

Abstract
Acute intermittent hypoxia (AIH) elicits a form of respiratory plasticity known as long-term facilitation (LTF). We hypothesized that: 1) daily AIH (dAIH) preconditioning enhances phrenic and hypoglossal (XII) LTF in a rat strain with low constitutive LTF expression; 2) dAIH induces brain-derived neurotrophic factor (BDNF), a critical protein for phrenic LTF (pLTF) in the cervical spinal cord; and 3) dAIH increases post-AIH extracellular regulated kinase (ERK) activation. Phrenic and XII motor output were monitored in anesthetized dAIH- or sham-treated Brown Norway rats with and without acute AIH. pLTF was observed in both sham (18 ± 9% baseline; 60 min post-hypoxia; p < 0.05; n = 18) and dAIH treated rats (37 ± 8%; p < 0.05; n = 14), but these values were not significantly different (p = 0.13). XII LTF was not observed in sham-treated rats (4 ± 5%), but was revealed in dAIH pretreated rats (48 ± 18%; p < 0.05). dAIH preconditioning increased basal ventral cervical BDNF protein levels (24 ± 8%; p < 0.05), but had no significant effect on ERK phosphorylation. AIH increased BDNF in sham (25 ± 8%; p < 0.05), but not dAIH-pretreated rats (−7 ± 4%), and had complex effects on ERK phosphorylation (ERK2 increased in shams whereas ERK1 increased in dAIH-treated rats). Thus, dAIH elicits metaplasticity in LTF, revealing XII LTF in a rat strain with no constitutive XII LTF expression. Increased BDNF synthesis may no longer be necessary for phrenic LTF following dAIH preconditioning since BDNF concentration is already elevated.
Keywords: respiratory plasticity, metaplasticity, phrenic, hypoglossal, intermittent hypoxia, brain derived neurotrophic factor, extracellular regulated kinase
The ability to alter the output of a physiological system in response to acute and/or chronic challenges is critical for optimal system performance. When challenges are prolonged, most neural systems exhibit lasting changes or plasticity, including the respiratory control system (Mitchell and Johnson, 2003). For example, acute exposure to intermittent hypoxia (AIH) elicits a form of respiratory plasticity known as respiratory long-term facilitation (LTF) (Mitchell et al., 2001; Powell et al., 1998). LTF is a progressive increase in ventilation or respiratory motor output (e.g., phrenic and/or hypoglossal) observed for hours post-AIH (Millhorn et al., 1980; Mitchell et al., 2001; Olson et al., 2001). While the physiological function of LTF remains unclear, it may serve as a compensatory mechanism to stabilize respiratory output following short bouts of hypoxia, such as that observed in patients with obstructive sleep apnea (Mahamed and Mitchell, 2007). In rats, the magnitude of AIH-induced respiratory LTF exhibits strain (Bavis et al., 2003) and sub-strain differences (Fuller et al., 2000; Fuller et al., 2001). For example, Brown Norway rats exhibit minimal phrenic or hypoglossal (XII) LTF relative to other inbred rat strains, such as Fischer 344 and Lewis rats (Bavis et al., 2003).
LTF in phrenic motor output (pLTF) requires new synthesis of brain-derived neurotrophic factor (BDNF), and activation of its high affinity receptor tyrosine kinase, TrkB (Baker-Herman et al., 2004). Other proteins likely to be necessary for pLTF include “downstream” signaling molecules from TrkB, including MAP kinases such as extracellular regulated kinases 1 and 2 (ERK1/2; (Errico et al., 2001; Gooney and Lynch, 2001; Huang and Reichardt, 2003; Lin et al., 2003; Ormond et al., 2004). ERKs are necessary in multiple forms of neural plasticity, including hippocampal long-term potentiation in rats (Coogan et al., 1999; Kelleher et al., 2004; Ying et al., 2002) and long-term facilitation in Aplysia (Liu et al., 2004; Ormond et al., 2004; Sharma et al., 2003). Since ERK1/2 are phosphorylated by BDNF-dependent TrkB activation and are necessary for multiple forms of synaptic plasticity (Gooney et al., 2002; Patterson et al., 2001; Slack et al., 2005; Slack et al., 2004), ERK1/2 are attractive candidate molecules in the mechanism of pLTF.
Respiratory LTF exhibits metaplasticity, since preconditioning with chronic intermittent hypoxia (7 days) enhances phrenic and XII LTF (Ling et al., 2001; McGuire et al., 2003; Zabka et al., 2003). Little is known concerning the cellular and synaptic mechanisms of metaplasticity in LTF following chronic intermittent hypoxia. Enhanced LTF following chronic intermittent hypoxia requires reactive oxygen species formation (Peng and Prabhakar, 2003) and contributions from novel serotonin receptors such as the 5-HT7 receptor (Ling et al., 2001; McGuire et al., 2004). However, the respective contributions of neurotrophins and protein kinases to this form of metaplasticity are not known. We hypothesize that metaplasticity is due in part to enhancement of the same cellular mechanisms that underlie pLTF in normal rats.
Here, we assessed the ability of a less “severe” model of preconditioning with intermittent hypoxia to elicit respiratory metaplasticity. In specific, we tested whether daily exposure to acute intermittent hypoxia (dAIH; 10 episodes) enhanced phrenic and XII LTF in a rat strain with low constitutive LTF, the Brown Norway rat. Furthermore, we investigated the effects of AIH and dAIH on spinal BDNF protein concentration and ERK1/2 phosphorylation in an initial attempt to investigate spinal mechanisms of enhanced pLTF following dAIH preconditioning. We hypothesized that dAIH would: 1) enhance phrenic and XII LTF, 2) increase ventral cervical BDNF expression and ERK1/2 phosphorylation, and 3) enhance changes in ventral cervical BDNF levels and ERK1/2 phosphorylation following subsequent AIH exposure.
MATERIALS AND METHODS
Experimental animals
Experiments were performed using 3–5 month old adult, male Brown Norway rats (Colony 217, Harlan Inc., Madison, WI). Animals were individually housed in a controlled environment (12 hr light/dark cycle), with food and water ad libitum. The Animal Care and Use Committee in the School of Veterinary Medicine at the University of Wisconsin approved all protocols.
Hypoxic conditioning
The day prior to the start of hypoxic preconditioning, rats were acclimated to custom-made Plexiglas chambers (1 rat per chamber, 12 in × 4.5 in × 4.5 in, gas flow through chamber = 4 L/min) under normoxic conditions (FIO2 = 0.21 ± 0.005). Over the next 7 days, rats were placed daily into the Plexiglas chambers, allowed a 20 min acclimation period under normoxic conditions, then exposed to daily acute intermittent hypoxia (dAIH; n = 20) or an equivalent duration of normoxia (control; n = 27). dAIH consisted of 10, 5-min episodes of hypoxia (FIO2 = 0.11) interspersed with 5-min normoxic intervals (total chamber time = 120 minutes). Normoxic and hypoxic conditions were established by mixing O2 and N2 gas via a custom-made computer-controlled system to obtain the desired inspired oxygen concentrations. Within the chambers, O2 levels were continuously monitored (S3A Oxygen Analyzer, Applied Electrochemistry Inc., Sunnyvale, CA). Changes in O2 levels within the chamber were reached within 60 ± 10 sec. The rats were poikilocapnic; thus, arterial CO2 levels were not controlled in this study.
