

Abstract
Proliferative lesions of the mammary gland are risk markers and potential precursors for the development of breast cancer in postmenopausal women. In this study we evaluated mammary epithelial proliferation and proliferative lesions in a group of 63 aged postmenopausal macaques randomized by social group to receive one of three experimental diets for 8 months: (1) control; (2) control with 17β-estradiol (E2) at the human equivalent dose of 1.0 mg/day; and (3) control with the soy phytoestrogen equol (EQ) at the human equivalent dose of 105 mg/day. In normal mammary epithelium, treatment with E2 but not EQ resulted in greater proliferation, epithelial area, and progesterone receptor expression (P < 0.05 for all). Mammary lesions included columnar cell change (26/63), columnar cell hyperplasia with and without atypia (13/63), atypical ductal hyperplasia (6/63), and atypical lobular hyperplasia (3/63). Lesions were most common within terminal ductal lobular units. The prevalence of columnar cell hyperplasia (total and atypical cases) was higher in animals treated with E2 compared to control (P < 0.05 for both). Compared to normal mammary epithelium, columnar cell lesions (CCLs) showed greater constitutive expression of estrogen receptor alpha across all groups (P < 0.001) and greater expression of progesterone receptor in response to E2 (P < 0.01). Independent of treatment, animals with CCLs on histology had greater gene expression of estrogen receptor alpha and markers of estrogen receptor activity (TFF1) and proliferation (MKI67) at a site contralateral to the CCL (P < 0.05 for all). These findings demonstrate that the terminal ductal lobular units of the postmenopausal mammary gland contain morphologically distinct cell populations which may hyperrespond to E2 exposure, resulting in specific types of hyperplastic lesions.
Keywords: breast cancer, columnar cell lesion, estradiol, estrogen receptor, equol, hyperplasia, progesterone receptor
Mammary gland hyperplasia and other forms of benign breast disease (BBD) confer increased risk for subsequent breast cancer in women.1–6 Columnar cell lesions (CCLs) are a specific type of proliferative change characterized by the presence of columnar epithelial cells within terminal ductal lobular units (TDLUs), frequently with cystic dilation of affected ducts.7,8 CCLs have emerged in recent years as potential risk markers for breast carcinoma, although the clinical significance of CCLs and their role as direct precursors to cancer remains unclear.8–11 Contributing to this uncertainty is a lack of standard terminology and diagnostic criteria regarding CCLs, which have previously been called, among other names, hyperplastic enlarged lobular units, atypical cystic lobules/ducts, clinging ductal carcinoma in situ, and monomorphic epithelial proliferations.8,12–14 Despite increased interest in CCLs as risk markers, the biologic behavior, long-term risk profile, and lifestyle determinants for these lesions are not well-established.
Current evidence suggests that CCLs may be associated with certain forms of breast cancer.7,15–17 These lesions occur more commonly in biopsy specimens containing ductal carcinoma in situ (DCIS) or invasive cancer15 and may show continuity with specific types of carcinoma.16–18 In one recent study, CCLs were colocalized with atypical ductal hyperplasia (ADH), DCIS, or tubular carcinoma in 85% of patients surveyed.16 Other studies have found genetic alterations such as loss of heterogeneity and allelic imbalance in CCLs similar to those in adjacent carcinomas,18–20 further suggesting that at least a subset of CCLs may progress to higher-grade lesions.
Estrogen exposure is a well-established determinant of certain types of breast cancer, particularly in postmenopausal women.21,22 The most bioactive physiologic mammalian estrogen is 17β-estradiol (E2), which may promote growth of responsive epithelial cells and possibly contribute to cellular transformation through genotoxic metabolites.21,23 Data from several trials suggest that estrogen status may influence the incidence and possibly the neoplastic progression of proliferative breast lesions.24–28 Prior studies of CCLs in particular have shown aberrant changes in sex steroid receptors,7,12,18,29–31 including increased expression of estrogen receptor alpha (ESR1) and its downstream marker progesterone receptor (PGR).12,18,29–31 This evidence suggests that abnormal estrogen signaling may have an important role in the development and promotion of certain types of proliferative breast lesions. To investigate this idea, in the current study we evaluated mammary changes in a randomized preclinical trial of older postmenopausal female macaques treated with placebo, E2, or equol (EQ), a soy isoflavone metabolite with reported phytoestrogenic properties.32 The primary goal of the study was to determine estrogen-related effects of E2 and EQ on mammary gland proliferation and prevalence of proliferative lesions.
MATERIALS AND METHODS
Animal subjects and design
We evaluated mammary gland tissue from 63 female cynomolgus macaques (Macaca fascicularis) with estimated ages ranging from 12.1 to 28.9 years (mean, 21.8 ± 0.5 years). Animals were originally imported from the Institut Pertanian Bogor in Bogor, Indonesia. All animals were considered multiparous based on historical data from the original colony, in which >90% of the adult females have had 2+ live births, and on myometrial evidence of prior pregnancy (expansion of venous adventitia). Macaques are Old World anthropoid primates with a high overall genetic coding sequence identity to humans,33 including important genes related to breast cancer susceptibility.34 Female macaques have a reproductive physiology highly similar to that of women, including a 28-day menstrual cycle and comparable ovarian hormone profile.35 Prior work from our laboratory and others have also demonstrated similarities between macaque and human mammary gland biology, including sex steroid receptor expression,36 responses to endogenous and exogenous sex steroids,37–39 and the presence of spontaneous hyperplastic and neoplastic mammary gland lesions.40
Animals were ovariectomized 4.5 years prior to this study and housed since this time in stable social groups of 3 – 4 animals each. Animals were randomized by social group to receive one of three diets containing a baseline casein/lactalbumin-based (isoflavone-free) control diet supplemented with (1) nothing (control, n = 21); (2) micronized E2 (Estrace, Mylan Pharmaceuticals; Morgantown, WV) at a dose of 66 μg/kg body weight (1.0 mg/1800 kcal) (n = 20); or (3) the soy isoflavonoid EQ at a dose of 7.2 mg/kg body weight (105 mg/1800 kcal) (n = 22). All animals received the control diet for at least 6 weeks prior to the start of the experiment. Dietary treatments were given for 8 months. The EQ supplement contained a 96.0% pure racemic mixture of S- and R-equol enantiomers, as determined by the provider using high performance liquid chromatography. Diets were otherwise the same in macronutrients, cholesterol, calcium, and phosphorus. Equol was generously provided by The Solae Company (St. Louis, MO, USA). Animals were fed 120 kcal/kg body weight (BW) (+10% extra to account for waste) once daily. Daily E2 and EQ doses were scaled to 1800 kcal of diet (the estimated daily intake for a U.S. woman) to account for differences in metabolic rates between monkeys and human subjects.41 The E2 dose approximated a standard hormone therapy dose taken by postmenopausal women (1.0 mg/day), while the EQ dose was designed to approximate the equivalent isoflavonoid exposure resulting from a maximal level of dietary soy intake (90 – 120 mg/day).42
All procedures involving these animals were conducted in compliance with State and Federal laws, standards of the U.S. Department of Health and Human Services, and guidelines established by the Wake Forest University Animal Care and Use Committee. The facilities and laboratory animal program of Wake Forest University are fully accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care.
