

Abstract
Cardiovascular disease, in which atherosclerosis is the major underlying cause, is currently the largest cause of death in the world. Atherosclerosis is an inflammatory disease characterized by the formation of arterial lesions over a period of several decades at sites of endothelial cell dysfunction. These lesions are composed of endothelial cells, vascular smooth muscle cells, monocytes/macrophages and T lymphocytes (CD4+). As the lesions progress some can become unstable and prone to disruption, resulting in thrombus formation and possibly a myocardial infarction or stroke depending upon the location. Although the exact triggers for plaque disruption remain unknown, much recent evidence has shown a link between the incidence of myocardial infarction and stroke and a recent respiratory tract infection. Interestingly, many reports have also shown a link between a family of pattern recognition receptors, the Toll-like receptors, and the progression of atherosclerosis, suggesting that infections may play a role in both the progression of atherosclerosis and in inducing the more severe complications associated with the disease.
Keywords: atherosclerosis, infection, inflammation, myocardial infarction, TLR
Introduction
Cardiovascular disease is currently the leading cause of death in the world and is predicted to remain so for many years to come, placing a huge financial burden on the world's health resources [1,2]. The two main causes of death are ischaemic heart disease and cerebrovascular disease, which together accounted for 27·2% of all deaths in high-income countries, and 21·3% of all deaths in low- or middle-income countries in 2001. The majority of these deaths are caused by underlying atherosclerosis, where disruption of arterial plaques within the coronary or carotid arteries prevents or severely reduces blood flow to the target organ.
Atherosclerosis
Atherosclerosis is a chronic inflammatory disease, characterized by the formation of lesions in the large- and medium-sized arteries, often at sites of disturbed blood flow. These arterial lesions, which can form over a period of several decades, are composed of endothelial cells, vascular smooth muscle cells (VSMCs), T lymphocytes (CD4+) and monocytes/macrophages. The currently accepted response-to-injury hypothesis for the development of atherosclerosis suggests that these lesions or plaques form at sites of endothelial cell dysfunction with subsequent monocyte recruitment [3]. There are many causes of endothelial cell dysfunction, such as the uptake of modified forms of low-density lipoprotein (LDL) [4], disturbed blood flow [5] and exposure to proinflammatory cytokines or pathogenic stimuli [6–8]. As the disease progresses, more inflammatory and smooth muscle cells are recruited to the growing lesion where they proliferate, further enhancing the size of the lesion. Outward arterial remodelling is associated with maintenance of the lumen size until a critical point is reached and the lesion begins to extend into the lumen [9], reducing blood flow. Mature plaques can become unstable, with rupture or erosion resulting in intravascular thrombosis and subsequent vascular occlusion, causing a myocardial infarction or stroke (Fig. 1).
Fig. 1.
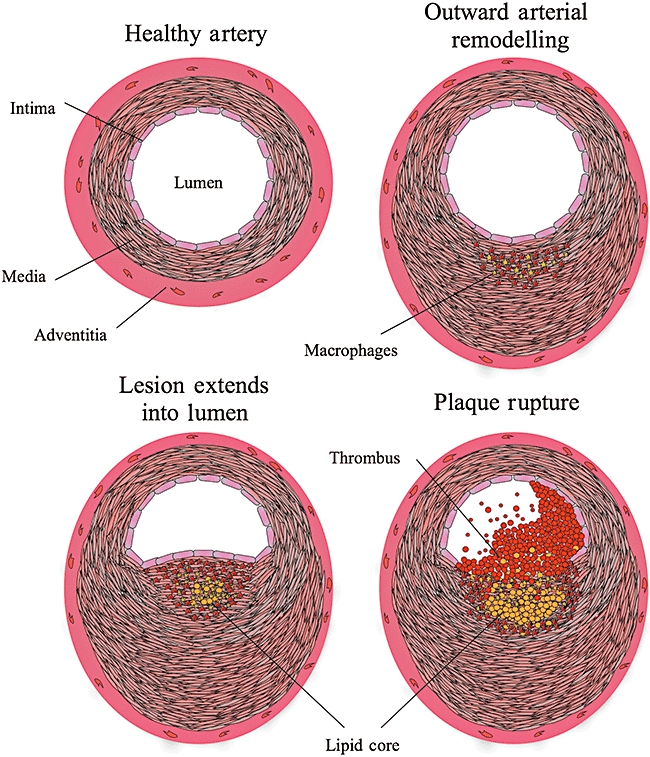
The progression of atherosclerosis from healthy artery to rupture of the plaque and thrombus formation. (a) Structure of a healthy artery; (b) thickening of the media due increased smooth muscle cell and macrophage invasion, although the lumen size is maintained by outward arterial remodelling; (c) when a critical point is reached the growing plaque extends into the lumen, reducing the blood flow; (d) plaque rupture exposes the prothrombotic necrotic/lipid core to the blood, inducing thrombus formation.
The inflammatory nature of atherosclerosis
During the past two decades work conducted by many laboratories, including our own, has defined atherosclerosis as a chronic inflammatory disease [3,10,11]. Indeed, increased circulating concentrations of C-reactive protein (CRP), an acute phase protein whose release is indicative of an inflammatory response, is predictive of future cardiovascular events [12–14]. A major risk factor for atherosclerosis is elevated levels of modified forms of LDL, which induce a variety of proinflammatory effects in the various cell types involved in atherosclerosis. These modified forms of LDL are formed when high plasma concentrations of native LDL induce its own transport across the endothelium into the subendothelial space and intima. Here it is retained through its interaction with matrix components [15] and undergoes modifications catalysed by products released from endothelial cells, VSMCs and macrophages [16]. Modified forms of LDL induce proinflammatory effects by binding to scavenger receptors such as CD36 and class A macrophage scavenger receptor (SR-A) on macrophages, and the lectin-like oxidized low-density lipoprotein scavenger receptor (LOX-1) on endothelial cells, macrophages, VSMCs and platelets [17].