Surgical Preparation and Nerve Isolation
Anesthetic induction of dAIH- and normoxia-conditioned Brown Norway rats was initiated with isoflurane in a closed chamber and then maintained with a nose cone (3.5% isoflurane in 50% O2, balance N2). The trachea was cannulated to permit pump-ventilation (tidal volume, 2 – 2.5 mL; FIO2 = 0.50; Rodent Respirator model 682, Harvard Apparatus, South Natick, MA) and tracheal pressure measurement. Isoflurane was subsequently administered via the inspired gas entering the ventilator. A bilateral vagotomy was performed at the mid-cervical level to prevent entrainment of respiratory motor output with the ventilator. Catheters were placed in the tail vein for fluid administration (1:11 by volume NaHCO3/lactated Ringer’s; 2.5 mL/hr) and the femoral artery for blood pressure measurement and to draw blood samples. Body temperature was maintained at 37.5 ± 1.0 °C using a rectal probe and heated table. The left phrenic and hypoglossal nerves were isolated using a dorsal approach, cut distally, desheathed, bathed in mineral oil and placed on bipolar silver electrodes. Rats were slowly converted to urethane anesthesia (1.6 g/kg, i.v.), then paralyzed with pancuronium bromide (2.5 mg/kg, i.v., supplemented as necessary) to prevent spontaneous breathing movements. End-tidal CO2 was measured throughout the experiment using a flow-through capnograph (Capnogard, Model 1265, Novametrix; Wallingford, CT) with sufficiently rapid response time to measure expiratory gas concentrations in rats.
LTF Protocol
The CO2 apneic and recruitment thresholds were determined by decreasing inspired CO2 levels and/or increasing the pump ventilator rate until phrenic activity ceased, then slowly decreasing the ventilator rate and/or increasing inspired CO2 levels until phrenic activity returned. End-tidal CO2 levels were set at 1–2 mmHg above the recruitment threshold for phrenic activity in all experiments. Stable phrenic and hypoglossal neurograms were established and an initial blood sample was taken to establish baseline PaO2, PaCO2, pH, and base excess values (0.3 ml in 0.5 ml heparinized glass syringe; ABL-500, Radiometer, Copenhagen, Denmark, unused blood was returned to the animal). Isocapnic conditions (± 1 mmHg from baseline PaCO2) were maintained throughout the protocol by adjusting ventilator frequency and/or inspired CO2. After stable baseline nerve activity was established, AIH was administered (3, 5-min hypoxic episodes; FIO2 = 0.11, PaO2 = 35 – 45 mmHg; 5 min intervals; FIO2 = 0.5, PaO2 > 150 mm Hg) to normoxia (n = 18) or dAIH (n = 14) treated rats. Phrenic and hypoglossal nerve activity was monitored for 60 min post-AIH. Arterial blood samples were drawn and analyzed during the first hypoxic episode, and 15, 30 and 60 min post-AIH. Additional rats treated similarly, but that did not receive AIH were used to investigate the stability of phrenic motor output over a similar time period (time controls; n = 9 and 6 for normoxia and dAIH-treated rats, respectively). Following protocols, rats were killed via exsanguination and the C4–C5 spinal segments were harvested, frozen ventral-side down on dry ice and then stored at −80°C for further analysis (see below).
Electrophysiological data analysis
Phrenic and hypoglossal nerve activity was amplified (× 10,000), band pass filtered (100 Hz to 10 kHz; Model 1700, A-M Systems, Inc., Carlsborg, WA), and integrated (time constant = 50 ms, Model MA-821RSP, CWE Inc., Ardmore, PA). Integrated signals were digitized and processed with commercially available software (WINDAQ software, DATAQ Instruments, Akron, OH). Peak integrated respiratory burst amplitude, burst frequency and mean arterial blood pressure were calculated over a 60 second period just prior to (baseline), at the end of the first hypoxic episode (short-term hypoxic response), and at 30 and 60 min post-treatment. Amplitude data were expressed as a percent change from baseline values. Data were compared within a treatment group with a one-sample t-test or between groups with a two-way repeated measures ANOVA unless otherwise indicated, followed by Fisher’s LSD post-hoc test (SigmaStat 2.03, SPSS Inc., Chicago, IL).
Cervical segment sample preparation
Frozen C4–C5 spinal segments were placed dorsal-side up on a freezing microtome and 50–100μm sections were removed until the central canal was visible. Ventral cervical spinal segments were thawed, quickly weighed, and then immediately homogenized (Tissue Tearor, Biospec Products Inc., Bartlesville, OK) in 500μL of radioimmunoprecipitation (RIPA) buffer (1% NP40, 0.1% SDS, 0.5% deoxycholate, 3mM phenylmethyl sulfonyl fluoride, 0.5mM sodium orthovanadate, 1.5 μM aprotinin, and 10.8 μM leupeptin in phosphate-buffered saline). Cell extracts were spun at 7,000 × g for 10 min, then the supernatant removed and aliquotted for ELISA, immunoblot, or total protein assays. Aliquots were immediately analyzed or stored at −80°C for later analysis.
Total protein quantification
Total protein concentrations for each sample were measured using the bicinchoninic acid method (Pierce Biotechnologies, Inc., Rockford, IL) according to manufacturer’s instructions.
BDNF protein analysis
BDNF protein concentrations were measured using an antibody sandwich ELISA (R&D Systems, Minneapolis, MN). BDNF protein levels were normalized to total protein concentration (see above) and expressed per gram of tissue (wet weight; data not shown). For each animal that received AIH, changes in BDNF concentration were expressed as a percent change relative to the mean BDNF protein concentration of the appropriate time control group (see above). BDNF protein data were expressed as a mean ± S.E.M. and compared with t-tests (one-sample or student’s, SPSS or SigmaStat 2.03, SPSS Inc., Chicago, IL).
ERK1/2 phosphorylation
An equal volume of sample buffer (125 mM Tris pH 6.8, 4% SDS, 10% glycerol, 0.006% bromophenol blue, 1.8% β-mercaptoethanol) was added to each sample and the sample boiled for 3 minutes. 25 μg of sample protein was electrophoresed at 120 volts (10% SDS-PAGE mini-gels, Biorad Laboratories, Hercules, CA), and transferred to a PVDF membrane (Immobilon P Transfer Membrane, Millipore Corp., Bedford, MA) at 50 mA for 2 hours. Membranes were incubated overnight in blocking buffer (5% non-fat dry milk, 10 mM Tris pH 7.5, 100 mM NaCl, 0.1% Tween 20) containing pERK1/2 antibody (1:2000, Cell Signaling Technologies, Inc., Beverly, MA). Membranes were washed (10 mM Tris pH 7.5, 100 mM NaCl, 0.1% Tween 20) several times, incubated in the appropriate secondary antibody, then washed several more times and pERK1/2 visualized using an enhanced chemiluminescence system (SuperSignal West Pico Chemiluminescent Substrate, Pierce Biotechnologies, Inc., Rockford, IL). Membranes were then stripped (Restore Western Blot Stripping Solution, Pierce Biotechnologies, Inc. Rockford, IL) and re-probed for total ERK1/2 (1:2500, Cell Signaling Technologies, Inc., Beverly, MA). Phospho- and total- ERK1/2 were quantified using densitometry (LabWorks software version 4.5, UVP BioImaging Systems, Upland, CA) and the ratio of phospho- to total- ERK1/2 used as a measure of ERK1/2 activation. Changes in ERK1/2 phosphorylation were calculated as the percent change from time controls run within the same gel. Differences in ERK1/2 phosphorylation relative to time controls were then averaged across gels. The effect of dAIH on basal ERK1/2 phosphorylation was assessed by calculating the percent change in the ratio of phosphorylated to total ERK1/2 protein in dAIH-treated animals versus normoxia-treated rats. For control or dAIH-treated animals that received AIH, changes in ERK1/2 phosphorylation were assessed by averaging percent change in ERK1/2 phosphorylation versus control or dAIH-treated time controls. The effects of dAIH (basal) and AIH in normoxia- and dAIH-treated rats on phosphorylated and total ERK1/2 were assessed by normalizing phosphorylated and total ERK1/2 to a loading control protein (Grb2, 1:3333, Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and calculating the percent change from the appropriate time control groups. Data are expressed as mean ± S.E.M. and were statistically analyzed with t-tests (one-sample or student’s, SPSS or SigmaStat 2.03, SPSS Inc., Chicago, IL).