Serum estradiol and equol concentrations
To confirm intake of dietary treatments, serum E2 and EQ concentrations were measured in blood samples collected by femoral venipuncture from a randomly selected subset of animals. Estradiol concentrations were measured at baseline and 3 weeks into treatment by radioimmunoassay using a commercially available kit and protocol from Diagnostic Systems Laboratories (E2, DSL-4800 ultra-sensitive; Webster, TX). Serum (0.5 ml) was extracted using ethyl ether, and extracts were dried and reconstituted with zero-standard serum. Equol concentrations were measured on serum collected 3 and 27 weeks into treatment by liquid chromatographic-photodiode array mass spectrometric analysis, using techniques described previously.43
Mammary gland collection and processing
Baseline mammary gland biopsies were collected from each animal prior to the treatment period. For this procedure, animals were anesthetized with ketamine and buprenorphine and a small (~0.3 gram) sample was removed from the left mammary gland by an experienced veterinary surgeon (SEA). Animals were monitored and given analgesia during recovery following approved clinical procedures. At the end of the treatment period, animals were sedated with ketamine and euthanized using sodium pentobarbital (100 mg/kg, intravenous), as recommended by the Panel on Euthanasia of the American Veterinary Medical Association. Euthanasia was performed for data collection related to cardiovascular endpoints (to be described elsewhere). Post-treatment samples from the right mammary gland were fixed at 4°C in fresh 4% paraformaldehyde solution, transferred to 70% ethanol 24 hours later, and trimmed to ~3 cm in length sagittal to the right nipple. Fixed samples were paraffin-embedded, sectioned, and stained with hematoxylin and eosin using standard histologic procedures. Corresponding samples from the left mammary gland (same location) were snap-frozen in liquid nitrogen and stored at −70°C for later gene expression analyses. For whole mount analysis, a portion of fixed mammary gland (craniolateral quadrant, ~4 × 4 cm2) from a subset of cases was dissected from the overlying skin and placed in acetone for two nights or until all fat was removed.39 Mammary gland tissues were then rehydrated in 70% ethanol, placed in deionized water for 30 min, stained in 0.28% Toluidine blue (0.25 gm diluted in 10 ml of 100% ethanol + 80 ml of water and aged for at least 2 days), soaked in 100% methanol for 90 min, cleared in 100% methyl salicylate, and photographed for qualitative assessment.39
Morphologic classification of mammary gland lesions
Mammary glands were evaluated for morphologic changes on single slides containing two histologic mammary sections from each animal spaced 20 – 40 μm apart. Lesions were classified as follows: columnar cell change (CCC, enlarged TDLUs with variably dilated acini lined by 1–2 layers of columnar epithelial cells with ovoid to elongate uniform nuclei oriented perpendicular to the basement membrane, evenly dispersed chromatin, and inconspicuous nucleoli); columnar cell hyperplasia (CCH, features of CCC but with stratification of more than two columnar cell layers); ADH (>2 layers of ductal epithelial cells with architectural complexity); atypical lobular hyperplasia (ALH, >2 layers of lobular epithelial cells with conspicuous cytologic or architectural atypia); or DCIS (cellular and architectural atypia without evidence of invasion or disruption of the basement membrane).8,9,44,45 DCIS lesions were graded using the modified Scarff-Bloom-Richardson system.46 ADH and DCIS lesions were often present in a context of CCC and/or CCH; in these cases lesion types were classified separately. Lesions consistent with human flat epithelial atypia (FEA, features of CCC or CCH but with low-grade monomorphic nuclear atypia; lacking complex architecture) were classified with CCC or CCH. Evaluations were performed blinded to experimental treatment by CEW in consultation with board-certified veterinary (JMC) and medical (KRG) pathologists. Prevalence values indicate the number of animals with a particular lesion type.
Immunohistochemistry
Mammary gland sections were immunostained using commercially-available primary monoclonal antibodies for ESR1 (NCL-ER-6F11, Novocastra; Newcastle–upon–Tyne, UK), PGR (NCL-PGR, Novocastra), and Ki67 (Ki67/MIB1, Dako; Carpinteria, CA). Antibodies were diluted for ESR1 (1:100), PGR (1:100), and Ki67 (1:50) in 1X Automation Buffer (Biomeda; Foster City, CA) containing 0.5% casein (Sigma; St. Louis, MO). Immunostaining procedures included antigen-retrieval with citrate buffer (pH 6.0), biotinylated rabbit anti-mouse Fc antibody as a linking reagent, alkaline phosphatase-conjugated streptavidin as the label, and Vector Red as the chromogen (Vector Laboratories; Burlingame, CA). Negative control slides were run for each immunostain using the same protocol as for study slides except with non-immune serum (from the same species as primary antibody) in place of the primary antibody. Cell labeling was quantified by a computer-assisted counting technique using a grid filter to select cells for counting and our modified procedure of cell selection, described previously.47 For normal mammary gland epithelium, 100 lobular cells (alveoli and TDLUs) and 100 ductal cells (extralobular large ducts) were counted for each slide. Lesions were sorted based on morphology, and 200 cells were counted for each particular lesion type over at least 3 microscopic fields. Immunolabeling counts were conducted blinded to experimental treatment and expressed and analyzed as a percentage of the total number of cells examined.
Histomorphometry
Mammary gland epithelial area was quantified by histomorphometry using methods described previously.38 Briefly, hematoxylin and eosin-stained slides were digitized using a Labophot 3 light microscope (Nikon Instruments; Melville, NY) and Infinity 3 digital camera (Lumenera; Ottawa, ON), and measurements were taken with Image Pro-Plus software (Media Cybernetics; Bethesda, MD). Three microscopic fields were randomly selected and examined at 20x magnification. Epithelial area was determined by manual tracing of lobuloalveolar units and expressed as a percentage of the total area examined.