During the initiation of atherosclerosis, the initial tethering of monocytes and T lymphocytes to the area of endothelial cell dysfunction is mediated through endothelial cell CD62P and CD62E interacting with their respective carbohydrate ligands [18]. More stable interactions between the monocytes or T lymphocytes and the endothelium are mediated through vascular cell adhesion molecule 1 (VCAM-1) binding its integrin ligand α4β1 (VLA-4) on the inflammatory cell [19,20]. Tightly bound monocytes and T lymphocytes can then enter the intima and subendothelial space by diapedesis at junctions between the endothelial cells. Following their transmigration into the subendothelial space and intima, monocytes differentiate into macrophages and up-regulate their scavenger receptor expression, allowing them to take up modified LDL [21]. This uptake leads to formation of lipoprotein-derived cholesterol and cholesterol esters leading to the development of foam cells, which along with T lymphocytes form the earliest type of lesion, the fatty streak [22].
As the atherosclerotic lesion progresses VSMCs migrate from the media into the intima, under the control of growth factors such as platelet-derived growth factor and insulin-like growth factor released by activated macrophages and endothelial cells [23]. Once within the intima, the VSMCs are able to proliferate and synthesize large amounts of extracellular matrix, increasing both the size and the stability of the plaque. This proliferation is controlled by other growth factors, including fibroblast growth factor, which is also released from activated macrophages and endothelial cells [24].
Cytokines, chemokines and inflammatory lipids in atherosclerosis
Monocyte recruitment to the intima and subendothelial space occurs by chemotaxis, mainly towards monocyte chemoattractant protein-1 (MCP-1/CCL2) [25], although a role for other chemokines such as regulated upon activation normal T cell expressed and secreted (CCL5) [26] and interleukin (IL)-8 (CXCL8) [27] has been suggested. T lymphocytes are recruited by the interferon (IFN)-γ-inducible cytokines inducible protein-10 (IP-10) (CXCL10), monokine induced by IFN-γ (MIG) (CXCL9) and IFN-inducible T cell alpha chemoattractant (I-TAC) (CXCL11) [28].
Uptake of modified LDL by macrophages induces the release of a plethora of proinflammatory mediators including IL-1β, tumour necrosis factor (TNF)-α, IL-6, IL-8, MCP-1 and several growth factors [29]. These mediators recruit further inflammatory cells to the lesion and induce the activation of endothelial cells and VSMCs, leading to the release of more proinflammatory mediators and up-regulation of adhesion molecules. Increased IL-6 enhances the release of acute phase proteins, including CRP, from the liver, enhancing inflammation further [30]. More cells are then recruited to the growing lesion, where they can be activated in this continuing cycle of inflammation.
One further axis with a key role in the progression of atherosclerosis is CD40/CD40L. Both the receptor and the ligand are expressed on many of the cell types associated with atherosclerosis [31]. CD40 is up-regulated on endothelial cells in response to IL-1β, TNF-α and IFN-γ, and interaction with CD40L can induce the expression of VCAM-1 and CD62E and the release of IL-6 and IL-8 [31]. CD40/CD40L interactions also increase the release of IL-1β and TNF-α from monocytes and macrophages [32] and the expression of tissue factor on macrophages, endothelial cells and VSMCs [31].
Recently there has been much interest in the role of lipids in the progression of atherosclerosis, especially in the prostanoids and lipoxins. Prostanoids include prostaglandins, thromboxanes and prostacylins, and are formed from arachidonic acid through a pathway involving cyclo-oxygenase [33]. Lipoxins are anti-inflammatory eicosanoids also derived from arachidonic acid through a pathway involving lipoxygenases [34]. The role of both cylo-oxygenases and lipoxygenases in atherosclerosis is complex because of their ability to metabolize arachidonic acid into both pro- and anti-inflammatory mediators, capable of influencing the various stages of the disease [33,35].
The IL-1β is a key mediator involved in the progression of atherosclerosis. Along with its naturally occurring antagonist, IL-1ra, it is expressed in endothelial cells, VSMCs and macrophages. Atherosclerotic plaques express significantly raised levels of IL-1β[36] and IL-1ra [37] compared with control arteries, particularly within the endothelium, and there are increased concentrations of circulating IL-1β in patients suffering from unstable angina [38]. Secretion of IL-1β is regulated tightly and efficient release requires a primary stimulation which induces up-regulation of pro-IL-1β, followed by a second stimulation such as ATP accumulation to induce activation of the inflammasome and processing of pro-IL-1β to the mature biologically active form [39]. IL-1β induces adhesion molecule expression, vascular permeability, leucocyte migration, macrophage activation and VSMC proliferation [40], ultimately driving atherosclerosis and plaque instability.
The IL-1β has been implicated in the progression of atherosclerosis using animal models, with reduced atherosclerosis observed in IL-1β[41] and IL-1 receptor-deficient mice [11], while IL-1ra-deficient mice exhibit increased neointima formation [42]. IL-1Ra administration to Apo E-deficient mice or porcine arterial injury models reduced neointima formation significantly [11,43] as well as reducing vascular remodelling following acute myocardial infarction in the rat [44]. Recently, small-scale human trials using anakinra, the recombinant form of IL-1ra, showed improved vascular and left ventricular function and reduced endothelial nitro-oxidative stress in rheumatoid arthritis patients [45]. The potential for IL-1ra to reduce inflammatory markers after a non-ST elevation myocardial infarction is under investigation in a multi-centre study [46].