RESULTS
dAIH effects on body mass, apneic threshold, blood gases, mean arterial blood pressure, and respiratory burst frequency
Body mass was not significantly different between normoxia (n = 27) and dAIH treated rats (n = 20) prior to (normoxia: 281 ± 4 g; dAIH: 279 ± 6 g) or following (normoxia: 288 ± 4 g; dAIH: 285 ± 5 g) chamber treatment (p = 0.97); increases in body mass over the one-week exposure were not different between groups (p = 0.95). Apneic threshold did not differ between normoxia (44 ± 1 mmHg) and dAIH-treated rats (44 ± 1 mmHg; p = 0.58) nor were there significant treatment effects on mean arterial blood pressure, PaCO2, PaO2 or respiratory burst frequency during baseline conditions (p > 0.05, Table 2).
Table 2.
Temporal changes in mean arterial blood pressure (MABP), respiratory burst frequency, PaCO2, PaO2, pH and base excess (SEBc) in normoxia (Nx) or daily acute intermittent hypoxia (dAIH) treated Brown Norway rats that were then surgically prepared and treated with acute intermittent hypoxia (AIH) or no hypoxia (time controls, TC)
| Group | Baseline | Treatment | 30min post-treatment | 60min post-treatment | |
|---|---|---|---|---|---|
| Normoxia | MABP | 88 ± 5 | 86 ± 5 | 93 ± 4 | 81 ± 3* |
| TC | Burst Frequency | 38 ± 1 | 37 ± 1 | 36 ± 2 | 39 ± 1 |
| PaCO2 | 49 ± 1 | 49 ± 1 | 49 ± 1 | 50 ± 1 | |
| PaO2 | 205 ± 15 | 207 ± 12 | 185 ± 11 | 168 ± 10* | |
| pH | 7.36 ± 0.01 | 7.35 ± 0.01 | 7.36 ± 0.01 | 7.36 ± 0.01 | |
| SBEc | 1.8 ± 0.6 | 1.7 ± 0.6 | 2.2 ± 0.6 | 2.4 ± 0.6 | |
| Normoxia | MABP | 85 ± 3 | 62 ± 3* | 84 ± 2 | 77 ± 2* |
| AIH | Burst Frequency | 36 ± 1 | 38 ± 1 | 35 ± 1 | 37 ± 1 |
| PaCO2 | 49 ± 1 | 48 ± 1 | 49 ± 1 | 48 ± 1 | |
| PaO2 | 216 ± 8 | 40 ± 1*† | 178 ± 8* | 177 ± 8* | |
| pH | 7.36 ± 0.01 | 7.36 ± 0.01 | 7.35 ± 0.01 | 7.36 ± 0.01 | |
| SBEc | 1.8 ± 0.3 | 1.4 ± 0.4* | 1.1 ± 0.3 | 1.7 ± 0.4 | |
| dAIH | MABP | 89 ± 5 | 86 ± 5 | 85 ± 6 | 83 ± 6 |
| TC | Burst Frequency | 41 ± 1 | 40 ± 1 | 43 ± 1 | 42 ± 2 |
| PaCO2 | 49 ± 1 | 49 ± 1 | 49 ± 1 | 50 ± 1 | |
| PaO2 | 222 ± 20 | 228 ± 16 | 195 ± 19* | 193 ± 19* | |
| pH | 7.35 ± 0.01 | 7.35 ± 0.01 | 7.35 ± 0.01 | 7.35 ± 0.02 | |
| SBEc | 1.6 ± 0.6 | 1.1 ± 0.5 | 1.1 ± 0.8 | 1.5 ± 0.5 | |
| dAIH | MABP | 89 ± 4 | 72 ± 4* | 89 ± 3 | 81 ± 4* |
| AIH | Burst Frequency | 39 ± 2 | 40 ± 1 | 38 ± 1 | 40 ± 1 |
| PaCO2 | 47 ± 1 | 47 ± 1 | 47 ± 1 | 48 ± 1 | |
| PaO2 | 221 ± 8 | 40 ± 1*† | 190 ± 10* | 174 ± 7* | |
| pH | 7.36 ± 0.01 | 7.36 ± 0.01 | 7.34 ± 0.01 | 7.35 ± 0.01 | |
| SBEc | 0.9 ± 0.5 | 0.4 ± 0.5* | 0.0 ± 0.5 | 0.6 ± 0.5 |
p < 0.05 relative to baseline
p < 0.001 relative to time control
Short-term hypoxic responses
Typical integrated phrenic and XII neurograms before, during and 60 minutes post-AIH are shown in Figure 1. During hypoxia, phrenic burst amplitude increased similarly in normoxia (110 ± 14 % baseline, n = 18) and dAIH-treated rats (113 ± 14 % baseline, n = 14; Table 1). Phrenic burst amplitude did not increase during an equivalent time point in animals that did not receive hypoxia (i.e., time controls; normoxia: −5 ± 2 % baseline, n = 9; dAIH: −1 ± 3 % baseline, n = 6; Table 1).
Figure 1.

Integrated phrenic and hypoglossal neurograms from representative rats treated with seven days of normoxia or daily acute intermittent hypoxia (dAIH) and then exposed to acute intermittent hypoxia (AIH) or no hypoxia (time control). Baseline, hypoxic (or sham) treatment, and 30 and 60min time points are indicated. Each neurogram represents approximately 100 min of integrated respiratory motor output.
Table 1.
Short-term hypoxic response in phrenic and hypoglossal burst amplitude and respiratory burst frequency from rats treated with seven days of normoxia (control) or intermittent hypoxia (daily acute intermittent hypoxia, dAIH).
| Short-term Hypoxic Response | |||
|---|---|---|---|
| Treatment | Δ burst amplitude (% baseline) | Δ burst frequency (from baseline) | |
| Phrenic | XII | ||
| Control | 110 ± 14* | 196 ± 18* | 2 ± 1 |
| dAIH | 113 ± 12* | 225 ± 22* | 1 ± 3 |
p < 0.05 relative to baseline
XII burst amplitude increased during hypoxia in normoxia and dAIH-treated animals (196 ± 18 % baseline, n = 17 and 225 ± 22 % baseline, n = 11, respectively, Table 1). Similar to phrenic motor output, normoxia- and dAIH-treated time controls did not show an increase in XII burst amplitude during an equivalent time point (normoxia: −2 ± 3 % baseline, n = 17; dAIH: −2 ± 3 % baseline, n = 11; Table 1).
During hypoxia, phrenic burst frequency did not significantly change from baseline in normoxia (2 ± 1 bursts/min from baseline) or dAIH-treated rats (1 ± 3 bursts/min from baseline; both p > 0.05, Table 1). Minimal breathing frequency changes during hypoxia are characteristic of Brown Norway rats (Strohl et al., 1997). Additionally, phrenic burst frequency did not change during an equivalent time point in time control animals that did not receive hypoxia (normoxia: −1 ± 1 bursts/min from baseline, dAIH: 1 ± 3 bursts/min from baseline; both p > 0.05, Table 1).
