

Abstract
Amyloid-β (Aβ) peptide accumulation in the brain is central to the pathogenesis of Alzheimer’s disease (AD). Aβ is produced through proteolytic processing of a transmembrane protein, β-amyloid precursor protein (APP), by β- and γ-secretases. Mounting evidence has demonstrated that alterations in APP cellular trafficking and localization directly impact its processing to Aβ. Members of the low-density lipoprotein receptor family, including LRP, LRP1B, SorLA/LR11, and apoER2, interact with APP and regulate its endocytic trafficking. Additionally, APP trafficking and processing are greatly affected by cellular cholesterol content. In this review, we summarize the current understanding of the roles of lipoprotein receptors and cholesterol in APP trafficking and processing and their implication for AD pathogenesis and therapy.
Keywords: lipoprotein receptors, cholesterol, amyloid precursor protein, amyloid-β peptide, Alzheimer’s disease
1. Amyloid-β peptide and Alzheimer’s disease
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly. The characteristic pathological lesions found in AD are the deposition of extracellular amyloid plaques and intracellular neurofibrillary tangles [1]. The major component of the amyloid plaques is the ~4 kDa amyloid-β (Aβ) peptide, which is a cleavage product of the β-amyloid precursor protein (APP) [2, 3]. Aβ ranges in size from 37 to 43 amino acids; however, Aβ42(43) may act as a pathogenic seed for Aβ aggregation and amyloid plaque formation because they are more hydrophobic compared to the shorter Aβ peptides. One current hypothesis known as the “amyloid hypothesis” postulates that increased Aβ production or reduced Aβ clearance results in the formation of aggregated Aβ deposits leading to AD dementia [1, 4, 5].
Non-amyloid assemblies of Aβ are now considered as the primary cause of neuronal injury, synaptic loss, and the eventual dementia associated with AD. Soluble Aβ42, isolated from brain, plasma, and cerebral-spinal fluid (CSF), correlates with the severity of neurodegeneration in AD [6, 7]. In vitro, soluble Aβ is neurotoxic and inhibits electrophysiological activity that may be necessary for the formation and maintenance of memory [8–11]. Although the importance of soluble Aβ species in neuronal and synaptic toxicity in AD is well documented/supported, the precise biochemical form in which toxic Aβ assemblies exist remains controversial. For example, a soluble, SDS-stable dodecamer known as Aβ*56 was identified as the toxic species of Aβ in brain extracts of certain APP transgenic mice [12], yet Aβ dimers isolated directly from human AD brains were found to be the only toxic species of Aβ that impairs synaptic plasticity and memory [13]. The relevance of these Aβ assemblies in the pathogenesis of AD, particularly their in vivo roles, requires further investigation.
Genetic studies have revealed that the processing of APP to Aβ is important for AD pathogenesis [4, 14]. Mutations in the APP gene, as well as presenilin 1 (PS1) and PS2 genes, whose products are major constitutes of the γ-secretase complex, can directly result in familial, often early-onset, AD (FAD). However, although FAD genetics and mouse models have generated tremendous insights into AD pathogenesis, the vast majority of AD cases are sporadic with late-onset. For this reason, it is of great interest to study the proteins, lipids, and microenvironments that modulate APP processing to Aβ.
2. APP biology and processing
2.1. APP structure and function
APP is a type I transmembrane protein with characteristics of a cell surface receptor despite the lack of a known bona fide ligand. The function of APP is further complicated by the presence of two APP-related genes, APLP1 and APLP2 [3]. Deletion of Aplp2 and either App or Aplp1 in mice results in early postnatal lethality [3, 15], suggesting redundancy between APLP2 and the other two family members. Only APP contains the Aβ region and produce the AD-associated Aβ peptide. The APP gene is alternatively spliced to produce three major isoforms of 695, 751, and 770 amino acids in length. The longer APP isoforms, APP751 and APP770, contain a 56 amino acid Kunitz Protease Inhibitor (KPI) homology domain within their extracellular regions. APP is expressed throughout the body, but APP695, which lacks the KPI domain, is the predominant form found in neurons. APP ectodomain has been shown to participate in cell adhesion, neurite outgrowth, and synaptogenesis [3]. The APP intracellular domain (AICD), featuring a motif that interacts with an array of adaptor proteins that modulates cell migration, axonal transport, and cell signaling [3, 16]. By modulating APP expression in developing embryos, a recent study has demonstrated a critical role of APP in neuronal migration during development [17].
2.2. APP processing to Aβ
APP has a relatively short half life [18, 19], largely due to its proteolytic processing through two alternative pathways [1, 3]. In the amyloidogenic pathway, APP is first cleaved at a β-secretase site by the enzyme BACE (β-site APP cleaving enzyme) [20], which releases a soluble β-cleaved APP fragment (sAPPβ) and leaves a 99 amino acid C-terminal fragment (CTF), known as C99, attached to the membrane. C99 is subsequently cleaved by a γ-secretase complex [21] within its intramembrane region to release the Aβ peptide. In the non-amyloidogenic pathway, APP is processed by an α-secretase [22] that clips within the Aβ region, which results in the release of a soluble ~110–120 kDa α-cleaved APP fragment (sAPPα). This pathway also releases a CTF that is 83 amino acids in length known as C83. C83 can also be cleaved by γ-secretase to release p3. In both the amyloidogenic and non-amyloidogenic pathways, the γ-secretase cleavage of APP also releases an APP intracellular domain fragment (AICD). The processing of APP to these various components may have important consequences in both disease and normal physiology [3, 16].
2.3. Endocytic trafficking of APP
APP and APP cleavage products are found in clathrin-coated vesicles, suggesting that the amyloidogenic processing of APP could occur in the endocytic pathway [23]. In 1994, Koo and Squazzo showed that cell surface radiolabeled APP releases Aβ and that endocytosis of APP is also necessary for Aβ production. Inhibiting endocytosis of cell surface APP by potassium depletion, which disrupts the formation of clathrin lattices, or by C-terminal deletion of the APP tail, which removes important internalization motifs, leads to a decrease in Aβ production as well as an increase in cell surface APP and sAPPα secretion [24]. By site-directed mutagenesis analysis, it was found that the dominant endocytosis motif within the APP tail is the tetrapeptide YENP [19]. Cell secreted Aβ was greatly reduced when this endocytosis motif was functionally disrupted.
A tight correlation between APP endocytosis and Aβ secretion was further established based on studies that show secretase localization/compartmentalization. β-secretase (BACE) localizes to the Golgi and endosomes in HEK cells and has optimal activity at the acidic pH found within the endosomal compartments [20, 25]. Components of the γ-secretase complex have been localized to the endoplasmic reticulum (ER), TGN, cell surface and also in endosomes and lysosomes [26–28], whereas α-secretase activity is found primarily at the cell surface [29]. Since the secretases responsible for APP proteolysis have optimal enzymatic activity or distribution within specific cellular compartments, shifting APP to these compartments leads to an increased probability that APP will be cleaved by that secretase. Therefore, when cell surface APP is internalized into endosomes, it is cleaved primarily at the β-secretase site by BACE. Subsequent cleavage by γ-secretase produces Aβ. On the other hand, if APP accumulates at the cell surface, it is more likely to be cleaved by α-secretase to sAPPα via the non-amyloidogenic pathway.
Although a large amount of work has been devoted to the study of APP trafficking within the endocytic pathway, there is only emerging evidence that APP-interacting proteins and lipids such as cholesterol can affect its trafficking and processing. The remainder of this review will focus on discussing the roles of several members of the low-density lipoprotein receptor (LDLR) family and cholesterol in the cellular localization and trafficking of APP and how these events in turn affect APP processing to Aβ.
3. Roles of LDLR family members in APP trafficking and processing
3.1. The LDLR family
The LDLR family consists of a large class of cell surface receptors of diverse function [30, 31]. The family includes the LDLR, LDLR-related protein 1 (LRP1, also known as LRP), LDL receptor-related protein 1B (LRP1B), megalin/LRP2, the very low density lipoprotein receptor (VLDLR), apoE receptor 2 (apoER2), LRP4/MEGF7, LRP5, LRP6, and sorting protein-related receptor containing LDLR class A repeats (sorLA) or LR11 (Figure 1). Although members of the LDLR family perform a variety of functions from cholesterol metabolism to cellular signaling, they share several structural and functional features: 1) ligand-binding complement-type repeats, 2) epidermal growth factor (EGF) receptor-like repeats, 3) YWTD β-propeller domains, 4) one or more endocytic motifs within their cytoplasmic domains, and 5) binding of apolipoprotein E (apoE), a protein involved in cholesterol transport. Interestingly, apoE exists in three isoforms (E2, E3, and E4) in humans, and the presence of the ε4 allele represents a genetic risk factor for late-onset AD [32]. Ligand binding to LDLR family members is antagonized by receptor-associated protein (RAP), an ER chaperone that assists their proper folding and prevents premature ligand binding to these receptors during their trafficking along the secretory pathway [33]. Recombinant RAP is an excellent pharmacological tool because its exogenous application was found to universally antagonize ligand binding to LDLR family members. At least four members of the LDLR family interact with APP and regulate its trafficking - LRP1, LRP1B, SorLA/LR11, and apoER2.
Fig. 1.

Schematic representation of members of the LDLR family. Members of the LDLR family have diverse functions from cholesterol metabolism, Reelin and Wnt signaling, to intracellular transport. Despite multiple functions, they share common structural motifs, including ligand-binding repeats, epidermal growth factor repeats, YWTD spacer domains, a single transmembrane domain, and a short cytoplasmic domain containing conserved endocytic motifs. Four APP-interacting receptors, LRP1, LRP1B, apoER2, and sorLA/LR11, interact with APP and are discussed in this review.