Atherosclerotic plaque disruption
The presentation of atherosclerotic disease as myocardial infarction or stroke is caused by disruption of atherosclerotic plaques which can become unstable, and have either ruptured their fibrous caps, exposing blood to the prothrombotic contents, or when the surface of the plaque has become eroded, exposing the prothrombotic matrix. Under basal or stable conditions the atherosclerotic plaque is composed of lipid contained within macrophages (now called foam cells) or within a necrotic core, VSMCs which synthesize collagen to give the cap mechanical strength, a few T lymphocytes and an overlying endothelial cell layer [10].
Disruption of the plaque involves a proinflammatory switch within the cells of the plaque. This switch matches well with a T helper type 1 proinflammatory phenotype when IFN-γ can activate an M1 phenotype in macrophages and causes inhibition of VSMC proliferation [47]. Macrophages within an activated or vulnerable plaque (vulnerable to rupture or causing thrombosis) express matrix metalloproteinases capable of digesting the fibrous cap [48,49], as well as tissue factor, on their cell surface [50]. Tissue factor is the essential co-factor for activation of the coagulation cascade and will produce thrombin upon exposure to blood following rupture.
Plaque erosion may be responsible for up to 30% of clinical presentations of atherosclerosis [51]. Here the cap of the plaque becomes eroded in such a way that subendothelial matrix is exposed to blood and produces thrombin, presumably because of expression of platelet adhesion molecules and tissue factor. The mechanism of plaque erosion is unknown, but apoptotic endothelial cells are known to express tissue factor [52]. The pathophysiological inducer of endothelial cell apoptosis is unknown.
Infections can increase the risk of myocardial infarction and stroke
Although many of the factors that predispose atherosclerotic plaques to rupture have been identified, the exact triggers for plaque disruption are as yet unknown. An early indication that infections may play a role in altering plaque stability was suggested by the seasonal distribution of myocardial infarctions. Analysis of data from 1400 hospitals in the United States of more than 250 000 cases of acute myocardial infarction reported that there was a 53% increase in hospital administrations because of myocardial infarction in the winter months compared with the summer [53]. This work confirmed earlier observations from a previous smaller study [54]. These results led to the hypothesis that severe respiratory tract infections, which occur at higher frequencies during the winter months, may play a role in inducing plaque disruption. Evidence supporting this hypothesis was provided by numerous reports showing that vaccination against influenza had a positive effect on reducing the incidences of ischaemic events [55]. One small study of 218 patients who had suffered a myocardial infarction reported lower levels of influenza vaccinations within the group that suffered further myocardial infarctions during the period of study, with vaccination associated with a 67% reduction in subsequent myocardial infarctions [56]. Similarly, it was also reported that there was a lower percentage of influenza vaccination among patients suffering out-of-hospital primary cardiac arrest [57], and among patients who had suffered an ischaemic stroke [58], when compared with the control groups. A large-scale study on the effects of influenza vaccination on ischaemic disease used the data from three large managed-care organizations of more than 280 000 patients who were aged at least 65 years. This study found that vaccination reduced the risk of stroke by 16–23% and the risk of cardiac disease by 19% [59]. These data imply clearly that influenza vaccination has the potential to prevent many deaths each year from myocardial infarction and stroke, with one report suggesting that up to 90 000 deaths could be prevented each year in the United States alone [55].
Additional evidence for the role of infection in inducing myocardial infarctions and strokes is provided by several studies showing an increased risk of these events following severe respiratory tract infections. The first of these analysed the United Kingdom General Practice Research Database (UKGP) and reported a transient but substantial increase [odds ratio (OR) 3·0] in the risk of myocardial infarction between 1 and 10 days after a reported respiratory tract infection, with this risk returning to normal after a period of several weeks [60]. Two more recent larger studies have confirmed the link between respiratory tract infections and myocardial infarctions. Again using the UKGP database, one research group analysed the records of more than 125 000 patients who had suffered a first or subsequent myocardial infarction or stroke. Patients who had a systemic respiratory tract infection had an increased risk of suffering their first myocardial infarction (OR 4·95) and stroke (OR 3·19) for the period of 3 days after the diagnosis, with the risk gradually falling and returning to normal after several weeks [61]. There was also an increased risk of a subsequent myocardial infarction (OR 3·14) and stroke (OR 2·57) during the 3-day period after diagnosis of a systemic respiratory infection. This study also found an increase in the risk of myocardial infarction or stroke following a urinary tract infection, although this was smaller than that following a respiratory tract infection, and has not been a universal finding [61]. Other work using the IMS Disease Analyser Mediplus database (IMS) studied the records of more than 20 000 patients who had suffered a first myocardial infarction or stroke [62]. This study showed an increased risk of myocardial infarction (OR 3·75) and stroke (OR 4·07) for the period of 3 days after a respiratory tract infection, which again returned to normal after several weeks. Further evidence for the role of infections in myocardial infarction was provided by analysis of autopsy-confirmed deaths in St Petersburg, Russia, from 1993 to 2000. For every year studied, the peak rate of deaths from myocardial infarction occurred during the influenza epidemic and peak acute respiratory disease activity [63].