Phrenic long-term facilitation (pLTF)
A trend for a treatment effect was observed between normoxia- and dAIH-treated animals that received acute intermittent hypoxia (AIH) or no hypoxia (time controls; p = 0.08, two-way repeated measures ANOVA). When compared to baseline, normoxia-treated animals that received AIH showed a small but significant increase in phrenic burst amplitude at 30 and 60 min post-AIH (21 ± 8% of baseline and 18 ± 9% of baseline, respectively, p < 0.01, n = 18, Figures 1 and 2A). The small magnitude of pLTF in normoxia-treated animals is consistent with previous findings in Brown Norway rats (Bavis et al., 2003). Time control rats that received seven days of normoxia (30 min: −1 ± 8% baseline; 60 min: 8 ± 6% baseline, n = 9, p > 0.31) without AIH did not show increased phrenic burst amplitude over a time course similar to rats treated with AIH (Figure 2A).
Figure 2.
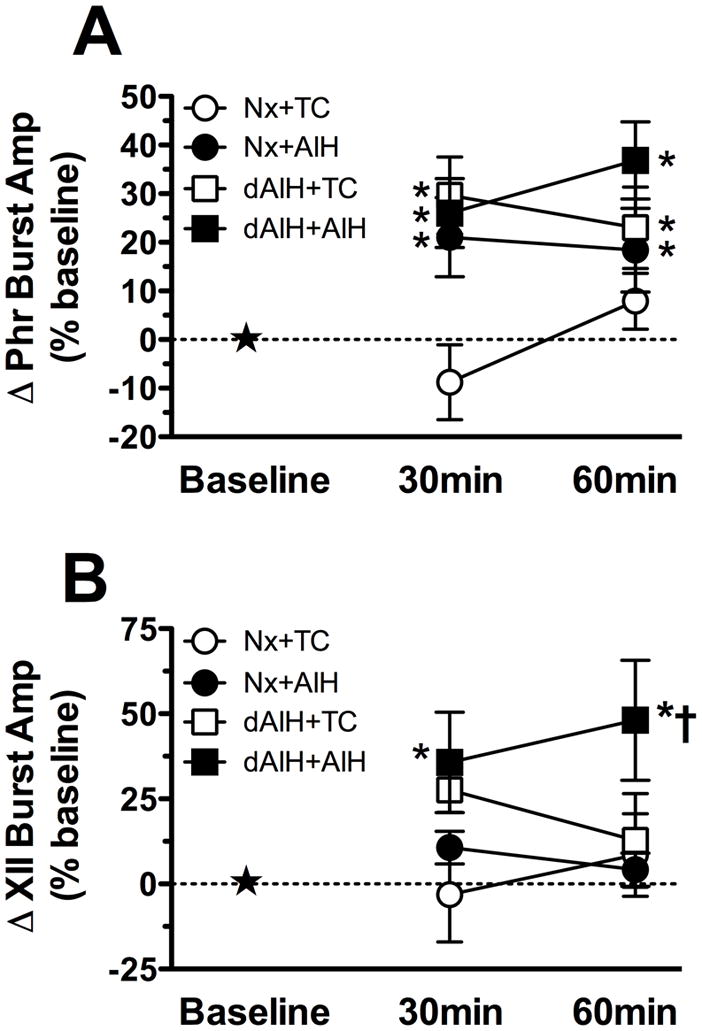
Percent change from baseline in (A) phrenic and (B) XII burst amplitude in adult Brown Norway rats treated for seven days with normoxia (Nx) or daily acute intermittent hypoxia (dAIH) and then treated with acute intermittent hypoxia (● Nx+AIH or ■ dAIH+AIH, respectively) or no hypoxia (○Nx+TC or □ dAIH+AIH, respectively). Data are presented as mean ± SEM. *Significantly different from baseline values (p < 0.05). †Significantly different from time control group (p < 0.05).
Significant pLTF was also observed in dAIH-treated rats at 30 and 60 min post-AIH when compared to baseline (26 ± 7% baseline and 37 ± 8% baseline, respectively, n = 14, p < 0.001, Figures 1 and 2A). However, dAIH-treated animals that did not receive hypoxia showed significant increases in phrenic burst amplitude over time as well (30 min: 30 ± 8% baseline; 60 min: 23 ± 8% baseline, p = 0.03). Thus, the presence of phrenic LTF in dAIH-treated animals treated with AIH should be interpreted with caution. One possibility is that dAIH-treated animals were experiencing respiratory plasticity prior to surgical preparation. However, baseline amplitude values were not significantly different between treatment groups (normoxia-time control: 2.9 ± 0.4V, normoxia-AIH: 2.8 ± 0.3V, dAIH-time control: 2.7 ± 0.5V, dAIH-AIH: 2.3 ± 0.3V p = 0.54), suggesting that dAIH-treated animals do not have enhanced phrenic motor output prior to an AIH or time control protocol.
XII long-term facilitation
A significant treatment effect was found between normoxia-and dAIH-treated animals that received AIH or no hypoxia (time controls; p = 0.05, two-way repeated measures ANOVA). In normoxia-treated rats, LTF was not evident in XII motor output at 30 or 60 min post-AIH (11 ± 5% baseline and 4 ± 5% baseline, respectively, p > 0.17, n=17, Figures 1 and 2B). In contrast, robust XII LTF was observed in dAIH-treated rats at 30 and 60 min post-AIH (36 ± 14% baseline and 48 ± 18% baseline, respectively, p < 0.001, n=11, Figures 1 and 2B), and this difference was significantly greater than in normoxia-treated rats (p = 0.01). Thus, dAIH induced XII LTF metaplasticity, revealing XII LTF in a rat strain that does not constitutively exhibit XII LTF.
Time control rats treated with normoxia (30 min: 3 ± 14% baseline, 60 min: 9 ± 12 %baseline; n = 8; p > 0.44) or dAIH (30 min: 27 ± 6% baseline, 60 min: 13 ± 14% baseline; n = 5; p > 0.05) without hypoxia did not show increased hypoglossal burst amplitude over a time course similar to hypoxia-treated rats (Figure 2B).
Frequency long-term facilitation
Phrenic burst frequency in normoxia-treated animals exposed to AIH was unchanged at 30 and 60 min post-AIH (baseline: 36 ± 1 bursts/min; 30 min: 35 ± 1 bursts/min; 60 min: 37 ± 1 bursts/min, p > 0.30, n = 18, Table 2). Similarly, there was no evidence for frequency LTF in dAIH-treated rats following AIH (baseline: 39 ± 2 bursts/min; 30 min: 35 ± 1 bursts/min 60 min: 40 ± 1 bursts/min, p > 0.18, n=14, Table 2). Time controls for normoxia (baseline: 38 ± 1 bursts/min; 30 min: 36 ± 2 bursts/min; 60 min: 39 ± 1 bursts/min, n = 9) and dAIH-treated (baseline: 41 ± 1 bursts/min; 30 min: 43 ± 1 bursts/min; 60 min: 42 ± 2 bursts/min, n = 6, Table 2) rats showed no change in phrenic burst frequency over time (p > 0.16 and p > 0.38, respectively). When all conditions were considered in the ANOVA, a very small, but statistically significant increase in phrenic burst frequency was found in dAIH-treated rats (p = 0.003).
