124/mol.63.5.1032. [DOI] [PubMed] [Google Scholar]
- 48.Ohta H, Sato K, Murata N, et al. Ki16425, a Subtype-Selective Antagonist for EDG-Family Lysophosphatidic Acid Receptors. Mol Pharmacol. 2003;64:994–1005. doi: 10.1124/mol.64.4.994. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.