Although evidence for the role of infections in increasing the risk of myocardial infarction and stroke is growing, the mechanisms by which this occurs remain unclear. Recent evidence suggests that the inflammation caused by a systemic infection may alter the structure of the plaque, causing an increase in the number of inflammatory cells and therefore making the plaque more prone to disruption. Whether this is through infection and activation of cells within the plaque, activation of cells which then enter the plaque or through the release of inflammatory mediators which activate cells within and around the plaque is unknown. Indeed, Apo E-deficient mice infected with influenza had increased numbers of inflammatory cells and smooth muscle cells in the subendothelial infiltrate, and increased superficial platelet aggregation when compared with uninfected Apo E-deficient mice [64]. Other studies have reported an alteration in human plaque structure in response to infections. Autopsy samples of atherosclerotic plaques from patients who were diagnosed with an acute systemic infection were analysed and compared with those who had no infection. This revealed increased numbers of macrophages in the adventitia, more T lymphocytes in the adventitia and periadventitial fat and more dendritic cells in the intima and media of those patients who had suffered an infection [65]. T lymphocytes isolated from atherosclerotic plaques were more responsive to influenza A virus than those purified from peripheral blood of the same donor, suggesting a mechanism of influenza-induced plaque inflammation [66]. It therefore seems as though infections may alter the structure of the atherosclerotic plaque, causing an increase in the number of inflammatory cells and reducing the stability.
Toll-like receptors in atherosclerosis
With atherosclerosis defined clearly as an inflammatory disease, and the suggested link between infections and ischaemic events, many groups have shown a link between many infectious organisms such as Chlamydia pneumoniae, Porphyromonas gingivalis, Helicobacter pylori and cytomegalovirus, and the progression of atherosclerosis. The roles for these organisms in atherosclerosis have been extensively reviewed elsewhere [67–71]. Recently, interest in the role of Toll-like receptors (TLRs) in the progression of atherosclerosis has increased. TLRs are a family of pattern recognition receptors that function to enable responses to a wide range of both pathogen-associated molecular patterns and damage-associated molecular patterns [72–88] (Fig. 2), and there is evidence of activation of their signalling pathways within the plaque [89]. TLRs are expressed on all cell types associated with atherosclerotic plaques, such as monocytes [90], endothelial cells [91], VSMCs [74] and platelets [92]. TLR expression is up-regulated on endothelial cells exposed to endogenous mediators of inflammation such as histamine [93,94], and also on macrophages and endothelial cells associated with plaques [95]. Similarly, the level of both TLR-2 and TLR-4 mRNA is increased with time in arterial tissue obtained from Apo E-deficient mice, and TLR-2 and TLR-4 expression on circulating monocytes is increased at 40 weeks in Apo E-deficient mice compared with controls [96]. Modified forms of LDL can enhance macrophage TLR-4 expression in atherosclerotic plaques [97], and agonists of TLR-3 can inhibit macrophage efflux of cholesterol [98], increasing foam cell formation. Animal studies have confirmed a role for TLRs in atherosclerosis, with atherosclerosis-prone mice deficient in TLR-2 or TLR-4 showing reduced atherosclerosis development when compared with those animals with functional receptors [99,100]. Recently, C. pneumoniae-induced enhancement of the progression of atherosclerosis in Apo E-deficient mice was shown to be dependent upon TLR-2, TLR-4 and MyD88 [101].
Fig. 2.
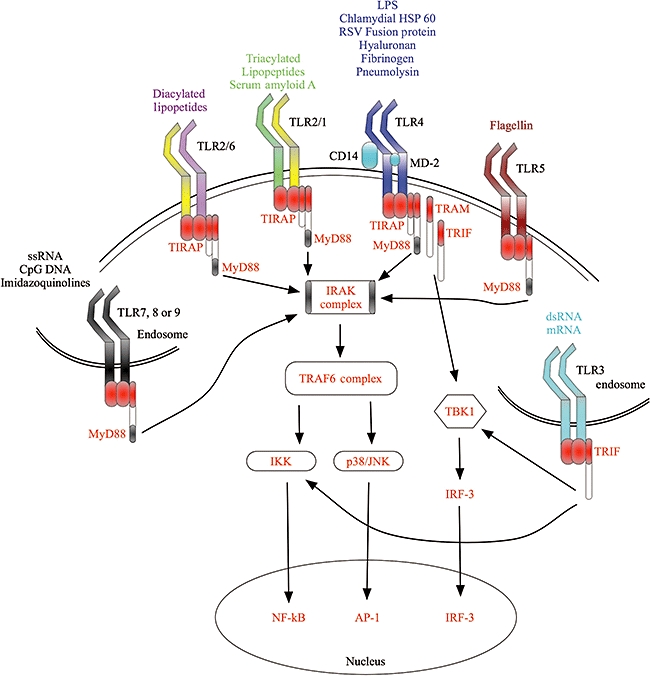
Toll-like receptor (TLR) activation. TLRs are activated by a number of different agonists, and are expressed either on the cell membrane or intracellularly on the membranes of endosomes. The extracellular receptors TLR-1, -2, -4, -5 and -6 respond predominantly to bacterial products as well as some endogenous agonists, whereas the intracellular receptors TLR-3, -7, -8 and -9 respond to nucleotides of viral and bacterial origin. The figure also shows a very simplified TLR signalling pathway, showing how many structurally different agonists can induce activation of similar pathways.