Mean arterial blood pressure (MABP)
Relative to baseline, normoxia-treated animals showed significant decreases in MABP during hypoxia (baseline: 85 ± 3 mmHg, hypoxia: 62 ± 3 mmHg, p < 0.001, Table 2). Similarly, MABP in dAIH-treated rats also decreased during hypoxia (baseline: 89 ± 4 mmHg, hypoxia: 72 ± 4 mmHg, p < 0.001, Table 2). These decreases were not significantly different between groups. No significant decreases in MABP were found in normoxia or dAIH treated animals that did not receive hypoxia at an equivalent time point (normoxia-baseline: 88 ± 5 mmHg, normoxia-no hypoxia: 86 ± 5 mmHg; dAIH-baseline: 89 ± 5 mmHg, dAIH-no hypoxia: 86 ± 5 mmHg; p > 0.47, Table 2). MABP was similar to baseline values at 30 min post-treatment in all experimental groups (p > 0.17, Table 2). Relative to baseline, MABP significantly decreased 60 min post-treatment in every experimental group with the exception of the dAIH-treated time controls (p < 0.04 for all experimental groups except dAIH-treated time controls where p = 0.15, Table 2). Overall, dAIH had no significant effect on MABP (p = 0.09).
BDNF protein levels
Since new BDNF synthesis is necessary for pLTF expression in normal Sprague-Dawley rats (Baker-Herman et al., 2004), we determined BDNF concentration in the ventral cervical spinal cords of normoxia- and dAIH-treated rats, with and without AIH exposure. BDNF concentration in dAIH-treated time control rats (188 ± 12 ng/g total protein, n = 6) was elevated 24 ± 8% versus normoxia-treated time control rats (152 ± 7 ng/g total protein, p = 0.01, n = 9, Figure 3). Thus, dAIH increases ventral spinal BDNF concentration in the region of the phrenic motor nucleus.
Figure 3.
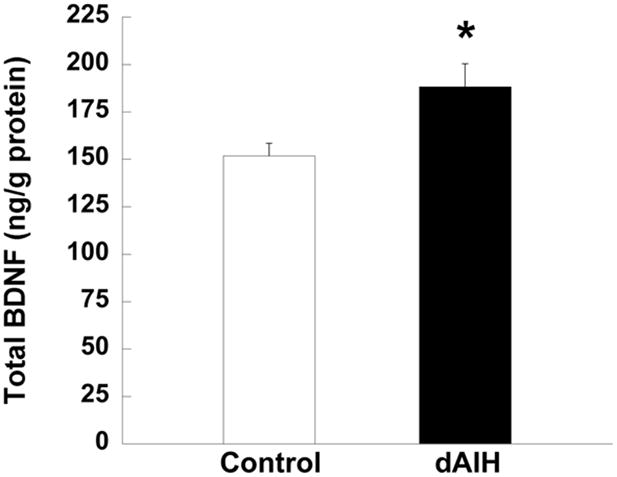
Change in basal levels of BDNF protein in ventral cervical spinal segments from rats treated with normoxia (control, unfilled bar) or dAIH (filled bar). Data are presented as mean ± S.E.M. *Significantly different from control group (p = 0.01).
In normoxia-treated rats, a small but significant increase in BDNF protein concentration was observed 60 min post-AIH relative to normoxia-treated time controls (25 ± 8% time control, p < 0.05, n = 18, Figure 4), consistent with the relatively small magnitude of pLTF in this rat strain. In contrast, there was no increase in BDNF protein concentration in dAIH-treated rats 60 min post-AIH relative to dAIH-treated time controls (−7 ± 4 % time control, p > 0.05, n = 14, Figure 4). Thus, whereas dAIH increases basal BDNF protein concentration in the ventral cervical spinal cord, subsequent increases in BDNF following AIH are no longer observed despite persistent (or even enhanced) pLTF in these same animals.
Figure 4.
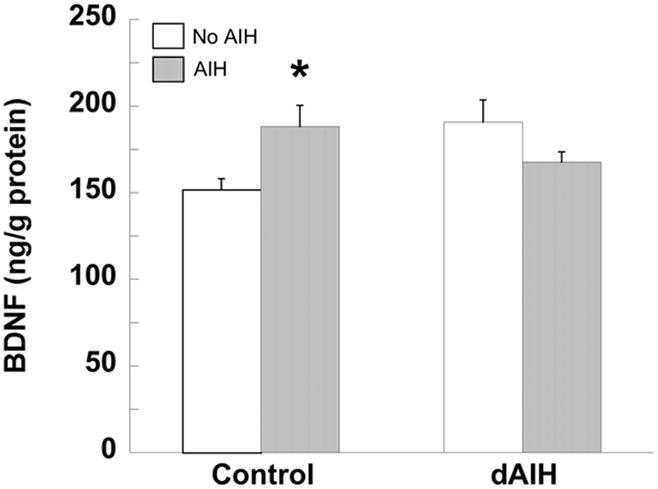
BDNF protein concentration 60 min following AIH (filled bars) or no hypoxia (time controls, unfilled bars) in ventral cervical spinal segments from rats pre-treated with normoxia (control) or dAIH. Data are presented as mean ± S.E.M. *Significantly different from normoxia or dAIH-treated time controls (p < 0.05).
ERK phosphorylation
Total and phosphorylated ERK1/2 were assessed in ventral cervical spinal segments of normoxia- and dAIH-treated time control rats; a subset of these data are presented in Figure 5. Although dAIH tended to increase the ratio of phosphorylated to total ERK1 (56 ± 28 %) and ERK2 (118 ± 28 %), neither apparent increase was significant (both p = 0.10; n = 6). Total ERK1 and ERK2 were also unaffected by dAIH when normalized to Grb2 (both p > 0.50). Thus, dAIH did not significantly alter total or phosphorylated ERK 1 or 2.
Figure 5.
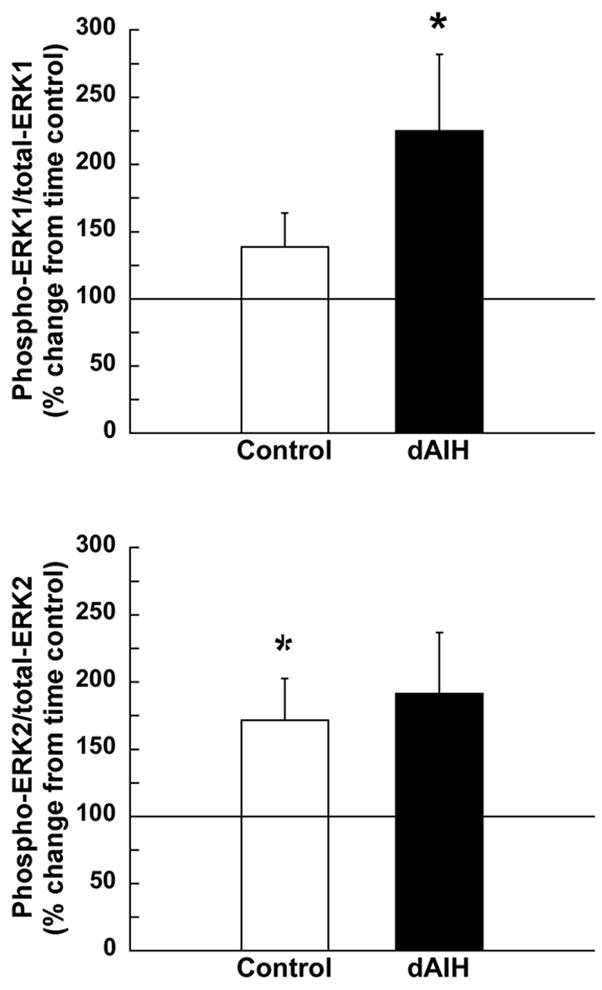
Change in the ratio of phosphorylated to total ERK 1 (A) and 2 (B) 60 min following AIH in ventral cervical spinal segments from rats pre-treated with normoxia (control, unfilled bars) or dAIH (filled bars). Data presented as mean percent change ± S.E.M. from the relevant time control group (i.e., normoxia and dAIH-treated rats that did not receive AIH; time controls set to 100%). *Significantly different from relevant time controls (p < 0.05).