Quantitative gene expression
Genes associated with estrogen receptor activity (ESR1; ESR2, estrogen receptor-beta; PGR; TFF1, trefoil factor 1, also known as pS2) and proliferation (MKI67, Ki67 antigen) were measured in mammary gland samples using quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR). Macaque-specific qRT-PCR primer-probe sets for internal control genes (GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ACTB, beta-actin) and other targets (ESR1, ESR2, TFF1) were generated through the Applied Biosystems (ABI) Taqman Assay-by-Design service (Foster City, CA),48 while pre-made human Taqman assays were used for PGR (Hs00172183_m1) and MKI67 (Hs00606991_m1). All probes spanned an exon-exon junction to eliminate genomic DNA contamination. Total RNA was extracted from frozen samples using Tri Reagent (Molecular Research Center; Cincinnati, OH), purified using RNeasy Mini kit (QIAGEN; Valencia, CA), and quantitated using a NanoDrop ND-1000 UV-vis spectrophotometer (NanoDrop; Wilmington, DE). RNA quality was confirmed using an Agilent 2100 Bioanalyzer (Wilmington, DE). Only samples with an RNA integrity number greater than 7.0 were used; three samples (all E2) were excluded due to poor RNA quality. Reactions (20 μl volume) were performed on an ABI Prism 7000 Sequence Detection System using standard Taqman reagents and thermocycling protocol.48 Relative expression was determined using the ΔΔCt method described in ABI User Bulletin #2 (available online). The Ct values for the control genes GAPDH and ACTB were averaged for use in internal calibration, while reference premenopausal mammary gland tissue RNA was run in parallel for plate-to-plate calibration. Calculations were performed using ABI Relative Quantification SDS Software v1.1.
Statistics
The power calculation for this study was based on breast epithelial proliferation determined by Ki67 labeling. For this measure, we estimated the minimum difference between control and E2 means to be 5.0% with a common intra-group standard deviation of 4.0%. The sample size in each group providing an 80% chance at a 0.05 significance level to detect a statistically significant difference was 16 after adjusting for multiple comparisons.
Data were analyzed using the SAS statistical package (version 8, SAS Institute; Cary, NC). All data were evaluated for normal distribution and homogeneity of variances among groups. A general linear model was used to determine mean values and calculate group differences for body weight, age, serum E2 and EQ, epithelial area, and qRT-PCR expression data. A two-tailed Fisher’s exact test was used to evaluate treatment group differences in lesion type prevalence. Five baseline biopsy samples (2 control, 1 E2, 2 EQ) lacked epithelial tissue on histology and were thus excluded from analysis. Immunolabeling between normal epithelium and CCLs (CCC and CCH) was evaluated within each treatment group using a nonparametric Kruskal-Wallis test followed by two-sided Wilcoxon Rank Sum pairwise analysis; for this comparison, the average values for ductal and lobular cells were used for normal epithelium. Gene expression data were log-transformed to improve distribution, and data were then retransformed to original scale and reported as fold-change of control with 90% confidence interval. All other data are reported as mean ± standard error. Treatment effects on gene expression were covaried by body weight to help correct for differences in adiposity across samples, while lesion status effects were covaried by treatment group. All pairwise P-values were adjusted for the number of pairwise tests using a Bonferroni correction. A two-tailed significance level of 0.05 was chosen for all comparisons.
RESULTS
Treatment group characteristics
No group differences were noted in age, body weight, or serum E2 at baseline (P > 0.1 for all) (Table 1). During 8 months of treatment, body weight increased ~2 – 3% in control and EQ groups, while the E2 group on average lost ~5% body weight (P = 0.003 compared to control). At 3 weeks of treatment, serum E2 was higher in the E2 group (P = 0.002) and serum EQ was higher in the EQ group (P < 0.0001) compared to the control group. Serum E2 concentrations were in the daily range of postmenopausal women taking an equivalent 1 mg/day dose of oral micronized E2,49 while EQ concentrations were consistent with isoflavonoid concentrations reported in women consuming a high-soy meal.50
Table 1.
Treatment group characteristics.*
| con | E2 | EQ | |
|---|---|---|---|
| Age (yrs) | |||
| baseline | 20.8 ± 0.8 | 21.2 ± 0.8 | 23.3 ± 0.8 |
| Body weight (kg) | |||
| baseline | 3.36 ± 0.14 | 3.32 ± 0.15 | 3.51 ± 0.14 |
| 8 months | 3.42 ± 0.15 | 3.14 ± 0.15 | 3.61 ± 0.14 |
| change | 0.06 ± 0.06 | −0.18 ± 0.06† | 0.10 ± 0.06 |
| Serum estradiol (pg/ml) | |||
| baseline | <5 | 6.1 ± 0.5 | 5.1 ± 0.4 |
| 3 weeks | <5 | 77.9 ± 14.1† | 5.5 ± 13.2 |
| Serum equol (ng/ml) | |||
| 3 weeks | <2 | <2 | 129.5 ± 8.9‡ |
| 27 weeks | ND | ND | 133.5 ± 22.7 |
con = control casein/lactalbumin-based diet; E2 = control diet with the human equivalent of 1 mg oral micronized 17β-estradiol per day; EQ = control diet with the human equivalent of 105 mg racemic equol per day; ND, not determined.
Values represent means ± SE. E2 and EQ were measured in a randomly selected subset of animals. Respective group numbers for con, E2, and EQ groups were 8, 12, and 18 (serum E2 baseline); 7, 7, and 8 (serum E2 3 weeks); 7, 7, and 22 (serum EQ 3 weeks); and 0, 0, and 22 (serum EQ 27 weeks). For conversion to SI units, multiply by 3.70 for E2 (pmol/L) and 4.13 for equol (nmol/L).
Significantly different from control group at P < 0.01.
Significantly different from control group at P < 0.0001.
Treatment effects on normal mammary gland epithelium
Mammary glands from control and EQ animals were diffusely atrophic with small scattered lobular units. In contrast, E2 treatment resulted in increased lobular size on qualitative assessment of whole mounts (Figure 1a) and greater epithelial density on histomorphometry (P = 0.02 compared to control group) (Figure 1b). Within normal epithelium, positive nuclear immunolabeling for ESR1 averaged 15 – 30% among lobular cells and 10 – 20% in extralobular ductal cells and did not differ among treatment groups (P > 0.1 for lobules and ducts) (Figure 1c). In contrast, PGR labeling was low (<3% of lobular and ductal cells) in control and EQ groups and significantly higher in the E2 group (7 – 9% positive lobular cells, P = 0.001; 10 – 12% positive ductal cells, P = 0.0004). Expression of the proliferation marker Ki67 was also low (<3% positive lobular cells, <1% positive ductal cells) in control and EQ groups and significantly higher in the E2 group for intralobular (6 – 10% positive cells, P = 0.0004) but not ductal epithelium (1 – 2% positive cells, P = 0.12).
Figure 1.

Treatment effects on normal mammary gland epithelium. (a), whole mount images showing atrophic postmenopausal mammary gland epithelium with small lobular size in the control (con) and equol (EQ) groups compared to the estradiol (E2) group; arrowhead indicates a focus of cystic change; bars = 1.5 cm. (b), effects of E2 and EQ on epithelial density, measured by histomorphometry. (c), immunolabeling of normal epithelial cells for estrogen receptor alpha (ESR1), progesterone receptor (PGR), and the proliferation marker Ki67. Vertical bars indicate standard errors. * P < 0.05 and *** P < 0.001 vs. respective control group by Wilcoxon Rank Sum test.