The TLR-4 is the receptor for lipopolysaccharide (LPS), and many studies have shown that this may influence the progression of atherosclerosis. In the Bruneck study, which looked at the number and size of atherosclerotic plaques in the carotid artery over a 5-year period, it was found that circulating LPS was a risk factor for atherosclerosis progression in smokers and patients with a chronic infection [102]. Similarly, two studies reported that weekly injections of LPS into rabbits fed on a high cholesterol diet enhanced atherosclerosis progression [103,104] and that LPS treatment of the adventia enhanced cellular migration into the media and intima, enhancing lesion size [105]. The TLR-2 agonist, Pam3CSK4, also enhanced lesion development in Apo E-deficient mice [106].
Conclusions
There is a clear role for inflammation and infections in both the progression of atherosclerosis and in the serious complications resulting from plaque disruption such as myocardial infarction and stroke. Many of the inflammatory mediators involved in mounting an effective inflammatory response are also involved in the exacerbation of atherosclerosis, so it is unsurprising that a family of pattern recognitions receptors, the TLRs, have been implicated in the disease. A key molecule involved in atherosclerosis is IL-1β, and TLR agonists are some of the most potent inducers of IL-1β release known. As we understand more about the complex signalling mechanisms that act to enhance inflammation, then we may be able to target novel, specific pathways that influence strongly the progression of atherosclerosis.
In terms of plaque disruption, it is clear that a severe infection increases the risk of myocardial infarction or stroke dramatically, but more research is required to determine the exact mechanism so that we can intervene therapeutically. Work from our group has indicated that far more complex signalling networks exist between endothelial cells and monocytes in a simple co-culture model of acute inflammation (data in preparation). The challenge is to dampen down the inflammatory response sufficiently around the time of infection to reduce the risk of ischaemic events, while not allowing the pathogen to take advantage of an impaired inflammatory response.
Acknowledgments
This work was supported by a BHF project grant (FS/06/004) (to J. R. W) and an MRC Senior Clinical Fellowship (G116/170) (to I. S.).
References
- 1.Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet. 2006;367:1747–57. doi: 10.1016/S0140-6736(06)68770-9. [DOI] [PubMed] [Google Scholar]
- 2.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 4.Rangaswamy S, Penn MS, Saidel GM, Chisolm GM. Exogenous oxidized low-density lipoprotein injures and alters the barrier function of endothelium in rats in vivo. Circ Res. 1997;80:37–44. doi: 10.1161/01.res.80.1.37. [DOI] [PubMed] [Google Scholar]
- 5.Tsao PS, Lewis NP, Alpert S, Cooke JP. Exposure to shear stress alters endothelial adhesiveness. Role of nitric oxide. Circulation. 1995;92:3513–9. doi: 10.1161/01.cir.92.12.3513. [DOI] [PubMed] [Google Scholar]
- 6.Morris GE, Parker LC, Ward JR, et al. Cooperative molecular and cellular networks regulate Toll-like receptor-dependent inflammatory responses. FASEB J. 2006;20:2153–5. doi: 10.1096/fj.06-5910fje. [DOI] [PubMed] [Google Scholar]
- 7.Cohen Tervaert JW, 23 Cohen Tervaert JW. Translational Mini-Review Series on Immunology of Vascular Disease: Accelerated atherosclerosis in vasculitis. Clin Exp Immunol. doi: 10.1111/j.1365-2249.2009.03885.x. doi: 10.1111/j.1365-2249.2009.03885.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maugeri N, Rovere-Querini P, Baldini M, Sabbadini MG, Manfredi AA. Translational Mini-Review Series on Immunology of Vascular Disease: Mechanisms of vascular inflammation and remodelling in systemic vasculitis. Clin Exp Immunol. doi: 10.1111/j.1365-2249.2009.03921.x. doi: 10.1111/j.1365-2249.2009.03921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316:1371–5. doi: 10.1056/NEJM198705283162204. [DOI] [PubMed] [Google Scholar]
- 10.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 11.Chamberlain J, Evans D, King A, et al. Interleukin-1beta and signaling of interleukin-1 in vascular wall and circulating cells modulates the extent of neointima formation in mice. Am J Pathol. 2006;168:1396–403. doi: 10.2353/ajpath.2006.051054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rifai N, Ridker PM. Proposed cardiovascular risk assessment algorithm using high-sensitivity C-reactive protein and lipid screening. Clin Chem. 2001;47:28–30. [PubMed] [Google Scholar]
- 13.Armani A, Becker RC. The biology, utilization, and attenuation of C-reactive protein in cardiovascular disease: part II. Am Heart J. 2005;149:977–83. doi: 10.1016/j.ahj.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 14.Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 15.Nievelstein PF, Fogelman AM, Mottino G, Frank JS. Lipid accumulation in rabbit aortic intima 2 hours after bolus infusion of low density lipoprotein. A deep-etch and immunolocalization study of ultrarapidly frozen tissue. Arterioscler Thromb. 1991;11:1795–805. doi: 10.1161/01.atv.11.6.1795. [DOI] [PubMed] [Google Scholar]