Although dAIH did not significantly affect basal ERK expression, AIH increased ERK phosphorylation more in dAIH versus normoxia-treated rats. In this analysis, normoxia and dAIH-treated animals exposed to AIH were compared with their corresponding time-controls. While AIH did not increase the ratio of phosphorylated to total ERK1 in normoxia-treated animals (60 min post-AIH: 38 ± 25 %, p > 0.05, n = 18), it significantly increased ERK1 phosphorylation in dAIH-treated rats (125 ± 57 %, p < 0.05, n = 13, Figure 5A). In this analysis, total ERK1 was unchanged (data not shown).
In contrast to ERK1, AIH increased the ratio of phosphorylated to total ERK2 in control (72 ± 31 %, p < 0.05), but an apparent increase in ERK2 in dAIH-treated rats was only marginally significant (60 min post-AIH: 91 ± 45 %, p = 0.06, Figure 5B). Increased ERK2 activation in control rats following AIH was due to decreased total ERK2 levels, without change in phosphorylated ERK2 (data not shown).
DISCUSSION
An understanding of mechanisms underlying respiratory plasticity and metaplasticity may provide the rationale to develop new pharmacological treatments for ventilatory control disorders, including sleep-disordered breathing and respiratory insufficiency following cervical spinal injury or during motor neuron disease. However, severe protocols of chronic intermittent hypoxia elicit pathologies including systemic hypertension (Fletcher, 2000; Fletcher, 2001; Prabhakar et al., 2005; Zielinski, 2005) and hippocampal apoptosis with learning deficits (Gozal et al., 2001; Li et al., 2003; Row et al., 2003; Xu et al., 2004). Less severe protocols of chronic intermittent hypoxia (e.g. dAIH) may elicit beneficial (compensatory) plasticity without morbidity associated with severe protocols, and may have important therapeutic potential in the treatment of ventilatory control disorders (Mahamed and Mitchell, 2007; Mitchell, 2007). Thus, dAIH versus more severe protocols of chronic intermittent hypoxia (Ling et al., 2001; Peng et al., 2001; Row et al., 2003) were used in this study.
We report that dAIH elicits respiratory metaplasticity without significantly altering resting ventilatory drive, the short-term hypoxic response or mean arterial blood pressure. Notably, dAIH induced the capacity for XII LTF in a rat strain with little or no constitutive LTF expression. Although dAIH did not significantly enhance phrenic LTF expression, it did alter the regulation of key molecules thought to play critical roles in the cellular mechanism underlying phrenic LTF (BDNF and ERK1/2).
Respiratory metaplasticity: central vs. peripheral mechanisms
Metaplasticity following chronic intermittent hypoxia appears to involve plasticity in both peripheral and central neural elements. For example, chronic intermittent hypoxia increases basal carotid chemosensory discharge (Peng and Prabhakar, 2004; Rey et al., 2004) and enhances carotid body discharge up to 60 min post-AIH (e.g., sensory long-term facilitation). Similar sensory LTF is not present in “naïve,” normoxia-treated rats (Peng et al., 2001; Peng et al., 2003; Peng and Prabhakar, 2003; Peng and Prabhakar, 2004). The effect of dAIH on carotid chemosensory function is not known.
Preconditioning with chronic intermittent hypoxia also elicits central neural plasticity. For example, phrenic responses to carotid sinus nerve stimulation are amplified, indicating plasticity at sites other than the carotid body chemoreceptors (Ling et al., 2001). Chronic intermittent hypoxia also alters synaptic transmission in the nucleus of the solitary tract, a critical region for integration of carotid chemoafferent inputs (de Paula et al., 2007; Kline et al., 2007). Chronic intermittent hypoxia amplifies the hypoxic phrenic response, at least in part, by strengthening synaptic connections between respiratory pre-motoneurons and spinal phrenic motoneurons (Fuller et al., 2003). Although dAIH effects on synaptic transmission in the nucleus of the solitary tract are not known, recent studies indicate that spinal synaptic pathways are similarly enhanced by dAIH, at least under certain conditions (Lovett-Barr et al., 2007).
Although the present study does not directly address the respective contributions of central versus peripheral mechanisms in dAIH-induced metaplasticity of AIH-induced LTF, indirect evidence suggests a prominent role for central neural contributions. For example, enhanced LTF following dAIH is observed only in XII, with lesser effects on phrenic motor output that did not, in fact, attain statistical significance. Sensory mechanisms are unlikely to underlie such differential responses in these motor outputs since carotid chemoreceptor activation increases the activity in both nerves. Further, preconditioning with dAIH alters proteins in spinal regions associated with the phrenic motor nucleus, including BDNF and the capacity to increase ERK1/2 phosphorylation following AIH. Taken together, these data provide strong, suggestive evidence that central neural mechanisms underlie at least part of the metaplasticity observed following dAIH in Brown Norway rats.
Effect of dAIH preconditioning and AIH on spinal BDNF and ERK1/2
Phrenic LTF requires spinal serotonin receptor activation, new synthesis of BDNF, and TrkB receptor activation in normal Sprague Dawley and Fischer 344 rats (Baker-Herman et al., 2004; Feldman et al., 2003; Mahamed and Mitchell, 2006). Whereas these same elements have yet to be confirmed in Brown Norway rats, the 25% increase in BDNF following AIH in normoxia-treated rats is consistent with a similar role for BDNF synthesis and release in this strain. Furthermore, the percentage increase in BDNF (25%) and the magnitude of AIH-induced pLTF (18%) are proportionately lower than values previously reported in Sprague-Dawley rats (60% and 54%, respectively; Baker-Herman et al., 2004). Since the magnitude of pLTF is strongly correlated with increases in BDNF in Sprague-Dawley rats (Baker-Herman et al., 2004), pLTF may be constrained by a relative lack of AIH-induced BDNF synthesis in Brown Norway rats, possibly accounting for strain differences in pLTF magnitude.
In dAIH-treated rats, AIH no longer increased ventral cervical BDNF levels, yet pLTF was expressed (and may have even increased). If new BDNF synthesis is indeed necessary for pLTF (Baker-Herman et al., 2004), this observation raises a question: how can AIH elicit pLTF without any increase in ventral spinal BDNF concentration? We hypothesize that dAIH-induced increases in ventral spinal BDNF concentration mitigated the need for additional BDNF synthesis following AIH. Elevated BDNF levels during baseline conditions in dAIH-treated rats may have been sufficient to enable BDNF release and utilization without new synthesis. This hypothesis is consistent with elevated ERK1/2 phosphorylation following AIH in dAIH-treated rats since TrkB activation increases ERK phosphorylation (Gooney et al., 2002; Patterson et al., 1996; Slack et al., 2005).
AIH-induced ERK phosphorylation is consistent with a role in the mechanism of AIH-induced pLTF. To our knowledge, this is the first study suggesting a role for ERK1/2 in any form of respiratory plasticity. ERK activation directly contributes to several forms of synaptic plasticity, regulating local protein synthesis, synaptic receptor trafficking, and/or gene expression (Impey et al., 1999; Sweatt, 2004). Although BDNF/TrkB interactions are expected to increase ERK phosphorylation, TrkB activates other kinases as well (e.g. Akt). Thus, an understanding of the specific role played by ERK phosphorylation in pLTF awaits experiments to differentiate among these candidate molecules.