Prevalence of mammary gland lesions
Histologic abnormalities identified on histology included CCC, CCH with and without atypia, ADH, and ALH (Figure 2). Baseline mammary biopsies contained 9 CCC lesions (5 control, 1 E2, 3 EQ) and single cases of CCH (EQ) and ADH (E2). Following treatment, the overall prevalence of mammary lesions on routine histology was 41% (26/63). Of these, 19 cases included elements of hyperplasia. Thirteen cases of CCH (3 control, 9 E2, 1 EQ) either with or without atypia were found (Figure 3). Two CCH cases (both E2) included areas of FEA (P = 0.23 compared to control). Six cases of ADH were identified (1 control, 4 E2, 1 EQ), and 3 of these cases (all E2) exhibited extensive cytologic and architectural atypia characteristic of DCIS. All DCIS lesions had a well-differentiated histologic grade and either a micropapillary or solid growth pattern. All ADH cases included features of CCH. Three cases of ALH were noted (2 control, 1 E2). The prevalence of total CCH lesions (P = 0.04) and CCH with atypia (P = 0.04) was higher in animals treated with E2 compared to control, while CCC, ALH, and ADH/DCIS prevalence did not differ among treatments. Incidental histologic included moderate to marked lobular enlargement (1 control, 7 E2, 2 EQ) (P = 0.02 for E2 compared to control), apocrine-type metaplasia (4 control, 2 E2, 3 EQ), secretory change (7 control, 1 E2, 7 EQ) (P = 0.04 for E2 compared to control), and focal lymphoplasmacytic mastitis (1 control, 1 E2).
Figure 2.
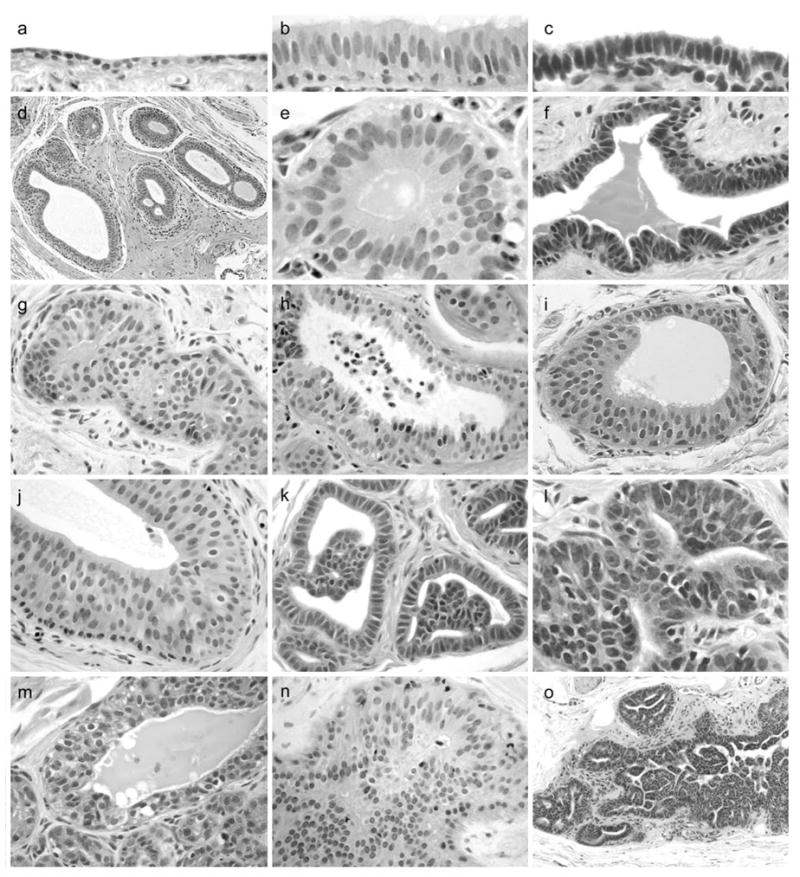
Morphologic features of mammary gland epithelial lesions. (a), normal duct with flattened cuboidal epithelium (left). (b) – (c), columnar cell change (CCC) variants showing luminal cells with ovoid to elongate nuclei aligned perpendicular to the basement membrane. (d), terminal ductal lobular unit (TDLU) with dilation of terminal ducts lined by columnar luminal cells. (e) – (f), CCC variants without atypia. (g), columnar cell hyperplasia (CCH) without atypia showing luminal cells in >2 layers with partial luminal obstruction. (h) CCH with tufting and apical secretory blebs (consistent with human “columnar alteration with prominent secretory snouts,” or CAPSS lesion). (i) – (j) CCH with low-grade monomorphic nuclear atypia and a flat apical surface (consistent with human “flat epithelial atypia,” or FEA lesion). (k) – (l), atypical ductal hyperplasia (ADH) lesions with micropapillary-type architecture. (m), atypical lobular hyperplasia (ALH) showing multiple layers of secretory acinar cells, bordered below by small normal lobuloalveolar acini. (n) – (o), ductal carcinoma in situ (DCIS) lesions with cells arranged in sheets, micropapillae, and fenestrated bridges.
Figure 3.

Prevalence of mammary epithelial lesions following control (con), estradiol (E2), and equol (EQ) treatment. CCC, columnar cell change; CCH, columnar cell hyperplasia; ADH, atypical ductal hyperplasia; ALH, atypical lobular hyperplasia. Numbers on bars indicate the number of animals identified with each type of lesion. * P < 0.05 vs. control group by Fisher’s exact test.
Sex steroid receptor expression and proliferation within mammary lesions
Immunolabeling of mammary epithelium was stratified by lesion type and treatment group. Expression of ESR1 was detected in ~50 – 55% of cells within CCLs, which was higher than normal ductal epithelium within each treatment group (P < 0.01 for all) (Figure 4a). Expression of PGR was also higher in CCLs compared to normal epithelium in the E2 (P = 0.003) and EQ (P = 0.048) groups but not the control group (P = 0.11). Ki67 expression was not increased in CCC or CCH lesions compared to normal ductal epithelium (P > 0.1 for all groups). Qualitatively, ESR1, PGR, and Ki67 labeling in normal epithelial cells was nuclear and scattered in distribution (Figure 4b). In comparison, CCLs showed more diffuse nuclear ESR1 and PGR staining and rare nuclear staining for Ki67. ADH and DCIS lesions had more variable nuclear ESR1 staining that was patchy in overall distribution, scant nuclear PGR labeling, and scattered nuclear Ki67 staining that was generally more prevalent in basilar cells compared to intraluminal micropapillary cells.
Figure 4.