- 16.Palinski W, Rosenfeld ME, Yla-Herttuala S, et al. Low density lipoprotein undergoes oxidative modification in vivo. Proc Natl Acad Sci USA. 1989;86:1372–6. doi: 10.1073/pnas.86.4.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy JE, Tedbury PR, Homer-Vanniasinkam S, Walker JH, Ponnambalam S. Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis. 2005;182:1–15. doi: 10.1016/j.atherosclerosis.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 18.Nimrichter L, Burdick MM, Aoki K, et al. E-selectin receptors on human leukocytes. Blood. 2008;112:3744–52. doi: 10.1182/blood-2008-04-149641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jonjic N, Jilek P, Bernasconi S, et al. Molecules involved in the adhesion and cytotoxicity of activated monocytes on endothelial cells. J Immunol. 1992;148:2080–3. [PubMed] [Google Scholar]
- 20.Rose DM, Grabovsky V, Alon R, Ginsberg MH. The affinity of integrin alpha(4)beta(1) governs lymphocyte migration. J Immunol. 2001;167:2824–30. doi: 10.4049/jimmunol.167.5.2824. [DOI] [PubMed] [Google Scholar]
- 21.Huh HY, Pearce SF, Yesner LM, Schindler JL, Silverstein RL. Regulated expression of CD36 during monocyte-to-macrophage differentiation: potential role of CD36 in foam cell formation. Blood. 1996;87:2020–8. [PubMed] [Google Scholar]
- 22.Jaakkola O, Nikkari T. Lipoprotein degradation and cholesterol esterification in primary cell cultures of rabbit atherosclerotic lesions. Am J Pathol. 1990;137:457–65. [PMC free article] [PubMed] [Google Scholar]
- 23.Bornfeldt KE, Raines EW, Nakano T, Graves LM, Krebs EG, Ross R. Insulin-like growth factor-I and platelet-derived growth factor-BB induce directed migration of human arterial smooth muscle cells via signaling pathways that are distinct from those of proliferation. J Clin Invest. 1994;93:1266–74. doi: 10.1172/JCI117081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindner V, Reidy MA. Proliferation of smooth muscle cells after vascular injury is inhibited by an antibody against basic fibroblast growth factor. Proc Natl Acad Sci USA. 1991;88:3739–43. doi: 10.1073/pnas.88.9.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu L, Okada Y, Clinton SK, et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–81. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- 26.Veillard NR, Kwak B, Pelli G, et al. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ Res. 2004;94:253–61. doi: 10.1161/01.RES.0000109793.17591.4E. [DOI] [PubMed] [Google Scholar]
- 27.Gerszten RE, Garcia-Zepeda EA, Lim YC, et al. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. 1999;398:718–23. doi: 10.1038/19546. [DOI] [PubMed] [Google Scholar]
- 28.Mach F, Sauty A, Iarossi AS, et al. Differential expression of three T lymphocyte-activating CXC chemokines by human atheroma-associated cells. J Clin Invest. 1999;104:1041–50. doi: 10.1172/JCI6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janabi M, Yamashita S, Hirano K, et al. Oxidized LDL-induced NF-kappa B activation and subsequent expression of proinflammatory genes are defective in monocyte-derived macrophages from CD36-deficient patients. Arterioscler Thromb Vasc Biol. 2000;20:1953–60. doi: 10.1161/01.atv.20.8.1953. [DOI] [PubMed] [Google Scholar]
- 30.Moshage H. Cytokines and the hepatic acute phase response. J Pathol. 1997;181:257–66. doi: 10.1002/(SICI)1096-9896(199703)181:3<257::AID-PATH756>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 31.Mach F, Schonbeck U, Libby P. CD40 signaling in vascular cells: a key role in atherosclerosis. Atherosclerosis. 1998;137(Suppl.):S89–95. doi: 10.1016/s0021-9150(97)00309-2. [DOI] [PubMed] [Google Scholar]
- 32.Alderson MR, Armitage RJ, Tough TW, Strockbine L, Fanslow WC, Spriggs MK. CD40 expression by human monocytes: regulation by cytokines and activation of monocytes by the ligand for CD40. J Exp Med. 1993;178:669–74. doi: 10.1084/jem.178.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cipollone F, Cicolini G, Bucci M. Cyclooxygenase and prostaglandin synthases in atherosclerosis: recent insights and future perspectives. Pharmacol Ther. 2008;118:161–80. doi: 10.1016/j.pharmthera.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 34.O'Meara SJ, Rodgers K, Godson C. Lipoxins: update and impact of endogenous pro-resolution lipid mediators. Rev Physiol Biochem Pharmacol. 2008;160:47–70. doi: 10.1007/112_2006_0606. [DOI] [PubMed] [Google Scholar]
- 35.Wittwer J, Hersberger M. The two faces of the 15-lipoxygenase in atherosclerosis. Prostaglandins Leukot Essent Fatty Acids. 2007;77:67–77. doi: 10.1016/j.plefa.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 36.Galea J, Armstrong J, Gadsdon P, Holden H, Francis SE, Holt CM. Interleukin-1 beta in coronary arteries of patients with ischemic heart disease. Arterioscler Thromb Vasc Biol. 1996;16:1000–6. doi: 10.1161/01.atv.16.8.1000. [DOI] [PubMed] [Google Scholar]
- 37.Dewberry R, Holden H, Crossman D, Francis S. Interleukin-1 receptor antagonist expression in human endothelial cells and atherosclerosis. Arterioscler Thromb Vasc Biol. 2000;20:2394–400. doi: 10.1161/01.atv.20.11.2394. [DOI] [PubMed] [Google Scholar]
- 38.Steppich BA, Moog P, Matissek C, et al. Cytokine profiles and T cell function in acute coronary syndromes. Atherosclerosis. 2007;190:443–51. doi: 10.1016/j.atherosclerosis.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 39.Ferrari D, Pizzirani C, Adinolfi E, et al. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176:3877–83. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- 40.Raines EW, Dower SK, Ross R. Interleukin-1 mitogenic activity for fibroblasts and smooth muscle cells is due to PDGF-AA. Science. 1989;243:393–6. doi: 10.1126/science.2783498. [DOI] [PubMed] [Google Scholar]