Although dAIH did not significantly alter basal ERK phosphorylation after one week of exposure, it is unclear if repetitive AIH would alter ERK phosphorylation at other time points. On the other hand, increased ERK phosphorylation following AIH in dAIH-treated rats further supports a role for TrkB-mediated ERK activation in pLTF.
One limitation of this study is that we did not assess neurochemical changes in the XII motor nucleus since we originally thought that we could investigate mechanisms of enhanced LTF in the phrenic motor nucleus more effectively; it came as somewhat of a surprise that dAIH-enhanced LTF was observed in the XII, but not the phrenic, in this rat strain. Further investigations concerning mechanistic similarities and differences between phrenic and XII LTF are necessary before we can appreciate the cellular mechanisms of dAIH enhanced XII LTF.
Significance
dAIH-induced metaplasticity in XII LTF in a rat strain where XII LTF is not constitutively expressed has interesting implications for humans. In sleeping patients with limited airflow through the upper airway (i.e., snorers), upper airway resistance is decreased following AIH, suggesting LTF in upper airway dilator muscles (Aboubakr et al., 2001; Shkoukani et al., 2002). In agreement, direct recordings of genioglossus EMG activity demonstrate LTF following AIH in sleeping patients (Chowdhuri et al., 2008). Collectively, these findings suggest that the ability of dAIH to enhance XII or genioglossal LTF may be harnessed to regulate upper airway tone over a longer time scale in human patients with limited airflow (i.e., snorers), potentially minimizing snoring or delaying the onset of overt sleep apnea symptoms (Mahamed and Mitchell, 2007).
Since dAIH is an effective means of eliciting respiratory metaplasticity without confounding effects of hypertension or (presumably) other sequelae that complicate interpretation, dAIH may be a useful model to study cellular mechanisms of respiratory metaplasticity. Regardless of its specific physiological significance, an understanding of cellular mechanisms underlying respiratory metaplasticity may provide the rationale for novel pharmacological interventions in patients with sleep-disordered breathing.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant HL80209. J.E.R.W. was supported by NIH Training Grant HL 07654. We thank past and present members of the Mitchell laboratory for expert technical assistance and many helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aboubakr SE, Taylor A, Ford R, Siddiqi S, Badr MS. Long-term facilitation in obstructive sleep apnea patients during NREM sleep. J Appl Physiol. 2001;91:2751–2757. doi: 10.1152/jappl.2001.91.6.2751. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Bavis RW, Baker-Herman TL, Zabka AG, Golder FJ, Fuller DD, Mitchell GS. Respiratory long-term facilitation differs among inbred rat strains. 2003 Experimental Biology meeting abstracts; 2003. Abstract #824. [Google Scholar]
- Chowdhuri S, Pierchala L, Aboubakr SE, Shkoukani M, Badr MS. Long-term facilitation of genioglossus activity is present in normal humans during NREM sleep. Respir Physiol Neurobiol. 2008;160:65–75. doi: 10.1016/j.resp.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coogan AN, O’Leary DM, O’Connor JJ. P42/44 MAP kinase inhibitor PD98059 attenuates multiple forms of synaptic plasticity in rat dentate gyrus in vitro. J Neurophysiol. 1999;81:103–110. doi: 10.1152/jn.1999.81.1.103. [DOI] [PubMed] [Google Scholar]
- de Paula PM, Tolstykh G, Mifflin S. Chronic intermittent hypoxia alters NMDA and AMPA-evoked currents in NTS neurons receiving carotid body chemoreceptor inputs. Am J Physiol Regul Integr Comp Physiol. 2007;292:R2259–2265. doi: 10.1152/ajpregu.00760.2006. [DOI] [PubMed] [Google Scholar]
- Errico M, Crozier RA, Plummer MR, Cowen DS. 5-HT(7) receptors activate the mitogen activated protein kinase extracellular signal related kinase in cultured rat hippocampal neurons. Neuroscience. 2001;102:361–367. doi: 10.1016/s0306-4522(00)00460-7. [DOI] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher EC. Effect of episodic hypoxia on sympathetic activity and blood pressure. Respir Physiol. 2000;119:189–197. doi: 10.1016/s0034-5687(99)00114-0. [DOI] [PubMed] [Google Scholar]
- Fletcher EC. Invited review: Physiological consequences of intermittent hypoxia: systemic blood pressure. J Appl Physiol. 2001;90:1600–1605. doi: 10.1152/jappl.2001.90.4.1600. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Bach KB, Baker TL, Kinkead R, Mitchell GS. Long term facilitation of phrenic motor output. Respir Physiol. 2000;121:135–146. doi: 10.1016/s0034-5687(00)00124-9. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Baker TL, Behan M, Mitchell GS. Expression of hypoglossal long-term facilitation differs between substrains of Sprague-Dawley rat. Physiol Genomics. 2001;4:175–181. doi: 10.1152/physiolgenomics.2001.4.3.175. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Johnson SM, Olson EB, Jr, Mitchell GS. Synaptic pathways to phrenic motoneurons are enhanced by chronic intermittent hypoxia after cervical spinal cord injury. J Neurosci. 2003;23:2993–3000. doi: 10.1523/JNEUROSCI.23-07-02993.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooney M, Lynch MA. Long-term potentiation in the dentate gyrus of the rat hippocampus is accompanied by brain-derived neurotrophic factor-induced activation of TrkB. J Neurochem. 2001;77:1198–1207. doi: 10.1046/j.1471-4159.2001.00334.x. [DOI] [PubMed] [Google Scholar]
- Gooney M, Shaw K, Kelly A, O’Mara SM, Lynch MA. Long-term potentiation and spatial learning are associated with increased phosphorylation of TrkB and extracellular signal-regulated kinase (ERK) in the dentate gyrus: evidence for a role for brain-derived neurotrophic factor. Behav Neurosci. 2002;116:455–463. doi: 10.1037//0735-7044.116.3.455. [DOI] [PubMed] [Google Scholar]
- Gozal D, Daniel JM, Dohanich GP. Behavioral and anatomical correlates of chronic episodic hypoxia during sleep in the rat. J Neurosci. 2001;21:2442–2450. doi: 10.1523/JNEUROSCI.21-07-02442.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Storm DR. Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron. 1999;23:11–14. doi: 10.1016/s0896-6273(00)80747-3. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Kline DD, Ramirez-Navarro A, Kunze DL. Adaptive depression in synaptic transmission in the nucleus of the solitary tract after in vivo chronic intermittent hypoxia: evidence for homeostatic plasticity. J Neurosci. 2007;27:4663–4673. doi: 10.1523/JNEUROSCI.4946-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li RC, Row BW, Gozal E, Kheirandish L, Fan Q, Brittian KR, Guo SZ, Sachleben LR, Jr, Gozal D. Cyclooxygenase 2 and intermittent hypoxia-induced spatial deficits in the rat. Am J Respir Crit Care Med. 2003;168:469–475. doi: 10.1164/rccm.200211-1264OC. [DOI] [PubMed] [Google Scholar]
- Lin SL, Johnson-Farley NN, Lubinsky DR, Cowen DS. Coupling of neuronal 5-HT7 receptors to activation of extracellular-regulated kinase through a protein kinase A-independent pathway that can utilize Epac. J Neurochem. 2003;87:1076–1085. doi: 10.1046/j.1471-4159.2003.02076.x. [DOI] [PubMed] [Google Scholar]