3.2. LRP1 promotes APP endocytosis and its processing to Aβ
LRP1 is a 600 kDa multi-functional endocytic receptor that is highly expressed in the brain. LRP1 is synthesized as a single polypeptide precursor and is cleaved by furin in the TGN to produce a non-covalently associated heterodimer: a heavy chain (515 kDa) containing four putative ligand binding domains, and a light chain (85 kDa) containing the transmembrane domain and the cytoplasmic tail [30, 34]. LRP1 was initially identified as a receptor for apoE [35] and activated alpha-2-macroglobulin (α2M) [36]. Since then, LRP1 has been shown to bind and endocytose over 40 structurally and functionally diverse ligands [30, 37]. The function of these ligands can be divided into many classes, including lipoproteins, proteinases, proteinase-inhibitor complexes, blood coagulation factors, growth factors, extracellular matrix proteins, chaperones, and bacteria/viral proteins. The majority of LRP1 ligands, including apoE and RAP, have been shown to bind to Domains II and IV of LRP1 with similar affinities [38–40].
A unique feature of LRP1 is its rapid endocytosis rate, with half of the receptors at the cell surface able to internalize within 30 seconds (t1/2<0.5 min) [41]. Site-directed mutagenesis revealed that the YATL sequence and the distal di-leucine are the major endocytic motifs within the LRP1 tail [41]. LRP1 rapidly endocytoses a wide variety of ligands suggesting that it primarily functions as a cargo transporter; however, several studies have found that LRP1 also regulates several signaling pathways that impact diverse cellular functions such as cell proliferation, synaptic plasticity, and glutamate receptor scaffolding [30, 37].
LRP1 has been linked to AD in several ways. First, LRP1 binds to several ligands that are genetically related to AD including α2M, lactoferrin, APP, and Aβ in addition to apoE [30]. Second, LRP1 mediates the clearance of Aβ in vitro either by binding to Aβ itself or Aβ complexed to these ligands [42, 43]. Third, LRP1 and its ligands are found in amyloid plaques in AD brains [44]. Finally, several polymorphisms within the LRP1 gene have been linked to the risk for AD [45, 46]. Together, these findings suggest that LRP1 could play a pivotal role in AD pathogenesis.
Mounting evidence demonstrates that LRP1 is a major APP binding partner and regulates it trafficking and processing to Aβ. Initial studies reported that both sAPP and cell surface APP containing the KPI domain bind to and are internalized by LRP1 [47, 48]. An intracellular interaction also exists between LRP1 and non-KPI containing APP through the cytoplasmic adaptor protein, FE65. Specifically, pull-down experiments demonstrated that the amino-terminal PTB1 of FE65 binds to LRP1 and the carboxyl-terminal PTB2 of FE65 binds to APP [49], suggesting that FE65 likely acts as an adaptor to complex these two proteins. A cytoplasmic interaction between APP and LRP1, bridged by FE65, could further strengthen the association between LRP1 and KPI-containing forms of APP and also account for an association between non-KPI containing APP and LRP1. These interactions between APP and LRP1 have been substantiated with cell surface biotinylation, co-immunoprecipitation, and fluorescence resonance energy transfer (FRET) techniques [50, 51].
A report by Ulery et al. [52] showed that long-term treatment of cells with RAP to disrupt the extracellular interaction between LRP1 and APP increases cell surface APP and decreases Aβ production. In the same study, co-transfection of APP and LRP1 in LRP1-deficient cells led to a ~3-fold increase in Aβ levels in the media compared to media from cells transfected with APP alone. These data demonstrate that LRP1 expression and function can influence APP processing to Aβ. The role of LRP1 in promoting APP processing to Aβ is further confirmed in several subsequent studies [53, 54]. Most importantly, the rapid endocytosis rate of LRP1 was found to be directly responsible for increased APP internalization and processing to Aβ [53–55]. Interestingly, studies by Ye et. al (2005) demonstrated that application of apoE4 to cells expressing KPI-lacking APP increases APP endocytosis and Aβ levels in a manner that depends on LRP1 function [56]. Although this study provides an interesting link between the pathogenic alleles of apoE, LRP1, and APP, it is unclear how the binding of apoE4 to LRP1 influences APP processing. Future studies are needed to determine if apoE binding to LRP1 alters LRP1 endocytosis, localization, or its ability to interact with APP.
LRP1 interacts with BACE and presenilin 1 and is a substrate for β- andγ-secretase [57–60], suggesting that the alterations in APP processing by LRP1 could be more complex than originally considered. LRP1 may influence APP access to secretases through interactions with the secretases themselves or by changing the compartmentalization of APP. In the case of γ-secretase activity, LRP1 C-terminal fragments may compete with APP as a substrate for cleavage [59]. In the case of β-secretase activity, LRP1 may cooperatively aid interactions between APP and BACE [57]. Together, these studies demonstrate that LRP1 and APP not only interact with one another but also are functionally related in cellular trafficking and processing pathways.
3.3. LRP1B inhibits APP endocytosis and reduces Aβ production
LRP1B was first identified as a LDLR family member with extensive homology to LRP1 [61]. LRP1B shares 59% amino acid identity with LRP1 and the two receptors also exhibit similar overall structural modules (Figure 1). There are two major structural differences between LRP1 and LRP1B. First, LRP1B contains one additional ligand binding repeat within its fourth ligand binding domain. Second, LRP1B has a unique 33 amino acid sequence within its cytoplasmic tail [61, 62]. LRP1B was initially named LRP-deleted in tumors (LRP-DIT) because the LRP1B gene was deleted or inactivated in 40% of the non-small cell lung cancer cell lines (NSCLC) [61]. Since then, inactivation of the LRP1B gene has been described in several types of human cancers [63, 64], suggesting a role for LRP1B as a tumor suppressor. Indeed, studies have shown that LRP1B impairs urokinase receptor regeneration on the cell surface and inhibits cell migration [65]. By using LRP1B minireceptor containing its fourth ligand binding domain, the Bu laboratory has demonstrated that several LRP1 ligands also bind to LRP1B [62]; however, LRP1B has a very slow endocytosis rate (t1/2~8 min) compared to LRP1 [62] likely by using a less efficient endocytosis signal [66]. The distinction between endocytosis properties of LRP1B and LRP1 suggests that these two receptors likely antagonize each other’s function by competing for binding of common ligands and thereby inhibit or promote their cellular catabolism.
Because of high degree of homology between LRP1B and LRP1, we investigated whether LRP1B could also interact with APP. Using an LRP1B minireceptor, we found that mLRP1B4 and APP form an immunoprecipitable complex [67]. Furthermore, mLRP1B4 bound and facilitated the degradation of a soluble form of APP containing a KPI domain. A functional consequence of mLRP1B4 expression was a significant accumulation of APP at the cell surface, which is likely related to the slow endocytosis rate of LRP1B, and a concomitant reduction of Aβ production [67]. Using a LRP1B specific antibody, we confirmed the expression LRP1B at the protein level in the cortex, hippocampus, and cerebellum [67]. Since these studies suggest its expression may decrease the extracellular release of Aβ, examination of the regulation of LRP1B may have important applications to AD therapy.
3.4. SorLA/LR11 regulates APP trafficking and reduces its processing to Aβ
SorLA/LR11 is a ~250 kDa receptor containing ligand-binding complement-type repeats and YWTD domains. Unique in the LDLR family, it contains a vacuolar protein sorting 10 protein (vps10p) domain, which is homologous to a yeast receptor that transports proteins between the late Golgi and a prevacuolar endosome-like compartment [68]. Abundant mRNA expression of sorLA was found in human brain, spinal cord, and testis [69]. SorLA shares similarity with other members of the LDLR family by binding to RAP, apoE, and lipoprotein lipase [69, 70]; however, its endocytosis rate is much slower than LRP1. Since sorLA is also part of the family of VPS10 domain-containing receptors, its main role may be to chaperone proteins as an intracellular sorting receptor. SorLA does not appear to play an important role in development since sorLA receptor-deficient mice were viable and fertile [71].
The first clue that sorLA might play an important role in AD pathogenesis was the finding that expression of sorLA/LR11 is significantly reduced in AD brains [72]. Soon after, studies by Andersen et al. [71] demonstrated a direct interaction between sorLA/LR11 and APP. Similar to LRP1 binding to APP, the extracellular cluster of ligand-binding repeats in sorLA is required for this interaction, although the KPI domain is not required [54, 73]. Over-expression of sorLA in neurons shifted APP redistribution to the Golgi compartment and decreased its processing to Aβ. More importantly, a deletion of the Sorla gene in mice resulted in increased levels of Aβ in the brain, reminiscent of what was observed in AD patients [71]. Thus, sorLA acts as a sorting receptor that protects APP from processing into Aβ. SorLA expression also significantly reduced Aβ levels in cells expressing endogenous APP [74]. Interestingly, sorLA also interacts with BACE and competes with interactions between APP and BACE in the Golgi apparatus [75]. In addition, sorLA is also proteolytically processed by the γ-secretase and this intramembrane proteolysis likely modulates a sorLA-mediated signaling event [76]. Altogether these findings indicate that sorLA regulates APP trafficking into discrete intracellular compartments and also influences its interactions with secretases. Decreased levels of sorLA as found in AD could result in increased interactions between APP and BACE which would enhance its processing to Aβ. More direct evidence that links sorLA to AD came from a recent study demonstrating that inherited variants in the SORL1 gene are associated with late-onset AD [77]. Taken together, although the exact mechanism by which sorLA expression is decreased in AD brains is still not clear, sorLA is clearly a potential target that can be explored for AD therapy.