- 41.Kirii H, Niwa T, Yamada Y, et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:656–60. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- 42.Isoda K, Shiigai M, Ishigami N, et al. Deficiency of interleukin-1 receptor antagonist promotes neointimal formation after injury. Circulation. 2003;108:516–8. doi: 10.1161/01.CIR.0000085567.18648.21. [DOI] [PubMed] [Google Scholar]
- 43.Morton AC, Arnold ND, Gunn J, et al. Interleukin-1 receptor antagonist alters the response to vessel wall injury in a porcine coronary artery model. Cardiovasc Res. 2005;68:493–501. doi: 10.1016/j.cardiores.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 44.Abbate A, Salloum FN, Vecile E, et al. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation. 2008;117:2670–83. doi: 10.1161/CIRCULATIONAHA.107.740233. [DOI] [PubMed] [Google Scholar]
- 45.Ikonomidis I, Lekakis JP, Nikolaou M, et al. Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation. 2008;117:2662–9. doi: 10.1161/CIRCULATIONAHA.107.731877. [DOI] [PubMed] [Google Scholar]
- 46.Crossman DC, Morton AC, Gunn JP, et al. Investigation of the effect of Interleukin-1 receptor antagonist (IL-1ra) on markers of inflammation in non-ST elevation acute coronary syndromes (The MRC-ILA-HEART Study) Trials. 2008;9:8. doi: 10.1186/1745-6215-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geng YJ, Wu Q, Muszynski M, Hansson GK, Libby P. Apoptosis of vascular smooth muscle cells induced by in vitro stimulation with interferon-gamma, tumor necrosis factor-alpha, and interleukin-1 beta. Arterioscler Thromb Vasc Biol. 1996;16:19–27. doi: 10.1161/01.atv.16.1.19. [DOI] [PubMed] [Google Scholar]
- 48.Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–503. doi: 10.1172/JCI117619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sukhova GK, Schonbeck U, Rabkin E, et al. Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation. 1999;99:2503–9. doi: 10.1161/01.cir.99.19.2503. [DOI] [PubMed] [Google Scholar]
- 50.Hatakeyama K, Asada Y, Marutsuka K, Sato Y, Kamikubo Y, Sumiyoshi A. Localization and activity of tissue factor in human aortic atherosclerotic lesions. Atherosclerosis. 1997;133:213–9. doi: 10.1016/s0021-9150(97)00132-9. [DOI] [PubMed] [Google Scholar]
- 51.Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47:C13–18. doi: 10.1016/j.jacc.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 52.Greeno EW, Bach RR, Moldow CF. Apoptosis is associated with increased cell surface tissue factor procoagulant activity. Lab Invest. 1996;75:281–9. [PubMed] [Google Scholar]
- 53.Spencer FA, Goldberg RJ, Becker RC, Gore JM. Seasonal distribution of acute myocardial infarction in the second National Registry of Myocardial Infarction. J Am Coll Cardiol. 1998;31:1226–33. doi: 10.1016/s0735-1097(98)00098-9. [DOI] [PubMed] [Google Scholar]
- 54.Spielberg C, Falkenhahn D, Willich SN, Wegscheider K, Voller H. Circadian, day-of-week, and seasonal variability in myocardial infarction: comparison between working and retired patients. Am Heart J. 1996;132:579–85. doi: 10.1016/s0002-8703(96)90241-0. [DOI] [PubMed] [Google Scholar]
- 55.Madjid M, Naghavi M, Litovsky S, Casscells SW. Influenza and cardiovascular disease: a new opportunity for prevention and the need for further studies. Circulation. 2003;108:2730–6. doi: 10.1161/01.CIR.0000102380.47012.92. [DOI] [PubMed] [Google Scholar]
- 56.Naghavi M, Barlas Z, Siadaty S, Naguib S, Madjid M, Casscells W. Association of influenza vaccination and reduced risk of recurrent myocardial infarction. Circulation. 2000;102:3039–45. doi: 10.1161/01.cir.102.25.3039. [DOI] [PubMed] [Google Scholar]
- 57.Siscovick DS, Raghunathan TE, Lin D, et al. Influenza vaccination and the risk of primary cardiac arrest. Am J Epidemiol. 2000;152:674–7. doi: 10.1093/aje/152.7.674. [DOI] [PubMed] [Google Scholar]
- 58.Lavallee P, Perchaud V, Gautier-Bertrand M, Grabli D, Amarenco P. Association between influenza vaccination and reduced risk of brain infarction. Stroke. 2002;33:513–18. doi: 10.1161/hs0202.102328. [DOI] [PubMed] [Google Scholar]
- 59.Nichol KL, Nordin J, Mullooly J, Lask R, Fillbrandt K, Iwane M. Influenza vaccination and reduction in hospitalizations for cardiac disease and stroke among the elderly. N Engl J Med. 2003;348:1322–32. doi: 10.1056/NEJMoa025028. [DOI] [PubMed] [Google Scholar]
- 60.Meier CR, Jick SS, Derby LE, Vasilakis C, Jick H. Acute respiratory-tract infections and risk of first-time acute myocardial infarction. Lancet. 1998;351:1467–71. doi: 10.1016/s0140-6736(97)11084-4. [DOI] [PubMed] [Google Scholar]
- 61.Smeeth L, Thomas SL, Hall AJ, Hubbard R, Farrington P, Vallance P. Risk of myocardial infarction and stroke after acute infection or vaccination. N Engl J Med. 2004;351:2611–18. doi: 10.1056/NEJMoa041747. [DOI] [PubMed] [Google Scholar]
- 62.Clayton TC, Thompson M, Meade TW. Recent respiratory infection and risk of cardiovascular disease: case-control study through a general practice database. Eur Heart J. 2008;29:96–103. doi: 10.1093/eurheartj/ehm516. [DOI] [PubMed] [Google Scholar]