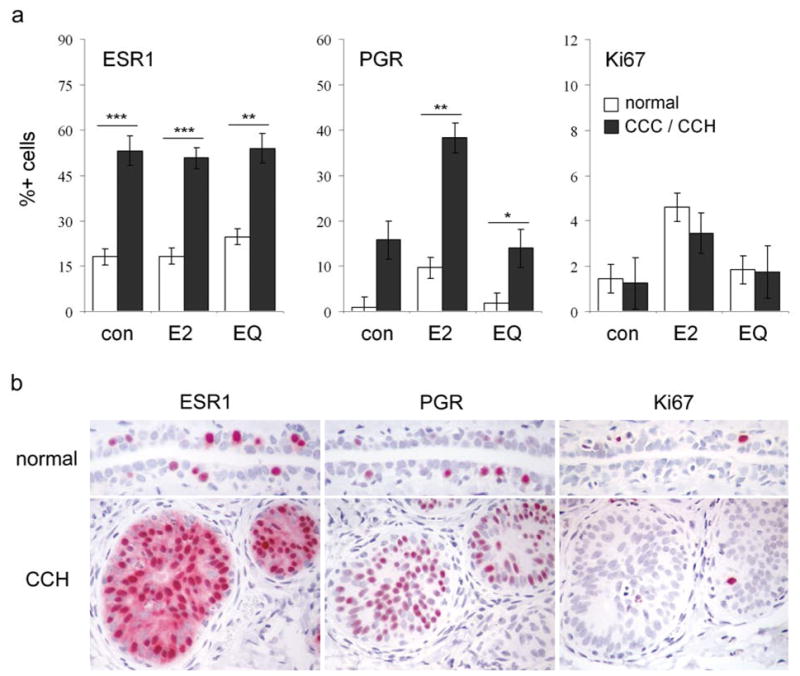
Immunolabeling of columnar cell lesions for estrogen receptor alpha (ESR1), progesterone receptor (PGR), and the proliferation marker Ki67. (a), labeling indices across treatments, stratified by lesion type. CCLs included columnar cell change (CCC) and columnar cell hyperplasia (CCH) lesions. Vertical bars indicate standard errors. * P < 0.05, ** P < 0.01, and *** P < 0.001 compared to respective normal epithelium values by Wilcoxon Rank Sum test. (b), representative immunostaining of normal duct (top row) and CCH (bottom row) for ESR1, PGR, and Ki67 in an E2-treated animal.
Intramammary gene expression
Lastly, we evaluated expression of gene markers related to estrogen receptor activity and proliferation. No differences in ESR1 or ESR2 expression were observed among treatment groups, while the E2 group showed increased expression of ESR1 activity markers (P < 0.001 for PGR and TFF1 compared to control) and a trend for higher MKI67 expression (ANCOVA P = 0.06) (Figure 5a). To examine lesion status effects on these same markers, animals were stratified based on the presence of CCLs in either baseline or post-treatment samples. This classification resulted in 30 lesion-negative (9 control, 7 E2, 14 EQ) and 30 lesion-positive (12 control, 10 E2, 8 EQ) cases. After adjusting for treatment group, animals with CCLs on histology had greater expression of ESR1, TFF1, and MKI67, and lower expression of ESR2 (P < 0.05 for all) at the corresponding site in the contralateral mammary gland (Figure 5b). When analysis was limited to CCH cases only (45 lesion-negative compared to 15 lesion-positive), greater expression of PGR, TFF1, and MKI67 was found (P < 0.01 for all) (data not shown).
Figure 5.

Relative expression of genes related to estrogen receptor activity and proliferation in the mammary gland. (a), effects of estradiol (E2) and equol (EQ) treatment on expression of estrogen receptor alpha (ESR1), estrogen receptor beta (ESR2), progesterone receptor (PGR), trefoil factor 1 (TFF1), and Ki67 antigen (MKI67). (b), gene expression sorted by columnar cell lesion (CCL) status. All gene expression values were corrected for internal control gene expression and then expressed relative to control group values; lesion status effects were adjusted for any treatment group effects. * P < 0.05, ** P < 0.01, and *** P < 0.001 compared to respective control values.
DISCUSSION
The primary goal of this study was to evaluate the effects of pharmacologic (E2) and dietary (EQ) estrogens on proliferative changes in the mammary gland. Treatment with E2 resulted in higher proliferation and PGR expression within normal breast epithelium and increased prevalence of total CCH and CCH with atypia. These lesions had increased ESR1 expression across all groups and an exaggerated increase in PGR expression in response to E2. In contrast, a high dietary dose of EQ resulted in no detectable estrogenic effects in either normal or hyperplastic epithelium.
Results of this study provide direct experimental evidence that E2 may promote certain types of benign proliferative breast lesions. Prior data on this subject have been drawn largely from observational studies, which have been mostly inconclusive or found no effect of postmenopausal hormone therapy on cancer risk associated with BBD.13,51,52 In contrast, a large clinical trial reported a substantial reduction in BBD incidence and subsequent cancer risk associated with BBD in women taking tamoxifen, which is antiestrogenic in the breast.25 Results from the Women’s Health Initiative Estrogen-Alone Trial, a large prospective randomized clinical trial of postmenopausal women with prior hysterectomy, showed that use of oral conjugated equine estrogens (CEE) resulted in a marginal reduction in the incidence of ductal carcinoma despite an increased number of abnormal mammograms.26 Subgroup analyses revealed a significant interaction between CEE treatment and prior BBD in which CEE-treated women with no prior BBD had lower risk of invasive breast cancer than women with prior BBD. A recent follow-up study from this trial reported a significant increase in benign proliferative breast lesions (primarily those without atypia) in women receiving CEE,28 supporting the idea that estrogen therapy may promote at least a subset of benign breast lesions. It is unclear whether the increase in lesions with atypia observed in the current study reflects a potential difference between CEE and E2 therapies.
Increased CCL expression of ESR1 (overall) and PGR (induced) suggests that some forms of proliferative mammary lesions exhibit an exaggerated response to E2. This finding is consistent with several prior studies showing increased ESR1 and PGR expression in CCLs7,18,29–31 and a recent microarray study showing differences between CCLs and normal TDLUs in the expression of numerous ESR1-regulated genes.12 This latter study also found increased CCL expression of amphiregulin, an important developmental growth factor, and speculated that CCLs may represent a reactivation of pathways related to embryonic development. This “recapitulation” idea is consistent with prior observations noting morphologic similarities between developing ductal structures in the early pubertal mammary gland and CCLs in the postmenopausal breast.39
Features of CCLs suggest that these lesions may be early precursors or marker lesions for certain types of ESR1-positive carcinomas.31 In this study CCLs had high ESR1/PGR expression, often exhibited atypia, and co-localized with low-grade ESR1-positive ADH and DCIS, consistent with observations from several studies of human CCLs.15–18 CCLs were also located within TDLUs, where the majority of breast cancers are thought to originate.53 In addition, we observed greater expression of gene markers related to ESR1 activity and proliferation in sites contralateral to CCLs, suggesting that these lesions commonly have a bilateral/multifocal distribution or reflect a more general state of increased estrogen sensitivity. Further work characterizing the molecular profile of CCLs in relation to different types of mammary gland hyperplasia and carcinoma should help in evaluating these ideas.