3.5. ApoER2 reduces APP endocytosis and is able to promote or inhibit its processing
ApoER2 is highly expressed in the hippocampus and the cortex [78]. Along with the VLDLR, apoER2 has a pivotal role in neuronal migration during brain development [79]. In addition, apoER2 has important roles in adults as a receptor for soluble ligands apoE, reelin, and F-spondin [78, 80, 81], and interacts and/or regulates membrane proteins, such as APP [82, 83] and NMDAR [84, 85]. Reelin binding initiates a signaling pathway through the binding and phosphorylation of the adaptor protein Dab1 to the NPxY motif of apoER2, which activates SRC family tyrosine kinases [86] and PKB/AKT, and ultimately inactivating GSK3β and blocking tau hyperphosphorylation [79]. ApoE binding also mediates the entrance of cholesterol to the cell [78] as well as signaling pathways related to cell survival or apoptosis, depending on the apoE isoform [87–89]. APP levels and Aβ are significantly increased in the frontal cortex and hippocampus of apoER2 KO mice [90] and the apoER2 ligand reelin is significantly reduced in the enthorinal cortex of AD model of mice and in human AD brains [91]. Moreover, a genetic polymorphism in the LRP8 (APOER2) gene has been associated with AD [92].
ApoER2 is expressed as several splice variants in a tissue specific manner [93], which determines the ligand repertoire and cell responses. One of the splicing variants includes exon 19 that encodes for a 59 amino acid proline-rich insert in the cytoplasmic domain of the receptor that binds to JNK interacting proteins (JIPs) [87, 94, 95]. The presence of this variant is responsible for the positive role of reelin on the NMDAR activation and calcium entrance through the binding of PSD95 and of X11α/β/Mint1/2 [96]. ApoER2 strongly associates with lipid raft (LR) independent of the presence of the proline-rich insert [97, 98], and is constitutively endocytosed by the clathrin-mediated pathway, which is dependent on the NPxY motif in its cytoplasmic domain and the adaptor protein Dab2 [98]. However, compared with the other members of the LDLR family, its internalization efficiency is significantly slower [98, 99] and does not depend on the presence of the proline-rich insert in the cytoplasmic tail [98]. This indicates that a significant amount of apoER2 is normally present at the plasma membrane.
Like APP, apoER2 also undergoes proteolytic processing by α- and γ-secretases [83, 100]. The extent of this processing is regulated by ligand binding for both APP and apoER2 [82, 101, 102]. F-spondin binds simultaneously to APP and apoER2, inducing the formation of a complex that stabilizes the proteins at the cell surface, thus inhibiting APP amyloidogenic processing [82]. Reelin also induces APP and apoER2 processing, most likely by α-secretase [102]. The resulting apoER2 ICD is able to traffic to the nucleus and regulates transcription [100].
APP and apoER2 bind common cytoplasmic adaptor proteins. Dab1 binds through its phosphotyrosine-binding (PTB) domain to NPxY motif of both APP [103] and apoER2, and overexpression of the full-length Dab1 or of its PTB domain augments the α-secretase processing of both APP [102, 104] and apoER2 processing [102]. Similarly, Dab2 binds to the NPxY motif of APP and apoER2 and mediates their endocytosis [18, 98], leading to an increase in amyloidogenic processing [18]. X11/Mint family proteins containing PTB and PDZ domains bind to APP (to the YENPTY motif) [105] and apoER2 (to the sequence YDRPLW within the 59-residue insert [96]) but with different effects on APP processing and Aβ production. Expression of the neuron-specific X11α and β correlates with a stabilization of APP, an increase in APP’s half-life, and a reduction of Aβ production [105, 106]. However, in the presence of apoE, X11/APP complex is endocytosed along with BACE, thus increasing Aβ40. This effect is more pronounced with apoE4 and depends on the co-expression of apoER2 [96], thus creating a link between the disease related isoform of apoE with it receptor in the amyloidogenic pathway. Fe65 also binds to APP and apoER2, and probably connects them. The final effect is an increase of both proteins at the cell surface and the promotion of α-secretase processing [107].
ApoER2 and APP also interact directly in the absence of ligand binding and independently of the cytoplasmic domain of the proteins [83], giving an additional level of regulation. This could imply apoER2 in a yet described role in the neurotrophic effect of soluble APP [3]. As apoER2 exhibits a slower endocytic rate than APP, the co-expression of both proteins increases APP at the cell surface and decreases APP endocytosis rate [83]. However, the final effect of this co-expression is an increase in both Aβ40 and Aβ42 [83]. How could this observation be reconciled with the idea that a more efficient internalization would increase the amyloidogenic pathway that takes place intracellularly in endocytic compartments? Since apoER2 is an LR-associated protein, its interaction with APP results in a localization shift of APP to the LR domains [83], where the proteolytic enzymes BACE and γ-secretase are also present, thus favoring amyloidogenic processing (see below, section 4). In addition, the expression of apoER2 was found to significantly increase γ-secretase activity [83] and in the presence of apoE, induced the internalization of APP along with BACE [96].
4. Role of Cholesterol in APP trafficking and processing
A number of findings indicate a connection between lipid metabolism and AD [108]. The genetic association of apoE ε4 allele with late-onset AD was established more than a decade ago [109], but the molecular mechanisms involved are still unclear. Increased plasma cholesterol is an important risk factor for AD [110–112]. Statins, cholesterol-lowering drugs, decrease Aβ levels and plaque formation in vivo, and clinical studies suggest that these drugs decrease the risk of AD [110, 111, 113, 114]. However, conflicting data were also reported [115, 116], illustrating the complexity of cholesterol in Aβ metabolism. There is also evidence indicating an anti-inflammatory role of statins in AD [117, 118]. Furthermore, one recent study suggested that cholesterol homeostasis is altered in AD brains, resulting in higher β- and γ-secretase activity and total cholesterol levels [119]. In vitro studies, however, show that the γ-secretase product AICD downregulates LRP1 expression, thus decreasing cholesterol levels, indicating a regulatory role for APP and γ-secretase in cholesterol metabolism [120].
4.1. Linking the amyloidogenic pathway to the association of APP and the secretases to lipid rafts membrane domains
Several evidence indicates [121, 122] that the amyloidogenic pathway takes place in membrane microdomains rich in cholesterol and glycosphingolipids, or LR, contrary to the non-amyloidogenic pathway, which is mediated by α-secretase localized mostly at the cell surface and out of LR domains [123]. Amyloidogenic activity is linked to cholesterol levels; β-secretase [124] and γ-secretase [125] activity is positively regulated by cholesterol and inhibited by low levels of cholesterol [126].
As was previously discussed, both β-secretase and γ-secretase can be found in the endosomal compartments, including early [127] and late [128, 129] endosomes, as well as in recycling compartments [130]. Recycling endosomal compartments and multivesicular domains contain the majority of cholesterol in the endocytic pathways [131, 132]. Non-processed full-length APP is localized mostly in non-raft membrane [122], and under some conditions it is shifted to LR domains, thus increasing Aβ production [83]. Even if the majority of APP is in non-raft domains, once processed by β-secretase (which is found in raft and non-raft membranes [122]), the β-CTFs concentrate in LR domains [122], allowing efficient processing by β-secretase. According to this model, cholesterol depletion disrupts β- and γ-secretase association to LRs and causes a decrease in Aβ production [122, 133, 134], indicating again that LRs are relevant sites for amyloidogenic processing of APP. In addition, cholesterol reducing drugs increase sAPPα secretion with a concomitant decrease in Aβ formation, which might occur by shifting APP localization from LRs to α-secretase-containing regions [121, 123] and/or decreasing the activity of the amyloidogenic enzymes.
4.2. Cholesterol biosynthesis and its intermediates in the APP trafficking and processing
HMG-CoA reductase, the target of statins, is the rate-limiting enzyme in cholesterol biosynthesis that catalyzes the production of mevalonate [135]. Therefore the role of statins in lowering neosynthesized cholesterol has been implicated as their major mechanism that explains their anti-amyloidogenic effect. For example, in vitro studies showed that atorvastatin stimulates APP α-secretase processing in a neuroblastoma cell line [136], while in primary hippocampal and cortical neurons, lovastatin inhibits the amyloidogenic secretases (β and γ) but does not necessarily increase APP α-secretase derived products [126]. Mevalonate is also a precursor of isoprenoids, including farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) [135]. Low doses of statins that do not decrease cholesterol levels block isoprenoids synthesis, which results in an inhibition of the amyloidogenic APP processing by different mechanisms, one of which is to decrease the isoprenylation of small GTPases involved in vesicular trafficking [137]. As a result of this process, APP accumulates intracellularly. Low doses of statins also inhibit β- and γ-secretases [138, 139] and change APP distribution in LR [138]. Direct addition of GGPP increases γ-secretase activity as well as its distribution in LR [139]. Overall the evidence indicates that statins prevent the amyloidogenic process by inhibiting cholesterol and isoprenoid synthesis.
4.3. Subcellular distribution and transport of cholesterol in APP processing
4.3.1. Cholesterol intracellular distribution and trafficking in APP processing
Several lines of evidence implicate intracellular cholesterol distribution, rather than total cholesterol levels, as the relevant factor in the regulation of Aβ generation. Blocking the activity of the acyl-coenzyme A: cholesterol acyltransferase (ACAT), which regulates the equilibrium between free cholesterol and cholesteryl esters (CE), has an important effect on the amyloidogenic pathway, both in vitro and in vivo [140, 141]. Cholesterol overproduction, in the absence of ACAT activity, almost completely blocks Aβ production, while increases in CE levels have the opposite effect. The mechanisms involved in this dramatic effect of CE are not clear but could relate to the activity of secretases [141]. In vivo inhibition of ACAT with the drug CP-113,818 in AD model mice had a remarkable effect on the reduction of Aβ in brains. This correlates with some improvements in special learning, making ACAT a possible therapeutically target for AD treatment [140].