- Ling L, Fuller DD, Bach KB, Kinkead R, Olson EB, Jr, Mitchell GS. Chronic intermittent hypoxia elicits serotonin-dependent plasticity in the central neural control of breathing. J Neurosci. 2001;21:5381–5388. doi: 10.1523/JNEUROSCI.21-14-05381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Hu JY, Schacher S, Schwartz JH. The two regulatory subunits of aplysia cAMP-dependent protein kinase mediate distinct functions in producing synaptic plasticity. J Neurosci. 2004;24:2465–2474. doi: 10.1523/JNEUROSCI.4331-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Barr MR, Sibigtroth CM, Mitchell GS. Daily acute intermittent hypoxia improves respiratory function in rats with chronic cervical spinal hemisection. FASEB J. 2007;21:918. [Google Scholar]
- Mahamed S, Mitchell GS. Does simulated apnea elicit respiratory long-term facilitation?. 2006 Experimental Biology meeting abstracts [on CD-ROM]; San Francisco, CA. 2006. [Google Scholar]
- Mahamed S, Mitchell GS. Is there a link between intermittent hypoxia-induced respiratory plasticity and obstructive sleep apnoea? Exp Physiol. 2007;92:27–37. doi: 10.1113/expphysiol.2006.033720. [DOI] [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Chronic intermittent hypoxia enhances ventilatory long-term facilitation in awake rats. J Appl Physiol. 2003;95:1499–1508. doi: 10.1152/japplphysiol.00044.2003. [DOI] [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Serotonin receptor subtypes required for ventilatory long-term facilitation and its enhancement after chronic intermittent hypoxia in awake rats. Am J Physiol Regul Integr Comp Physiol. 2004;286:R334–341. doi: 10.1152/ajpregu.00463.2003. [DOI] [PubMed] [Google Scholar]
- Millhorn DE, Eldridge FL, Waldrop TG. Prolonged stimulation of respiration by a new central neural mechanism. Respir Physiol. 1980;41:87–103. doi: 10.1016/0034-5687(80)90025-0. [DOI] [PubMed] [Google Scholar]
- Mitchell GS. Respiratory plasticity following intermittent hypoxia: a guide for novel therapeutic approaches to ventilatory control disorders. In: Gaultier C, editor. Genetic Basis for Respiratory Control Disorders. Springer Publishing Company; New York: 2007. [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr Invited review: Intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Olson EB, Jr, Bohne CJ, Dwinell MR, Podolsky A, Vidruk EH, Fuller DD, Powell FL, Mitchel GS. Ventilatory long-term facilitation in unanesthetized rats. J Appl Physiol. 2001;91:709–716. doi: 10.1152/jappl.2001.91.2.709. [DOI] [PubMed] [Google Scholar]
- Ormond J, Hislop J, Zhao Y, Webb N, Vaillaincourt F, Dyer JR, Ferraro G, Barker P, Martin KC, Sossin WS. ApTrkl, a Trk-like receptor, mediates serotonin- dependent ERK activation and long-term facilitation in Aplysia sensory neurons. Neuron. 2004;44:715–728. doi: 10.1016/j.neuron.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER. Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron. 2001;32:123–140. doi: 10.1016/s0896-6273(01)00443-3. [DOI] [PubMed] [Google Scholar]
- Peng Y, Kline DD, Dick TE, Prabhakar NR. Chronic intermittent hypoxia enhances carotid body chemoreceptor response to low oxygen. Adv Exp Med Biol. 2001;499:33–38. doi: 10.1007/978-1-4615-1375-9_5. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci U S A. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Reactive oxygen species in the plasticity of respiratory behavior elicited by chronic intermittent hypoxia. J Appl Physiol. 2003;94:2342–2349. doi: 10.1152/japplphysiol.00613.2002. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol. 2004;96:1236–1242. doi: 10.1152/japplphysiol.00820.2003. discussion 1196. [DOI] [PubMed] [Google Scholar]
- Powell FL, Milsom WK, Mitchell GS. Time domains of the hypoxic ventilatory response. Respir Physiol. 1998;112:123–134. doi: 10.1016/s0034-5687(98)00026-7. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Peng YJ, Jacono FJ, Kumar GK, Dick TE. Cardiovascular alterations by chronic intermittent hypoxia: importance of carotid body chemoreflexes. Clin Exp Pharmacol Physiol. 2005;32:447–449. doi: 10.1111/j.1440-1681.2005.04209.x. [DOI] [PubMed] [Google Scholar]
- Rey S, Del Rio R, Alcayaga J, Iturriaga R. Chronic intermittent hypoxia enhances cat chemosensory and ventilatory responses to hypoxia. J Physiol. 2004;560:577–586. doi: 10.1113/jphysiol.2004.072033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Row BW, Liu R, Xu W, Kheirandish L, Gozal D. Intermittent hypoxia is associated with oxidative stress and spatial learning deficits in the rat. Am J Respir Crit Care Med. 2003;167:1548–1553. doi: 10.1164/rccm.200209-1050OC. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Sherff CM, Shobe J, Bagnall MW, Sutton MA, Carew TJ. Differential role of mitogen-activated protein kinase in three distinct phases of memory for sensitization in Aplysia. J Neurosci. 2003;23:3899–3907. doi: 10.1523/JNEUROSCI.23-09-03899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkoukani M, Babcock MA, Badr MS. Effect of episodic hypoxia on upper airway mechanics in humans during NREM sleep. J Appl Physiol. 2002;92:2565–2570. doi: 10.1152/japplphysiol.00938.2001. [DOI] [PubMed] [Google Scholar]
- Slack SE, Grist J, Mac Q, McMahon SB, Pezet S. TrkB expression and phospho-ERK activation by brain-derived neurotrophic factor in rat spinothalamic tract neurons. J Comp Neurol. 2005;489:59–68. doi: 10.1002/cne.20606. [DOI] [PubMed] [Google Scholar]
- Slack SE, Pezet S, McMahon SB, Thompson SW, Malcangio M. Brain-derived neurotrophic factor induces NMDA receptor subunit one phosphorylation via ERK and PKC in the rat spinal cord. Eur J Neurosci. 2004;20:1769–1778. doi: 10.1111/j.1460-9568.2004.03656.x. [DOI] [PubMed] [Google Scholar]
- Strohl KP, Thomas AJ, St Jean P, Schlenker EH, Koletsky RJ, Schork NJ. Ventilation and metabolism among rat strains. J Appl Physiol. 1997;82:317–323. doi: 10.1152/jappl.1997.82.1.317. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Xu W, Chi L, Row BW, Xu R, Ke Y, Xu B, Luo C, Kheirandish L, Gozal D, Liu R. Increased oxidative stress is associated with chronic intermittent hypoxia-mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. Neuroscience. 2004;126:313–323. doi: 10.1016/j.neuroscience.2004.03.055. [DOI] [PubMed] [Google Scholar]
- Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci. 2002;22:1532–1540. doi: 10.1523/JNEUROSCI.22-05-01532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabka AG, Mitchell GS, Olson EB, Jr, Behan M. Selected contribution: chronic intermittent hypoxia enhances respiratory long-term facilitation in geriatric female rats. J Appl Physiol. 2003;95:2614–2623. doi: 10.1152/japplphysiol.00476.2003. discussion 2604. [DOI] [PubMed] [Google Scholar]
- Zielinski J. Effects of intermittent hypoxia on pulmonary haemodynamics: animal models versus studies in humans. Eur Respir J. 2005;25:173–180. doi: 10.1183/09031936.04.00037204. [DOI] [PubMed] [Google Scholar]