A notable feature of CCLs is their heterogeneity, which has contributed to the many CCL synonyms in the literature. In this study we observed two main CCL subtypes (Figure 2). The most common type showed variable degrees of luminal cell stratification, ovoid nuclei with evenly dispersed chromatin, abundant finely granular pale eosinophilic cytoplasm, and in some cases rudimentary lumen formation and small distinct nucleoli. The other type was characterized by palisading cells with lesser amounts of cytoplasm and more densely-packed, hyperchromatic, and near-rectangular nuclei aligned in conspicuous “picket fence” rows. This second CCL type was consistently associated with micropapillary ADH. While case numbers were insufficient to conduct any type of stratified analyses, the first type of CCL tended to show higher ESR1 and PGR expression and lower Ki67 expression compared to the second CCL type; however, it is unclear at this point whether any such subtypes represent distinct lineages with different biologic behaviors and risk profiles.
We have previously documented similarities in morphologic and immunophenotypic patterns between human and macaque mammary carcinomas,40 and the CCL features described herein provide further support for the idea that carcinogenesis may occur along homologous pathways in these species. Similarities between human and macaque CCLs included location at TDLUs, nuclear morphology, cystic dilation of ducts, variable apical secretory blebbing, and frequently high ESR1 and PGR expression. Noted differences were the relative paucity of prominent secretory snouts and intralesional mineralization in macaque CCLs. We speculate that these differences may relate in part to the ovariectomized status of the animals and the resulting loss of mammary gland secretory activity in the absence of progesterone. This idea is supported by the observation that CCLs noted incidentally in a survey of older mostly intact animals more commonly exhibited secretory snouts and mineralization.40
One of the aims of this study was to evaluate the mammary gland effects of EQ, a phytoestrogenic compound produced by gut flora from the isoflavone daidzein in about one-third of people consuming soy.32,54 This daidzein-metabolizing phenotype has been associated with such traits as lower serum estrogens55 and lower breast cancer risk,56 although the direct role of EQ in these observations is unclear. Our data indicate that EQ, when given as a purified supplement at a high dietary dose for a period >6 months, lacks estrogenic effects on the normal postmenopausal mammary gland and has no apparent promotional activity for proliferative lesions. It remains unclear whether comparable doses of purified genistein or daidzein (the predominant soy isoflavonoids) have a similar lack of effect. A recent study of MCF-7 breast cancer cell transplants in nude mice showed no estrogenic effects of equol and minimal effects of daidzein,57 in contrast to prior data showing estrogen-related proliferation with high doses of genistein.58
Columnar cell lesions represent a morphologically distinct cell population often associated with hyperplasia, atypia, and carcinoma in the human breast. Data from this study indicate that these lesions exhibit a hyperresponsive phenotype in the presence of estrogen and may be promoted by exogenous E2 exposure. These observations are supported by prior studies in the human breast noting aberrant overexpression of estrogen-related markers within similar lesions. This information may be useful for postmenopausal women with a history of CCL or other BBD who are currently receiving (or considering) estrogen or antiestrogen therapy. Future study of CCLs may provide valuable information related to estrogen effects on benign or premalignant breast disease, the role of abnormal estrogen signaling in early stages of breast cancer development, and the usefulness of CCLs as risk biomarkers for subsequent breast cancer.
Acknowledgments
Supported by the National Institutes of Health (P01 HL 45666, K01 RR 021322-03, and T32 RR 07009).
The authors thank Joseph Finley, Lisa O’Donnell, Hermina Borgerink, Jean Gardin, and Maryanne Post for their technical contributions; Dr. Adrian A. Franke at the Cancer Research Center of Hawaii for serum equol measurements; and Dr. Haiying Chen in the Department of Biostatistical Sciences, Wake Forest University School of Medicine, for statistical advice. We also thank Dr. Thomas B. Clarkson for his work in the original design and implementation of this study. This work was supported by grants from the National Institutes of Health (NIH) National Center for Research Resources (NCRR) (K01 RR 021322-03 and T32 RR 07009) and National Heart, Lung, and Blood Institute (NHBLI) (P01 45666). The contents are solely the responsibility of the authors and do not necessarily represent the view of the NCRR, NHLBI, or NIH.
Abbreviations
- ABI
Applied Biosystems
- ACTB
beta-actin
- ADH
atypical ductal hyperplasia
- ALH
atypical lobular hyperplasia
- BBD
benign breast disease
- BW
body weight
- CCC
columnar cell change
- CCH
columnar cell hyperplasia
- CCL
columnar cell lesion
- CEE
conjugated equine estrogens
- Con
control
- DCIS
ductal carcinoma in situ
- E2
17β-estradiol
- EQ
equol
- ESR1
estrogen receptor alpha
- ESR2
estrogen receptor beta
- FEA
flat epithelial atypia
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- MKI67
gene for Ki67 antigen
- PGR
progesterone receptor
- qRT-PCR
quantitative real-time reverse transcriptase polymerase chain reaction
- TDLU
terminal ductal lobular unit
- TFF1
trefoil factor 1
References
- 1.Dupont WD, Page DL. Risk factors for breast cancer in women with proliferative breast disease. N Engl J Med. 1985;312:146–151. doi: 10.1056/NEJM198501173120303. [DOI] [PubMed] [Google Scholar]
- 2.Santen RJ, Mansel R. Benign breast disorders. N Engl J Med. 2005;353:275–285. doi: 10.1056/NEJMra035692. [DOI] [PubMed] [Google Scholar]
- 3.Hartmann LC, Sellers TA, Frost MH, et al. Benign breast disease and the risk of breast cancer. N Engl J Med. 2005;353:229–237. doi: 10.1056/NEJMoa044383. [DOI] [PubMed] [Google Scholar]
- 4.Carter CL, Corle DK, Micozzi MS, et al. A prospective study of the development of breast cancer in 16,692 women with benign breast disease. Am J Epidemiol. 1988;128:467–477. doi: 10.1093/oxfordjournals.aje.a114995. [DOI] [PubMed] [Google Scholar]