In the inherited neurodegenerative condition of Niemann-Pick C (NPC) disease, exogenous cholesterol internalized in lipoproteins is unable to incorporate into the metabolically active cholesterol pool [142, 143]. NPC1 or NPC2 mutant proteins are associated with cholesterol accumulation in late endosomes and lysosomes, with concomitant blocking of cholesterol esterification by ACAT in the ER [144]. A similar cholesterol accumulation is induced in vitro by using the agent U18666A [144]. Brains from NPC mice have Aβ40 and 42 levels subtly increased [145]. However, the most relevant alteration is the stimulation of γ-secretase along with a change in PS1 distribution from the ER, where most of the neuronal PS1 is found [146], to a rab5-endosomal compartment [145]. After loading neurons with LDL-cholesterol, PS1 and PS2 redistribute to a rab7 compartment, close to the late-endosomal compartment where cholesterol is trapped and the γ-secretase activity is increased [147]. The change of PS1 in NPC models is associated with an accumulation of insoluble Aβ42, [147, 148] consistent with the observed increase in intraneuronal Aβ in human NPC brains [149]. Interestingly, in human and mouse NPC brains, βCTFs are increased [145, 149] but the in vitro studies show differing effects on the β-secretase-mediated APP processing [147, 149]. Since β-secretase activity is related to cholesterol levels in neurons [150] and is more active when in LR [134], it is conceivable that an alteration in cholesterol distribution in NPC affects the β-secretase-mediated APP processing. Other proteins that regulate APP processing and are raft-associated (e.g. apoER2) could also be altered in its function or distribution. Overall, this evidence indicates that cholesterol intracellular trafficking and distribution are relevant in the APP processing pathways.
4.3.2. Oxysterols and their protein partners, LXR and OSBP1, in AD
Oxysterols are cholesterol-derived ligands of the liver X receptor (LXR) and of oxysterol binding protein 1 (OSBP1) that have protective roles in AD [151, 152]. The nuclear receptor LXR regulates the expression of several genes related to cholesterol transport and metabolism [153], and ultimately has an impact on APP processing and the inflammatory responses related to AD [154, 155]. Brains from AD patients have lower levels of LXRβ [119]. The in vivo protective role of LXR in AD pathogenesis is evidenced in the negative effect of the global deletion of LXRα or LXRβ in APP transgenic mice, which results in increased amyloid plaque load [154]. The genes for apoE and ABCA1 (ATP-binding cassette transporter A1), a major regulator of cholesterol efflux to apoAI and apoE as acceptors, are LXR targets [156, 157]. ABCA1 deficiency increases Aβ deposition in APP transgenic mice along with a significant reduction in total brain apoE [158, 159]. Brain overexpression of ABCA1 is related to an increase in the levels of lipidated-apoE and a reduction of Aβ deposits in APP transgenic mice [160]. Similarly, treating AD model mice with LXR agonists induces an upregulation of the expression of ABCA1 and apoE in astrocytes and microglia, and a reduction of Aβ levels as well as proteins related to inflammatory response and apoptosis [155]. In cells overexpressing hAPP, oxysterols decrease the cellular cholesterol content by the expression of ABCA1 and the induction of apoA-I-mediated cholesterol efflux, thereby significantly reducing Aβ levels [151, 152]. This reduction likely results from a blocking of both β- and γ-secretase-mediated processing of APP [151].
Oxysterols are also ligands for Oxysterol Binding Protein 1 (OSBP1), a cytosolic protein belonging to a family of sterol-binding proteins. OSBP1 binds both 25-hydroxy cholesterol and cholesterol, and has been linked to APP processing [161]. OSBP1 overexpression or silencing reduces or increases, respectively, APP processing, likely by regulating APP trafficking [161].
5. Summary and future directions
Alterations in APP processing to favor Aβ production and its accumulation in the brain are key pathogenic events in AD. A number of cell surface proteins, including members of the LDLR family discussed here, interact with APP and regulate its trafficking and processing to Aβ. Several APP-interacting intracellular adaptor proteins not discussed here, including FE65, X11, Dab1/2, and sorting nexin 17 [18], also regulate APP trafficking and processing to Aβ. A schematic diagram illustrating the roles of some of these proteins in APP trafficking and processing is shown in Figure 2. Although we have focused primarily on the APP trafficking roles, these LDLR family members themselves are also regulated by alternative splicing and subjected to proteolysis that can further influence their interactions with APP and intricate intracellular signaling pathways [60, 162]. Because the production of AD-related Aβ occurs in a very polarized cell type, the neurons, the distribution of associated proteins adds an additional level of complexity. This consideration in their polarized sorting and distribution should lead to a better understanding on precise roles of the LDLR family member in APP trafficking and processing. Future studies are needed to determine if interactions between these receptors, ligands, APP, and/or co-receptors can activate downstream signaling cascades that may ultimately affect the pathogenesis of AD. Further, the exact role of these receptors in Aβ metabolism in vivo requires unique animal models that delete these receptors in specific brain regions and/or cell types. Finally, the impact and the underlying mechanisms of cholesterol in APP trafficking and processing require further investigation. APP processing to Aβ is an inherently complex process. The study of LDLR family members and cholesterol that affect APP processing is an important step to uncover new therapies to reduce Aβ and its associated dementia.
Fig. 2.
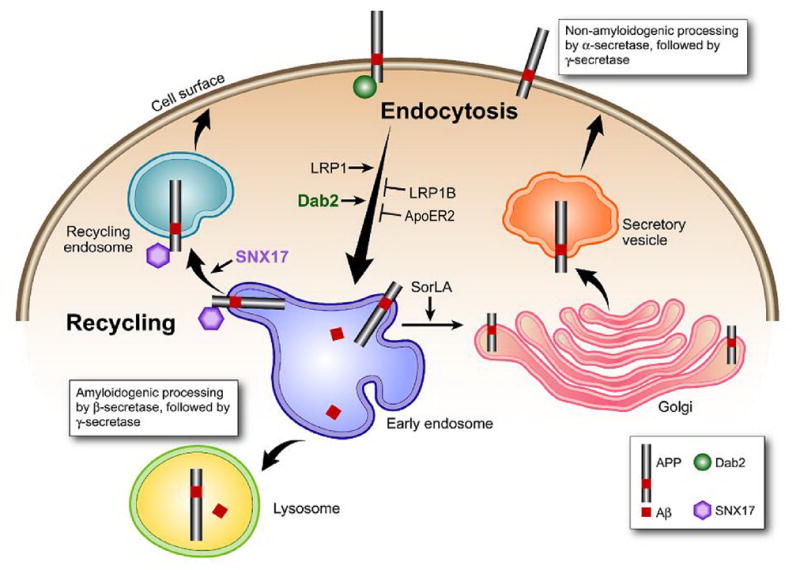
Model of APP processing pathways regulated by LDLR family members. LRP1 fast endocytosis enhances APP endocytosis and processing to Aβ, whereas LRP1B and apoER2 slow endocytosis rates retain APP at the cell surface. LRP1B decreases APP processing to Aβ whereas apoER2 has differential effects depending on ligand binding. SorLA/LR11 may shuttle APP to the Golgi compartments and reduce its processing by β-secretase in the early endosome, thus decreasing processing to Aβ. Adaptor proteins Dab2 and sorting nexin 17 (SNX17), which also regulate APP trafficking but not discussed in this review, are also depicted here.
Acknowledgments
Work in Bu’s laboratory is supported by grants from the National Institutes of Health, the Alzheimer’s Association and the American Health Assistance Foundation. Work in Marzolo’s laboratory is supported by Fondo de Investigación Avanzada en Areas Prioritarias (FONDAP) Grant 13980001 and the Millenium Institute for Fundamental and Applied Biology (MIFAB). We wish to thank Olivia Ying for her critical reading of this manuscript, Jiyeon Lee, Barbara Ramos and Pamela Farfán for their contributions in the illustrations.
Abbreviations
- LDLR
low-density lipoprotein receptor
- LRP
LDLR-related protein
- apoER2
apoE receptor 2
- sorLA
sorting protein-related receptor containing LDLR class A repeats
- AD
Alzheimer’s disease
- Aβ
amyloid-β peptide
- APP
β-amyloid precursor protein
- APLP1
amyloid precursor like proteins-1
- APLP2
amyloid precursor like proteins-2
- sAPP
soluble APP fragment
- CTF
c-terminal fragment
- LR
lipid rafts
- RAP
receptor-associated protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Selkoe DJ. Deciphering the genesis and fate of amyloid beta-protein yields novel therapies for Alzheimer disease. J Clin Invest. 2002;110:1375–81. doi: 10.1172/JCI16783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Cell biology of the amyloid b-protein precursor and the mechanism of Alzheimer’s disease. Annu Rev Cell Biol. 1994;10:373–403. doi: 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- 3.Zheng H, Koo E. The amyloid precursor protein: beyond amyloid. Mol Neurodegener. 2006;1:5. doi: 10.1186/1750-1326-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 5.Citron M. Strategies for disease modification in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:677–85. doi: 10.1038/nrn1495. [DOI] [PubMed] [Google Scholar]
- 6.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–62. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46:860–6. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 8.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 9.Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, et al. Soluble oligomers of beta amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924:133–40. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- 10.Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–53. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 11.Manelli AM, Stine WB, Van Eldik LJ, LaDu MJ. ApoE and Abeta1–42 interactions: effects of isoform and conformation on structure and function. J Mol Neurosci. 2004;23:235–46. doi: 10.1385/JMN:23:3:235. [DOI] [PubMed] [Google Scholar]
- 12.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–7. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 13.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008 doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 15.Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rulicke T, et al. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci. 2000;20:7951–63. doi: 10.1523/JNEUROSCI.20-21-07951.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muller T, Meyer HE, Egensperger R, Marcus K. The amyloid precursor protein intracellular domain (AICD) as modulator of gene expression, apoptosis, and cytoskeletal dynamics-Relevance for Alzheimer’s disease. Prog Neurobiol. 2008;85:393–406. doi: 10.1016/j.pneurobio.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 17.Young-Pearse TL, Bai J, Chang R, Zheng JB, LoTurco JJ, Selkoe DJ. A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J Neurosci. 2007;27:14459–69. doi: 10.1523/JNEUROSCI.4701-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee J, Retamal C, Cuitino L, Caruano-Yzermans A, Shin JE, van Kerkhof P, et al. Adaptor protein sorting nexin 17 regulates amyloid precursor protein trafficking and processing in the early endosomes. J Biol Chem. 2008;283:11501–8. doi: 10.1074/jbc.M800642200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez RG, Soriano S, Hayes JD, Ostaszewski B, Xia W, Selkoe DJ, et al. Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J Biol Chem. 1999;274:18851–6. doi: 10.1074/jbc.274.27.18851. [DOI] [PubMed] [Google Scholar]
- 20.Cole SL, Vassar R. The Alzheimer’s disease beta-secretase enzyme, BACE1. Mol Neurodegener. 2007;2:22. doi: 10.1186/1750-1326-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vetrivel K, Zhang Y-w, Xu H, Thinakaran G. Pathological and physiological functions of presenilins. Mol Neurodegener. 2006;1:4. doi: 10.1186/1750-1326-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allinson TM, Parkin ET, Turner AJ, Hooper NM. ADAMs family members as amyloid precursor protein alpha-secretases. J Neurosci Res. 2003;74:342–52. doi: 10.1002/jnr.10737. [DOI] [PubMed] [Google Scholar]
- 23.Nordstedt C, Caporaso GL, Thyberg J, Gandy SE, Greengard P. Identification of the Alzheimer beta/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. J Biol Chem. 1993;268:608–12. [PubMed] [Google Scholar]
- 24.Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994;269:17386–9. [PubMed] [Google Scholar]
- 25.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–41. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 26.Ray WJ, Yao M, Mumm J, Schroeter EH, Saftig P, Wolfe M, et al. Cell surface presenilin-1 participates in the gamma-secretase-like proteolysis of Notch. J Biol Chem. 1999;274:36801–7. doi: 10.1074/jbc.274.51.36801. [DOI] [PubMed] [Google Scholar]
- 27.Pasternak SH, Bagshaw RD, Guiral M, Zhang S, Ackerley CA, Pak BJ, et al. Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J Biol Chem. 2003;278:26687–94. doi: 10.1074/jbc.m304009200. [DOI] [PubMed] [Google Scholar]
- 28.Cupers P, Bentahir M, Craessaerts K, Orlans I, Vanderstichele H, Saftig P, et al. The discrepancy between presenilin subcellular localization and gamma-secretase processing of amyloid precursor protein. J Cell Biol. 2001;154:731–40. doi: 10.1083/jcb.200104045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parvathy S, Hussain I, Karran EH, Turner AJ, Hooper NM. Cleavage of Alzheimer’s amyloid precursor protein by alpha-secretase occurs at the surface of neuronal cells. Biochemistry. 1999;38:9728–34. doi: 10.1021/bi9906827. [DOI] [PubMed] [Google Scholar]
- 30.Herz J, Bock HH. Lipoprotein receptors in the nervous system. Annu Rev Biochem. 2002;71:405–34. doi: 10.1146/annurev.biochem.71.110601.135342. [DOI] [PubMed] [Google Scholar]
- 31.Herz J, Gotthardt M, Willnow TE. Cellular signalling by lipoprotein receptors. Curr Opin Lipidol. 2000;11:161–6. doi: 10.1097/00041433-200004000-00009. [DOI] [PubMed] [Google Scholar]
- 32.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–81. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bu G. Receptor-associated protein: a specialized chaperone and antagonist for members of the LDL receptor gene family. Curr Opin Lipidol. 1998;9:149–55. doi: 10.1097/00041433-199804000-00012. [DOI] [PubMed] [Google Scholar]
- 34.Herz J, Hamann U, Rogne S, Myklebost O, Gausepohl H, Stanley KK. Surface location and high affinity for calcium of a 500-kd liver membrane protein closely related to the LDL-receptor suggest a physiological role as lipoprotein receptor. EMBO J. 1988;7:4119–27. doi: 10.1002/j.1460-2075.1988.tb03306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beisiegel U, Weber W, Ihrke G, Herz J, Stanley KK. The LDL-receptor-related protein, LRP, is an apolipoprotein E-binding protein. Nature. 1989;341:162–4. doi: 10.1038/341162a0. [DOI] [PubMed] [Google Scholar]
- 36.Strickland DK, Ashcom JD, Williams S, Burgess WH, Migliorini M, Argraves WS. Sequence identity between the alpha 2-macroglobulin receptor and low density lipoprotein receptor-related protein suggests that this molecule is a multifunctional receptor. J Biol Chem. 1990;265:17401–4. [PubMed] [Google Scholar]
- 37.Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest. 2001;108:779–84. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Obermoeller-McCormick L, Li Y, Osaka H, FitzGerald D, Schwartz A, Bu G. Dissection of receptor folding and ligand-binding property with functional minireceptors of LDL receptor-related protein. J Cell Sci. 2001;114:899–908. doi: 10.1242/jcs.114.5.899. [DOI] [PubMed] [Google Scholar]
- 39.Croy JE, Brandon T, Komives EA. Two apolipoprotein E mimetic peptides, ApoE(130–149) and ApoE(141–155)2, bind to LRP1. Biochemistry. 2004;43:7328–35. doi: 10.1021/bi036208p. [DOI] [PubMed] [Google Scholar]
- 40.Croy JE, Shin WD, Knauer MF, Knauer DJ, Komives EA. All three LDL receptor homology regions of the LDL receptor-related protein bind multiple ligands. Biochemistry. 2003;42:13049–57. doi: 10.1021/bi034752s. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Marzolo MP, Kerkhof P, Strous GJ, Bu G. The YXXL motif, but not the two NPXY motifs, serves as the dominant endocytosis signal For LDL receptor-related protein (LRP) J Biol Chem. 2000;275:17187–94. doi: 10.1074/jbc.M000490200. [DOI] [PubMed] [Google Scholar]
- 42.Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–44. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 43.Zerbinatti CV, Wahrle SE, Kim H, Cam JA, Bales K, Paul SM, et al. Apolipoprotein E and low density lipoprotein receptor-related protein facilitate intraneuronal Abeta42 accumulation in amyloid model mice. J Biol Chem. 2006;281:36180–6. doi: 10.1074/jbc.M604436200. [DOI] [PubMed] [Google Scholar]
- 44.Rebeck GW, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron. 1993;11:575–80. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 45.Kang DE, Pietrzik CU, Baum L, Chevallier N, Merriam DE, Kounnas MZ, et al. Modulation of amyloid beta-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor-related protein pathway. J Clin Invest. 2000;106:1159–66. doi: 10.1172/JCI11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kang DE, Saitoh T, Chen X, Xia Y, Masliah E, Hansen LA, et al. Genetic association of the low-density lipoprotein receptor-related protein gene (LRP), an apolipoprotein E receptor, with late-onset Alzheimers disease. Neurology. 1997;49:56–61. doi: 10.1212/wnl.49.1.56. [DOI] [PubMed] [Google Scholar]