- 5.London SJ, Connolly JL, Schnitt SJ, et al. A prospective study of benign breast disease and the risk of breast cancer. JAMA. 1992;267:941–944. [PubMed] [Google Scholar]
- 6.Ashbeck EL, Rosenberg RD, Stauber PM, et al. Benign Breast Biopsy Diagnosis and Subsequent Risk of Breast Cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:467–472. doi: 10.1158/1055-9965.EPI-06-0394. [DOI] [PubMed] [Google Scholar]
- 7.Fraser JL, Raza S, Chorny K, et al. Columnar alteration with prominent apical snouts and secretions: a spectrum of changes frequently present in breast biopsies performed for microcalcifications. Am J Surg Pathol. 1998;22:1521–1527. doi: 10.1097/00000478-199812000-00009. [DOI] [PubMed] [Google Scholar]
- 8.Schnitt SJ, Vincent-Salomon A. Columnar cell lesions of the breast. Adv Anat Pathol. 2003;10:113–124. doi: 10.1097/00125480-200305000-00001. [DOI] [PubMed] [Google Scholar]
- 9.Nasser SM. Columnar cell lesions: current classification and controversies. Semin Diagn Pathol. 2004;21:18–24. doi: 10.1053/j.semdp.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Tan PH, Ho BC, Selvarajan S, et al. Pathological diagnosis of columnar cell lesions of the breast: are there issues of reproducibility? J Clin Pathol. 2005;58:705–709. doi: 10.1136/jcp.2004.025239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Datrice N, Narula N, Maggard M, et al. Do breast columnar cell lesions with atypia need to be excised? Am Surg. 2007;73:984–986. [PubMed] [Google Scholar]
- 12.Lee S, Medina D, Tsimelzon A, et al. Alterations of gene expression in the development of early hyperplastic precursors of breast cancer. Am J Pathol. 2007;171:252–262. doi: 10.2353/ajpath.2007.061010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schnitt SJ. Benign breast disease and breast cancer risk: morphology and beyond. Am J Surg Pathol. 2003;27:836–841. doi: 10.1097/00000478-200306000-00017. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein NS, Kestin LJ, Vicini FA. Monomorphic epithelial proliferations: characterization and evidence suggesting they are the pool of partially transformed lesions from which some invasive carcinomas arise. Am J Clin Pathol. 2007;128:1023–1034. doi: 10.1309/YU37DVCUFUHP8VPY. [DOI] [PubMed] [Google Scholar]
- 15.Oyama T, Iijima K, Takei H, et al. Atypical cystic lobule of the breast: an early stage of low-grade ductal carcinoma in-situ. Breast Cancer. 2000;7:326–331. doi: 10.1007/BF02966399. [DOI] [PubMed] [Google Scholar]
- 16.Abdel-Fatah TM, Powe DG, Hodi Z, et al. High frequency of coexistence of columnar cell lesions, lobular neoplasia, and low grade ductal carcinoma in situ with invasive tubular carcinoma and invasive lobular carcinoma. Am J Surg Pathol. 2007;31:417–426. doi: 10.1097/01.pas.0000213368.41251.b9. [DOI] [PubMed] [Google Scholar]
- 17.Kusama R, Fujimori M, Matsuyama I, et al. Clinicopathological characteristics of atypical cystic duct (ACD) of the breast: assessment of ACD as a precancerous lesion. Pathol Int. 2000;50:793–800. doi: 10.1046/j.1440-1827.2000.01121.x. [DOI] [PubMed] [Google Scholar]
- 18.Simpson PT, Gale T, Reis-Filho JS, et al. Columnar cell lesions of the breast: the missing link in breast cancer progression? A morphological and molecular analysis. Am J Surg Pathol. 2005;29:734–746. doi: 10.1097/01.pas.0000157295.93914.3b. [DOI] [PubMed] [Google Scholar]
- 19.Moinfar F, Man YG, Bratthauer GL, et al. Genetic abnormalities in mammary ductal intraepithelial neoplasia-flat type (“clinging ductal carcinoma in situ”): a simulator of normal mammary epithelium. Cancer. 2000;88:2072–2081. [PubMed] [Google Scholar]
- 20.Dabbs DJ, Carter G, Fudge M, et al. Molecular alterations in columnar cell lesions of the breast. Mod Pathol. 2006;19:344–349. doi: 10.1038/modpathol.3800538. [DOI] [PubMed] [Google Scholar]
- 21.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006;354:270–282. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 22.Key T, Appleby P, Barnes I, et al. Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J Natl Cancer Inst. 2002;94:606–616. doi: 10.1093/jnci/94.8.606. [DOI] [PubMed] [Google Scholar]
- 23.Russo J, Russo IH. The role of estrogen in the initiation of breast cancer. J Steroid Biochem Mol Biol. 2006;102:89–96. doi: 10.1016/j.jsbmb.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 1998;90:1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- 25.Tan-Chiu E, Wang J, Costantino JP, et al. Effects of tamoxifen on benign breast disease in women at high risk for breast cancer. J Natl Cancer Inst. 2003;95:302–307. doi: 10.1093/jnci/95.4.302. [DOI] [PubMed] [Google Scholar]
- 26.Stefanick ML, Anderson GL, Margolis KL, et al. Effects of conjugated equine estrogens on breast cancer and mammography screening in postmenopausal women with hysterectomy. JAMA. 2006;295:1647–1657. doi: 10.1001/jama.295.14.1647. [DOI] [PubMed] [Google Scholar]
- 27.Schnitt SJ. Benign breast disease and breast cancer risk: potential role for antiestrogens. Clin Cancer Res. 2001;7:4419S–4422S. [PubMed] [Google Scholar]
- 28.Rohan TE, Negassa A, Chlebowski RT, et al. Conjugated equine estrogen and risk of benign proliferative breast disease: a randomized controlled trial. J Natl Cancer Inst. 2008;100:563–571. doi: 10.1093/jnci/djn075. [DOI] [PubMed] [Google Scholar]
- 29.Dessauvagie BF, Zhao W, Heel-Miller KA, et al. Characterization of columnar cell lesions of the breast: immunophenotypic analysis of columnar alteration of lobules with prominent apical snouts and secretions. Hum Pathol. 2007;38:284–292. doi: 10.1016/j.humpath.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 30.Tremblay G, Deschenes J, Alpert L, et al. Overexpression of estrogen receptors in columnar cell change and in unfolding breast lobules. Breast J. 2005;11:326–332. doi: 10.1111/j.1075-122X.2005.21698.x. [DOI] [PubMed] [Google Scholar]
- 31.Lee S, Mohsin SK, Mao S, et al. Hormones, receptors, and growth in hyperplastic enlarged lobular units: early potential precursors of breast cancer. Breast Cancer Res. 2006;8:R6. doi: 10.1186/bcr1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Setchell KD, Brown NM, Lydeking-Olsen E. The clinical importance of the metabolite equol - a clue to the effectiveness of soy and its isoflavones. J Nutr. 2002;132:3577–3584. doi: 10.1093/jn/132.12.3577. [DOI] [PubMed] [Google Scholar]