- 47.Kounnas MZ, Chappell DA, Wong H, Argraves WS, Strickland DK. The cellular internalization and degradation of hepatic lipase is mediated by low density lipoprotein receptor-related protein and requires cell surface proteoglycans. J Biol Chem. 1995;270:9307–12. doi: 10.1074/jbc.270.16.9307. [DOI] [PubMed] [Google Scholar]
- 48.Knauer MF, Orlando RA, Glabe CG. Cell surface APP751 forms complexes with protease nexin 2 ligands and is internalized via the low density lipoprotein receptor-related protein (LRP) Brain Res. 1996;740:6–14. doi: 10.1016/s0006-8993(96)00711-1. [DOI] [PubMed] [Google Scholar]
- 49.Trommsdorff R, Borg JP, Margolis B, Herz J. Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J Biol Chem. 1998;273:33556–60. doi: 10.1074/jbc.273.50.33556. [DOI] [PubMed] [Google Scholar]
- 50.Cam J, Bu G. Modulation of beta-amyloid precursor protein trafficking and processing by the low density lipoprotein receptor family. Mol Neurodegener. 2006;1:8. doi: 10.1186/1750-1326-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bu G, Cam JA, Zerbinatti CV. LRP in Amyloid-beta Production and Metabolism. Ann NY Acad Sci. 2006;1086:35–53. doi: 10.1196/annals.1377.005. [DOI] [PubMed] [Google Scholar]
- 52.Ulery PG, Beers J, Mikhailenko I, Tanzi RE, Rebeck GW, Hyman BT, et al. Modulation of beta-amyloid precursor protein processing by the low density lipoprotein receptor-related protein (LRP). Evidence that lrp contributes to the pathogenesis of alzheimer’s disease. J Biol Chem. 2000;275:7410–5. doi: 10.1074/jbc.275.10.7410. [DOI] [PubMed] [Google Scholar]
- 53.Pietrzik CU, Busse T, Merriam DE, Weggen S, Koo EH. The cytoplasmic domain of the LDL receptor-related protein regulates multiple steps in APP processing. EMBO J. 2002;21:5691–700. doi: 10.1093/emboj/cdf568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cam JA, Zerbinatti CV, Li Y, Bu G. Rapid endocytosis of the low density lipoprotein receptor-related protein modulates cell surface distribution and processing of the beta-amyloid precursor protein. J Biol Chem. 2005;280:15464–70. doi: 10.1074/jbc.M500613200. [DOI] [PubMed] [Google Scholar]
- 55.Zerbinatti CV, Wozniak DF, Cirrito J, Cam JA, Osaka H, Bales KR, et al. Increased soluble amyloid-beta peptide and memory deficits in amyloid model mice overexpressing the low-density lipoprotein receptor-related protein. Proc Natl Acad Sci U S A. 2004;101:1075–80. doi: 10.1073/pnas.0305803101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ye S, Huang Y, Mullendorff K, Dong L, Giedt G, Meng EC, et al. Apolipoprotein (apo) E4 enhances amyloid beta peptide production in cultured neuronal cells: apoE structure as a potential therapeutic target. Proc Natl Acad Sci U S A. 2005;102:18700–5. doi: 10.1073/pnas.0508693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.von Arnim CA, Kinoshita A, Peltan ID, Tangredi MM, Herl L, Lee BM, et al. The low density lipoprotein receptor-related protein (LRP) is a novel beta-secretase (BACE1) substrate. J Biol Chem. 2005;280:17777–85. doi: 10.1074/jbc.M414248200. [DOI] [PubMed] [Google Scholar]
- 58.May P, Reddy YK, Herz J. Proteolytic processing of low density lipoprotein receptor-related protein mediates regulated release of its intracellular domain. J Biol Chem. 2002;277:18736–43. doi: 10.1074/jbc.M201979200. [DOI] [PubMed] [Google Scholar]
- 59.Lleo A, Waldron E, von Arnim CA, Herl L, Tangredi MM, Peltan ID, et al. Low density lipoprotein receptor-related protein (LRP) interacts with presenilin 1 and is a competitive substrate of the amyloid precursor protein (APP) for gamma-secretase. J Biol Chem. 2005;280:27303–9. doi: 10.1074/jbc.M413969200. [DOI] [PubMed] [Google Scholar]
- 60.Rebeck GW, Ladu MJ, Estus S, Bu G, Weeber EJ. The generation and function of soluble apoE receptors in the CNS. Mol Neurodegener. 2006;1:15. doi: 10.1186/1750-1326-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu CX, Musco S, Lisitsina NM, Forgacs E, Minna JD, Lisitsyn NA. LRP-DIT, a putative endocytic receptor gene, is frequently inactivated in non-small cell lung cancer cell lines. Cancer Res. 2000;60:1961–7. [PubMed] [Google Scholar]
- 62.Liu CX, Li Y, Obermoeller-McCormick LM, Schwartz AL, Bu G. The putative tumor suppressor LRP1B, a novel member of the low density lipoprotein (LDL) receptor family, exhibits both overlapping and distinct properties with the LDL receptor-related protein. J Biol Chem. 2001;276:28889–96. doi: 10.1074/jbc.M102727200. [DOI] [PubMed] [Google Scholar]
- 63.Langbein S, Szakacs O, Wilhelm M, Sukosd F, Weber S, Jauch A, et al. Alteration of the LRP1B gene region is associated with high grade of urothelial cancer. Lab Invest. 2002;82:639–43. doi: 10.1038/labinvest.3780458. [DOI] [PubMed] [Google Scholar]
- 64.Sonoda I, Imoto I, Inoue J, Shibata T, Shimada Y, Chin K, et al. Frequent silencing of low density lipoprotein receptor-related protein 1B (LRP1B) expression by genetic and epigenetic mechanisms in esophageal squamous cell carcinoma. Cancer Res. 2004;64:3741–7. doi: 10.1158/0008-5472.CAN-04-0172. [DOI] [PubMed] [Google Scholar]
- 65.Li Y, Knisely JM, Lu W, McCormick LM, Wang J, Henkin J, et al. Low density lipoprotein (LDL) receptor-related protein 1B impairs urokinase receptor regeneration on the cell surface and inhibits cell migration. J Biol Chem. 2002;277:42366–71. doi: 10.1074/jbc.M207705200. [DOI] [PubMed] [Google Scholar]
- 66.Knisely JM, Li Y, Griffith JM, Geuze HJ, Schwartz AL, Bu G. Slow endocytosis of the LDL receptor-related protein 1B: implications for a novel cytoplasmic tail conformation. Exp Cell Res. 2007;313:3298–307. doi: 10.1016/j.yexcr.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cam JA, Zerbinatti CV, Knisely JM, Hecimovic S, Li Y, Bu G. The low density lipoprotein receptor-related protein 1B retains beta-amyloid precursor protein at the cell surface and reduces amyloid-beta peptide production. J Biol Chem. 2004;279:29639–46. doi: 10.1074/jbc.M313893200. [DOI] [PubMed] [Google Scholar]
- 68.Yamazaki H, Bujo H, Kusonoki J, Seimiya K, Kanaki T, Morisaki N, et al. Elements of neural adhesion molecules and a yeast vacuolar protein sorting receptor are present in a novel mammalian low density lipoprotein receptor family member. J Biol Chem. 1996;271:24761–8. doi: 10.1074/jbc.271.40.24761. [DOI] [PubMed] [Google Scholar]
- 69.Jacobsen L, Madsen P, Moestrup SK, Lund AH, Tommerup N, Nykjaer A, et al. Molecular characterization of a novel human hybrid-type receptor that binds the alpha2-macroglobulin receptor-associated protein. J Biol Chem. 1996;271:31379–83. doi: 10.1074/jbc.271.49.31379. [DOI] [PubMed] [Google Scholar]
- 70.Jacobsen L, Madsen P, Jacobsen C, Nielsen MS, Gliemann J, Petersen CM. Activation and functional characterization of the mosaic receptor SorLA/LR11. J Biol Chem. 2001;276:22788–96. doi: 10.1074/jbc.M100857200. [DOI] [PubMed] [Google Scholar]
- 71.Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2005;102:13461–6. doi: 10.1073/pnas.0503689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scherzer CR, Offe K, Gearing M, Rees HD, Fang G, Heilman CJ, et al. Loss of apolipoprotein E receptor LR11 in Alzheimer disease. Arch Neurol. 2004;61:1200–5. doi: 10.1001/archneur.61.8.1200. [DOI] [PubMed] [Google Scholar]
- 73.Andersen OM, Schmidt V, Spoelgen R, Gliemann J, Behlke J, Galatis D, et al. Molecular dissection of the interaction between amyloid precursor protein and its neuronal trafficking receptor SorLA/LR11. Biochemistry. 2006;45:2618–28. doi: 10.1021/bi052120v. [DOI] [PubMed] [Google Scholar]
- 74.Offe K, Dodson SE, Shoemaker JT, Fritz JJ, Gearing M, Levey AI, et al. The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J Neurosci. 2006;26:1596–603. doi: 10.1523/JNEUROSCI.4946-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Spoelgen R, von Arnim CA, Thomas AV, Peltan ID, Koker M, Deng A, et al. Interaction of the cytosolic domains of sorLA/LR11 with the amyloid precursor protein (APP) and beta-secretase beta-site APP-cleaving enzyme. J Neurosci. 2006;26:418–28. doi: 10.1523/JNEUROSCI.3882-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bohm C, Seibel NM, Henkel B, Steiner H, Haass C, Hampe W. SorLA signaling by regulated intramembrane proteolysis. J Biol Chem. 2006;281:14547–53. doi: 10.1074/jbc.M601660200. [DOI] [PubMed] [Google Scholar]
- 77.Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–77. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim DH, Iijima H, Goto K, Sakai J, Ishii H, Kim HJ, et al. Human apolipoprotein E receptor 2. A novel lipoprotein receptor of the low density lipoprotein receptor family predominantly expressed in brain. J Biol Chem. 1996;271:8373–80. doi: 10.1074/jbc.271.14.8373. [DOI] [PubMed] [Google Scholar]
- 79.Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, et al. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–9. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- 80.D’Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24:471–9. doi: 10.1016/s0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 81.Hoe HS, Harris DC, Rebeck GW. Multiple pathways of apolipoprotein E signaling in primary neurons. J Neurochem. 2005;93:145–55. doi: 10.1111/j.1471-4159.2004.03007.x. [DOI] [PubMed] [Google Scholar]
- 82.Hoe HS, Wessner D, Beffert U, Becker AG, Matsuoka Y, Rebeck GW. F-spondin interaction with the apolipoprotein E receptor ApoEr2 affects processing of amyloid precursor protein. Mol Cell Biol. 2005;25:9259–68. doi: 10.1128/MCB.25.21.9259-9268.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fuentealba RA, Barria MI, Lee J, Cam J, Araya C, Escudero CA, et al. ApoER2 expression increases Abeta production while decreasing Amyloid Precursor Protein (APP) endocytosis: Possible role in the partitioning of APP into lipid rafts and in the regulation of gamma-secretase activity. Mol Neurodegener. 2007;2:14. doi: 10.1186/1750-1326-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hoe HS, Pocivavsek A, Chakraborty G, Fu Z, Vicini S, Ehlers MD, et al. Apolipoprotein E receptor 2 interactions with the N-methyl-D-aspartate receptor. J Biol Chem. 2006;281:3425–31. doi: 10.1074/jbc.M509380200. [DOI] [PubMed] [Google Scholar]
- 85.Beffert U, Weeber EJ, Durudas A, Qiu S, Masiulis I, Sweatt JD, et al. Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron. 2005;47:567–79. doi: 10.1016/j.neuron.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 86.Bock HH, Herz J. Reelin activates SRC family tyrosine kinases in neurons. Curr Biol. 2003;13:18–26. doi: 10.1016/s0960-9822(02)01403-3. [DOI] [PubMed] [Google Scholar]
- 87.Hoe HS, Pocivavsek A, Dai H, Chakraborty G, Harris DC, Rebeck GW. Effects of apoE on neuronal signaling and APP processing in rodent brain. Brain Res. 2006;1112:70–9. doi: 10.1016/j.brainres.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 88.Hoe HS, Freeman J, Rebeck GW. Apolipoprotein E decreases tau kinases and phospho-tau levels in primary neurons. Mol Neurodegener. 2006;1:18. doi: 10.1186/1750-1326-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beffert U, Durudas A, Weeber EJ, Stolt PC, Giehl KM, Sweatt JD, et al. Functional dissection of Reelin signaling by site-directed disruption of Disabled-1 adaptor binding to apolipoprotein E receptor 2: distinct roles in development and synaptic plasticity. J Neurosci. 2006;26:2041–52. doi: 10.1523/JNEUROSCI.4566-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Petit-Turcotte C, Aumont N, Beffert U, Dea D, Herz J, Poirier J. The apoE receptor apoER2 is involved in the maintenance of efficient synaptic plasticity. Neurobiol Aging. 2005;26:195–206. doi: 10.1016/j.neurobiolaging.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 91.Chin J, Massaro CM, Palop JJ, Thwin MT, Yu GQ, Bien-Ly N, et al. Reelin depletion in the entorhinal cortex of human amyloid precursor protein transgenic mice and humans with Alzheimer’s disease. J Neurosci. 2007;27:2727–33. doi: 10.1523/JNEUROSCI.3758-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ma SL, Ng HK, Baum L, Pang JC, Chiu HF, Woo J, et al. Low-density lipoprotein receptor-related protein 8 (apolipoprotein E receptor 2) gene polymorphisms in Alzheimer’s disease. Neurosci Lett. 2002;332:216–8. doi: 10.1016/s0304-3940(02)00942-4. [DOI] [PubMed] [Google Scholar]
- 93.Sun XM, Soutar AK. Expression in vitro of alternatively spliced variants of the messenger RNA for human apolipoprotein E receptor-2 identified in human tissues by ribonuclease protection assays. Euro J Biochem. 1999;262:230–9. doi: 10.1046/j.1432-1327.1999.00394.x. [DOI] [PubMed] [Google Scholar]
- 94.Stockinger W, Brandes C, Fasching D, Hermann M, Gotthardt M, Herz J, et al. The reelin receptor ApoER2 recruits JNK-interacting proteins-1 and -2. J Biol Chem. 2000;275:25625–32. doi: 10.1074/jbc.M004119200. [DOI] [PubMed] [Google Scholar]
- 95.Beffert U, Nematollah Farsian F, Masiulis I, Hammer RE, Yoon SO, Giehl KM, et al. ApoE receptor 2 controls neuronal survival in the adult brain. Curr Biol. 2006;16:2446–52. doi: 10.1016/j.cub.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 96.He X, Cooley K, Chung CH, Dashti N, Tang J. Apolipoprotein receptor 2 and X11 alpha/beta mediate apolipoprotein E-induced endocytosis of amyloid-beta precursor protein and beta-secretase, leading to amyloid-beta production. J Neurosci. 2007;27:4052–60. doi: 10.1523/JNEUROSCI.3993-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Riddell DR, Sun XM, Stannard AK, Soutar AK, Owen JS. Localization of apolipoprotein E receptor 2 to caveolae in the plasma membrane. J Lipid Res. 2001;42:998–1002. [PubMed] [Google Scholar]
- 98.Cuitino L, Matute R, Retamal C, Bu G, Inestrosa NC, Marzolo MP. ApoER2 is endocytosed by a clathrin-mediated process involving the adaptor protein Dab2 independent of its Rafts’ association. Traffic. 2005;6:820–38. doi: 10.1111/j.1600-0854.2005.00320.x. [DOI] [PubMed] [Google Scholar]
- 99.Li Y, Lu W, Marzolo MP, Bu G. Differential functions of members of the low density lipoprotein receptor family suggested by their distinct endocytosis rates. J Biol Chem. 2001;276:18000–6. doi: 10.1074/jbc.M101589200. [DOI] [PubMed] [Google Scholar]
- 100.May P, Bock HH, Nimpf J, Herz J. Differential glycosylation regulates processing of lipoprotein receptors by gamma-secretase. J Biol Chem. 2003;278:37386–92. doi: 10.1074/jbc.M305858200. [DOI] [PubMed] [Google Scholar]
- 101.Hoe HS, Rebeck GW. Regulation of ApoE receptor proteolysis by ligand binding. Brain Res Mol Brain Res. 2005;137:31–9. doi: 10.1016/j.molbrainres.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 102.Hoe HS, Tran TS, Matsuoka Y, Howell BW, Rebeck GW. DAB1 and Reelin effects on amyloid precursor protein and ApoE receptor 2 trafficking and processing. J Biol Chem. 2006;281:35176–85. doi: 10.1074/jbc.M602162200. [DOI] [PubMed] [Google Scholar]
- 103.Homayouni R, Rice DS, Sheldon M, Curran T. Disabled-1 binds to the cytoplasmic domain of amyloid precursor-like protein 1. J Neurosci. 1999;19:7507–15. doi: 10.1523/JNEUROSCI.19-17-07507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Parisiadou L, Efthimiopoulos S. Expression of mDab1 promotes the stability and processing of amyloid precursor protein and this effect is counteracted by X11alpha. Neurobiol Aging. 2007;28:377–88. doi: 10.1016/j.neurobiolaging.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 105.Borg JP, Ooi J, Levy E, Margolis B. The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol Cell Biol. 1996;16:6229–41. doi: 10.1128/mcb.16.11.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.King GD, Scott Turner R. Adaptor protein interactions: modulators of amyloid precursor protein metabolism and Alzheimer’s disease risk? Exp Neurol. 2004;185:208–19. doi: 10.1016/j.expneurol.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 107.Hoe HS, Magill LA, Guenette S, Fu Z, Vicini S, Rebeck GW. FE65 interaction with the ApoE receptor ApoEr2. J Biol Chem. 2006;281:24521–30. doi: 10.1074/jbc.M600728200. [DOI] [PubMed] [Google Scholar]
- 108.Sambamurti K, Granholm AC, Kindy MS, Bhat NR, Greig NH, Lahiri DK, et al. Cholesterol and Alzheimer’s disease: clinical and experimental models suggest interactions of different genetic, dietary and environmental risk factors. Curr Drug Targets. 2004;5:517–28. doi: 10.2174/1389450043345335. [DOI] [PubMed] [Google Scholar]
- 109.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 110.Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet. 2000;356:1627–31. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- 111.Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–43. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 112.Sjogren M, Mielke M, Gustafson D, Zandi P, Skoog I. Cholesterol and Alzheimer’s disease--is there a relation? Mech Ageing Dev. 2006;127:138–47. doi: 10.1016/j.mad.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 113.Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P, et al. Simvastatin strongly reduces levels of Alzheimer’s disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci U S A. 2001;98:5856–61. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas-Bryant T, et al. A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2001;8:890–9. doi: 10.1006/nbdi.2001.0422. [DOI] [PubMed] [Google Scholar]
- 115.Park I-H, Hwang EM, Hong HS, Boo JH, Oh SS, Lee J, et al. Lovastatin enhances A[beta] production and senile plaque deposition in female Tg2576 mice. Neurobiol Aging. 2003;24:637–43. doi: 10.1016/s0197-4580(02)00155-0. [DOI] [PubMed] [Google Scholar]
- 116.Abad-Rodriguez J, Ledesma MD, Craessaerts K, Perga S, Medina M, Delacourte A, et al. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J Cell Biol. 2004;167:953–60. doi: 10.1083/jcb.200404149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cordle A, Koenigsknecht-Talboo J, Wilkinson B, Limpert A, Landreth G. Mechanisms of statin-mediated inhibition of small G-protein function. J Biol Chem. 2005;280:34202–9. doi: 10.1074/jbc.M505268200. [DOI] [PubMed] [Google Scholar]
- 118.Paris D, Townsend KP, Humphrey J, Obregon DF, Yokota K, Mullan M. Statins inhibit A beta-neurotoxicity in vitro and A beta-induced vasoconstriction and inflammation in rat aortae. Atherosclerosis. 2002;161:293–9. doi: 10.1016/s0021-9150(01)00660-8. [DOI] [PubMed] [Google Scholar]
- 119.Xiong H, Callaghan D, Jones A, Walker DG, Lue LF, Beach TG, et al. Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol Dis. 2008;29:422–37. doi: 10.1016/j.nbd.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu Q, Zerbinatti CV, Zhang J, Hoe HS, Wang B, Cole SL, et al. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron. 2007;56:66–78. doi: 10.1016/j.neuron.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol. 2003;160:113–23. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vetrivel KS, Cheng H, Kim SH, Chen Y, Barnes NY, Parent AT, et al. Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J Biol Chem. 2005;280:25892–900. doi: 10.1074/jbc.M503570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc Natl Acad Sci U S A. 2001;98:5815–20. doi: 10.1073/pnas.081612998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kalvodova L, Kahya N, Schwille P, Ehehalt R, Verkade P, Drechsel D, et al. Lipids as modulators of proteolytic activity of BACE: Involvement of cholesterol, glycospingolipids, and anionic phospholipids in vitro. J Biol Chem. 2005;280:36815–23. doi: 10.1074/jbc.M504484200. [DOI] [PubMed] [Google Scholar]