- 33.Magness CL, Fellin PC, Thomas MJ, et al. Analysis of the Macaca mulatta transcriptome and the sequence divergence between Macaca and human. Genome Biol. 2005;6:R60. doi: 10.1186/gb-2005-6-7-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pavlicek A, Noskov VN, Kouprina N, et al. Evolution of the tumor suppressor BRCA1 locus in primates: implications for cancer predisposition. Hum Mol Genet. 2004;13:2737–2751. doi: 10.1093/hmg/ddh301. [DOI] [PubMed] [Google Scholar]
- 35.Stute P, Wood CE, Kaplan J, et al. Cyclic changes in the mammary gland of cynomolgus macaques. Fertil Steril. 2004;82:1160–1170. doi: 10.1016/j.fertnstert.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 36.Cheng G, Li Y, Omoto Y, et al. Differential regulation of estrogen receptor (ER)alpha and ERbeta in primate mammary gland. J Clin Endocrinol Metab. 2005;90:435–444. doi: 10.1210/jc.2004-0861. [DOI] [PubMed] [Google Scholar]
- 37.Cline JM, Wood CE. Hormonal effects on the mammary gland of postmenopausal nonhuman primates. Breast Dis. 2005–2006;24:59–70. doi: 10.3233/bd-2006-24105. [DOI] [PubMed] [Google Scholar]
- 38.Cline JM, Soderqvist G, von Schoultz E, et al. Effects of conjugated estrogens, medroxyprogesterone acetate, and tamoxifen on the mammary glands of macaques. Breast Cancer Res Treat. 1998;48:221–229. doi: 10.1023/a:1005984932268. [DOI] [PubMed] [Google Scholar]
- 39.Wood CE, Hester J, Cline JM. Mammary gland development in early pubertal female macaques. Toxicol Pathol. 2007;35:793–803. doi: 10.1080/01926230701584213. [DOI] [PubMed] [Google Scholar]
- 40.Wood CE, Usborne A, Tarara R, et al. Hyperplastic and neoplastic lesions of the mammary gland in macaques. Vet Pathol. 2006;43:471–483. doi: 10.1354/vp.43-4-471. [DOI] [PubMed] [Google Scholar]
- 41.Schneider K, Oltmanns J, Hassauer M. Allometric principles for interspecies extrapolation in toxicological risk assessment -- empirical investigations. Regul Toxicol Pharmacol. 2004;39:334–347. doi: 10.1016/j.yrtph.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 42.Messina M, Nagata C, Wu AH. Estimated Asian adult soy protein and isoflavone intakes. Nutr Cancer. 2006;55:1–12. doi: 10.1207/s15327914nc5501_1. [DOI] [PubMed] [Google Scholar]
- 43.Franke AA, Custer LJ, Wilkens LR, et al. Liquid chromatographic-photodiode array mass spectrometric analysis of dietary phytoestrogens from human urine and blood. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;777:45–59. doi: 10.1016/s1570-0232(02)00216-7. [DOI] [PubMed] [Google Scholar]
- 44.Tavassoli FA, Hoefler H, Rosai J, et al. Intraductal Proliferative Lesions. In: Tavassoli FA, Devilee P, editors. Pathology and Genetics: Tumours of the Breast and Female Genital Organs. Lyon: IARC Press; 2003. pp. 63–73. [Google Scholar]
- 45.Rosen PP. Rosen’s Breast Pathology. 2. Philadelphia: Lippincott Williams & Wilkins; 2001. Ductal Hyperplasia: Ordinary and Atypical; pp. 201–251. [Google Scholar]
- 46.Dalton LW, Page DL, Dupont WD. Histologic grading of breast carcinomas: a reproducibility study. Cancer. 1994;73:2765–2770. doi: 10.1002/1097-0142(19940601)73:11<2765::aid-cncr2820731119>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 47.Cline JM. Assessing the mammary gland of nonhuman primates: effects of endogenous hormones and exogenous hormonal agents and growth factors. Birth Defects Res B Dev Reprod Toxicol. 2007;80:126–146. doi: 10.1002/bdrb.20112. [DOI] [PubMed] [Google Scholar]
- 48.Wood CE, Register TC, Franke AA, et al. Dietary Soy Isoflavones Inhibit Estrogen Effects in the Postmenopausal Breast. Cancer Res. 2006;66:1241–1249. doi: 10.1158/0008-5472.CAN-05-2067. [DOI] [PubMed] [Google Scholar]
- 49.Slater CC, Hodis HN, Mack WJ, et al. Markedly elevated levels of estrone sulfate after long-term oral, but not transdermal, administration of estradiol in postmenopausal women. Menopause. 2001;8:200–203. doi: 10.1097/00042192-200105000-00009. [DOI] [PubMed] [Google Scholar]
- 50.Setchell KD, Brown NM, Desai PB, et al. Bioavailability, disposition, and dose-response effects of soy isoflavones when consumed by healthy women at physiologically typical dietary intakes. J Nutr. 2003;133:1027–1035. doi: 10.1093/jn/133.4.1027. [DOI] [PubMed] [Google Scholar]
- 51.Byrne C, Connolly JL, Colditz GA, et al. Biopsy confirmed benign breast disease, postmenopausal use of exogenous female hormones, and breast carcinoma risk. Cancer. 2000;89:2046–2052. doi: 10.1002/1097-0142(20001115)89:10<2046::aid-cncr3>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 52.Dupont WD, Page DL, Rogers LW, et al. Influence of exogenous estrogens, proliferative breast disease, and other variables on breast cancer risk. Cancer. 1989;63:948–957. doi: 10.1002/1097-0142(19890301)63:5<948::aid-cncr2820630527>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 53.Russo J, Hu YF, Yang X, et al. Developmental, cellular, and molecular basis of human breast cancer. J Natl Cancer Inst Monogr. 2000;27:17–37. doi: 10.1093/oxfordjournals.jncimonographs.a024241. [DOI] [PubMed] [Google Scholar]
- 54.Selvaraj V, Zakroczymski MA, Naaz A, et al. Estrogenicity of the isoflavone metabolite equol on reproductive and non-reproductive organs in mice. Biol Reprod. 2004;71:966–972. doi: 10.1095/biolreprod.104.029512. [DOI] [PubMed] [Google Scholar]
- 55.Duncan AM, Merz-Demlow BE, Xu X, et al. Premenopausal equol excretors show plasma hormone profiles associated with lowered risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2000;9:581–586. [PubMed] [Google Scholar]
- 56.Ingram D, Sanders K, Kolybaba M, et al. Case-control study of phytooestrogens and breast cancer. Lancet. 1997;350:990–994. doi: 10.1016/S0140-6736(97)01339-1. [DOI] [PubMed] [Google Scholar]
- 57.Ju YH, Fultz J, Allred KF, et al. Effects of dietary daidzein and its metabolite, equol, at physiological concentrations on the growth of estrogen-dependent human breast cancer (MCF-7) tumors implanted in ovariectomized athymic mice. Carcinogenesis. 2006;27:856–863. doi: 10.1093/carcin/bgi320. [DOI] [PubMed] [Google Scholar]
- 58.Ju YH, Allred CD, Allred KF, et al. Physiological concentrations of dietary genistein dose-dependently stimulate growth of estrogen-dependent human breast cancer (MCF-7) tumors implanted in athymic nude mice. J Nutr. 2001;131:2957–2962. doi: 10.1093/jn/131.11.2957. [DOI] [PubMed] [Google Scholar]