- 125.Grimm MOW, Grimm HS, Patzold AJ, Zinser EG, Halonen R, Duering M, et al. Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat Cell Biol. 2005;7:1118–23. doi: 10.1038/ncb1313. [DOI] [PubMed] [Google Scholar]
- 126.Grimm MO, Grimm HS, Tomic I, Beyreuther K, Hartmann T, Bergmann C. Independent inhibition of Alzheimer disease beta- and gamma-secretase cleavage by lowered cholesterol levels. J Biol Chem. 2008;283:11302–11. doi: 10.1074/jbc.M801520200. [DOI] [PubMed] [Google Scholar]
- 127.Kinoshita A, Fukumoto H, Shah T, Whelan CM, Irizarry MC, Hyman BT. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J Cell Sci. 2003;116:3339–46. doi: 10.1242/jcs.00643. [DOI] [PubMed] [Google Scholar]
- 128.Ni Y, Zhao X, Bao G, Zou L, Teng L, Wang Z, et al. Activation of beta2-adrenergic receptor stimulates gamma-secretase activity and accelerates amyloid plaque formation. Nat Med. 2006;12:1390–6. doi: 10.1038/nm1485. [DOI] [PubMed] [Google Scholar]
- 129.Vetrivel KS, Cheng H, Lin W, Sakurai T, Li T, Nukina N, et al. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J Biol Chem. 2004;279:44945–54. doi: 10.1074/jbc.M407986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang M, Haapasalo A, Kim DY, Mackenzie Ingano LA, Pettingell WH, Kovacs DM. Presenilin/gamma-secretase activity regulates protein clearance from the endocytic recycling compartment. FASEB J. 2006 doi: 10.1096/fj.05-5531fje. [DOI] [PubMed] [Google Scholar]
- 131.Gagescu R, Gruenberg J, Smythe E. Membrane dynamics in endocytosis: structure--function relationship. Traffic. 2000;1:84–8. doi: 10.1034/j.1600-0854.2000.010112.x. [DOI] [PubMed] [Google Scholar]
- 132.Mobius W, van Donselaar E, Ohno-Iwashita Y, Shimada Y, Heijnen HF, Slot JW, et al. Recycling compartments and the internal vesicles of multivesicular bodies harbor most of the cholesterol found in the endocytic pathway. Traffic. 2003;4:222–31. doi: 10.1034/j.1600-0854.2003.00072.x. [DOI] [PubMed] [Google Scholar]
- 133.Riddell DR, Christie G, Hussain I, Dingwall C. Compartmentalization of [beta]-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Current Biology. 2001;11:1288–93. doi: 10.1016/s0960-9822(01)00394-3. [DOI] [PubMed] [Google Scholar]
- 134.Cordy JM, Hussain I, Dingwall C, Hooper NM, Turner AJ. Exclusively targeting beta-secretase to lipid rafts by GPI-anchor addition up-regulates beta-site processing of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2003;100:11735–40. doi: 10.1073/pnas.1635130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Werner N, Nickenig G, Laufs U. Pleiotropic effects of HMG-CoA reductase inhibitors. Basic Res Cardiol. 2002;97:105–16. doi: 10.1007/s003950200000. [DOI] [PubMed] [Google Scholar]
- 136.Parvathy S, Ehrlich M, Pedrini S, Diaz N, Refolo L, Buxbaum JD, et al. Atorvastatin-induced activation of Alzheimer’s alpha secretase is resistant to standard inhibitors of protein phosphorylation-regulated ectodomain shedding. J Neurochem. 2004;90:1005–10. doi: 10.1111/j.1471-4159.2004.02521.x. [DOI] [PubMed] [Google Scholar]
- 137.Ostrowski SM, Wilkinson BL, Golde TE, Landreth G. Statins reduce amyloid-beta production through inhibition of protein isoprenylation. J Biol Chem. 2007;282:26832–44. doi: 10.1074/jbc.M702640200. [DOI] [PubMed] [Google Scholar]
- 138.Won JS, Im YB, Khan M, Contreras M, Singh AK, Singh I. Lovastatin inhibits amyloid precursor protein (APP) beta-cleavage through reduction of APP distribution in Lubrol WX extractable low density lipid rafts. J Neurochem. 2008;105:1536–49. doi: 10.1111/j.1471-4159.2008.05283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhou Y, Suram A, Venugopal C, Prakasam A, Lin S, Su Y, et al. Geranylgeranyl pyrophosphate stimulates gamma-secretase to increase the generation of Abeta and APP-CTFgamma. FASEB J. 2008;22:47–54. doi: 10.1096/fj.07-8175com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hutter-Paier B, Huttunen HJ, Puglielli L, Eckman CB, Kim DY, Hofmeister A, et al. The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease. Neuron. 2004;44:227–38. doi: 10.1016/j.neuron.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 141.Puglielli L, Konopka G, Pack-Chung E, Ingano LA, Berezovska O, Hyman BT, et al. Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat Cell Biol. 2001;3:905–12. doi: 10.1038/ncb1001-905. [DOI] [PubMed] [Google Scholar]
- 142.Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–31. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- 143.Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, et al. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290:2298–301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- 144.Liscum L, Faust JR. The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-beta-[2-(diethylamino)ethoxy]androst-5-en-17-one. J Biol Chem. 1989;264:11796–806. [PubMed] [Google Scholar]
- 145.Burns M, Gaynor K, Olm V, Mercken M, LaFrancois J, Wang L, et al. Presenilin redistribution associated with aberrant cholesterol transport enhances beta-amyloid production in vivo. J Neurosci. 2003;23:5645–9. doi: 10.1523/JNEUROSCI.23-13-05645.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Annaert W, De Strooper B. Presenilins: molecular switches between proteolysis and signal transduction. Trends Neurosci. 1999;22:439–43. doi: 10.1016/s0166-2236(99)01455-1. [DOI] [PubMed] [Google Scholar]
- 147.Runz H, Rietdorf J, Tomic I, de Bernard M, Beyreuther K, Pepperkok R, et al. Inhibition of intracellular cholesterol transport alters presenilin localization and amyloid precursor protein processing in neuronal cells. J Neurosci. 2002;22:1679–89. doi: 10.1523/JNEUROSCI.22-05-01679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yamazaki T, Chang TY, Haass C, Ihara Y. Accumulation and aggregation of amyloid beta-protein in late endosomes of Niemann-pick type C cells. J Biol Chem. 2001;276:4454–60. doi: 10.1074/jbc.M009598200. [DOI] [PubMed] [Google Scholar]
- 149.Jin LW, Shie FS, Maezawa I, Vincent I, Bird T. Intracellular accumulation of amyloidogenic fragments of amyloid-beta precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities. Am J Pathol. 2004;164:975–85. doi: 10.1016/s0002-9440(10)63185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci U S A. 1998;95:6460–4. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sun Y, Yao J, Kim TW, Tall AR. Expression of liver X receptor target genes decreases cellular amyloid beta peptide secretion. J Biol Chem. 2003;278:27688–94. doi: 10.1074/jbc.M300760200. [DOI] [PubMed] [Google Scholar]
- 152.Koldamova RP, Lefterov IM, Ikonomovic MD, Skoko J, Lefterov PI, Isanski BA, et al. 22R-hydroxycholesterol and 9-cis-retinoic acid induce ATP-binding cassette transporter A1 expression and cholesterol efflux in brain cells and decrease amyloid beta secretion. J Biol Chem. 2003;278:13244–56. doi: 10.1074/jbc.M300044200. [DOI] [PubMed] [Google Scholar]
- 153.Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. 2006;116:607–14. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, et al. Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc Natl Acad Sci U S A. 2007;104:10601–6. doi: 10.1073/pnas.0701096104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lefterov I, Bookout A, Wang Z, Staufenbiel M, Mangelsdorf D, Koldamova R. Expression profiling in APP23 mouse brain: inhibition of Abeta amyloidosis and inflammation in response to LXR agonist treatment. Mol Neurodegener. 2007;2:20. doi: 10.1186/1750-1326-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Laffitte BA, Repa JJ, Joseph SB, Wilpitz DC, Kast HR, Mangelsdorf DJ, et al. LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc Natl Acad Sci U S A. 2001;98:507–12. doi: 10.1073/pnas.021488798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Liang Y, Lin S, Beyer TP, Zhang Y, Wu X, Bales KR, et al. A liver X receptor and retinoid X receptor heterodimer mediates apolipoprotein E expression, secretion and cholesterol homeostasis in astrocytes. J Neurochem. 2004;88:623–34. doi: 10.1111/j.1471-4159.2004.02183.x. [DOI] [PubMed] [Google Scholar]
- 158.Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem. 2005;280:43224–35. doi: 10.1074/jbc.M504513200. [DOI] [PubMed] [Google Scholar]
- 159.Wahrle SE, Jiang H, Parsadanian M, Hartman RE, Bales KR, Paul SM, et al. Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem. 2005;280:43236–42. doi: 10.1074/jbc.M508780200. [DOI] [PubMed] [Google Scholar]
- 160.Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118:671–82. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zerbinatti CV, Cordy JM, Chen CD, Guillily M, Suon S, Ray WJ, et al. Oxysterol-binding protein-1 (OSBP1) modulates processing and trafficking of the amyloid precursor protein. Mol Neurodegener. 2008;3:5. doi: 10.1186/1750-1326-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hoe HS, Rebeck GW. Regulated proteolysis of APP and ApoE receptors. Mol Neurobiol. 2008;37:64–72. doi: 10.1007/s12035-008-8017-0. [DOI] [PubMed] [Google Scholar]