- 63.Madjid M, Miller CC, Zarubaev VV, et al. Influenza epidemics and acute respiratory disease activity are associated with a surge in autopsy-confirmed coronary heart disease death: results from 8 years of autopsies in 34 892 subjects. Eur Heart J. 2007;28:1205–10. doi: 10.1093/eurheartj/ehm035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Naghavi M, Wyde P, Litovsky S, et al. Influenza infection exerts prominent inflammatory and thrombotic effects on the atherosclerotic plaques of apolipoprotein E-deficient mice. Circulation. 2003;107:762–8. doi: 10.1161/01.cir.0000048190.68071.2b. [DOI] [PubMed] [Google Scholar]
- 65.Madjid M, Vela D, Khalili-Tabrizi H, Casscells SW, Litovsky S. Systemic infections cause exaggerated local inflammation in atherosclerotic coronary arteries: clues to the triggering effect of acute infections on acute coronary syndromes. Tex Heart Inst J. 2007;34:11–18. [PMC free article] [PubMed] [Google Scholar]
- 66.Keller TT, van der Meer JJ, Teeling P, et al. Selective expansion of influenza A virus-specific T cells in symptomatic human carotid artery atherosclerotic plaques. Stroke. 2008;39:174–9. doi: 10.1161/STROKEAHA.107.491282. [DOI] [PubMed] [Google Scholar]
- 67.Campbell LA, Kuo CC. Chlamydia pneumoniae – infectious risk factor for atherosclerosis. Nat Rev Microbiol. 2004;2:23–32. doi: 10.1038/nrmicro796. [DOI] [PubMed] [Google Scholar]
- 68.Gibson FC, III, Yumoto H, Takahashi Y, Chou HH, Genco CA. Innate immune signaling and Porphyromonas gingivalis-accelerated atherosclerosis. J Dent Res. 2006;85:106–21. doi: 10.1177/154405910608500202. [DOI] [PubMed] [Google Scholar]
- 69.Gibson FC, III, Genco CA. Porphyromonas gingivalis mediated periodontal disease and atherosclerosis: disparate diseases with commonalities in pathogenesis through TLRs. Curr Pharm Des. 2007;13:3665–75. doi: 10.2174/138161207783018554. [DOI] [PubMed] [Google Scholar]
- 70.Gasbarrini A, Cremonini F, Armuzzi A, et al. The role of Helicobacter pylori in cardiovascular and cerebrovascular diseases. J Physiol Pharmacol. 1999;50:735–42. [PubMed] [Google Scholar]
- 71.Adam E, Melnick JL, DeBakey ME. Cytomegalovirus infection and atherosclerosis. Cent Eur J Public Health. 1997;5:99–106. [PubMed] [Google Scholar]
- 72.Sabroe I, Read RC, Whyte MK, Dockrell DH, Vogel SN, Dower SK. Toll-like receptors in health and disease: complex questions remain. J Immunol. 2003;171:1630–5. doi: 10.4049/jimmunol.171.4.1630. [DOI] [PubMed] [Google Scholar]
- 73.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–8. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 74.Sasu S, LaVerda D, Qureshi N, Golenbock DT, Beasley D. Chlamydia pneumoniae and chlamydial heat shock protein 60 stimulate proliferation of human vascular smooth muscle cells via toll-like receptor 4 and p44/p42 mitogen-activated protein kinase activation. Circ Res. 2001;89:244–50. doi: 10.1161/hh1501.094184. [DOI] [PubMed] [Google Scholar]
- 75.Kurt-Jones EA, Popova L, Kwinn L, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 76.Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–84. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 77.Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001;167:2887–94. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- 78.Malley R, Henneke P, Morse SC, et al. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci USA. 2003;100:1966–71. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takeuchi O, Kawai T, Muhlradt PF, et al. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol. 2001;13:933–40. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- 80.Takeuchi O, Sato S, Horiuchi T, et al. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. 2002;169:10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 81.Cheng N, He R, Tian J, Ye PP, Ye RD. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J Immunol. 2008;181:22–6. doi: 10.4049/jimmunol.181.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hayashi F, Smith KD, Ozinsky A, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 83.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 84.Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004;279:12542–50. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- 85.Diebold SS, Kaisho T, Hemmi H, Akira S, Reise Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–31. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 86.Heil F, Hemmi H, Hochrein H, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–9. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 87.Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 88.Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 89.Brand K, Page S, Rogler G, et al. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J Clin Invest. 1996;97:1715–22. doi: 10.1172/JCI118598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Visintin A, Mazzoni A, Spitzer JH, Wyllie DH, Dower SK, Segal DM. Regulation of Toll-like receptors in human monocytes and dendritic cells. J Immunol. 2001;166:249–55. doi: 10.4049/jimmunol.166.1.249. [DOI] [PubMed] [Google Scholar]
- 91.Faure E, Equils O, Sieling PA, et al. Bacterial lipopolysaccharide activates NF-kappaB through toll-like receptor 4 (TLR-4) in cultured human dermal endothelial cells. Differential expression of TLR-4 and TLR-2 in endothelial cells. J Biol Chem. 2000;275:11058–63. doi: 10.1074/jbc.275.15.11058. [DOI] [PubMed] [Google Scholar]
- 92.Ward JR, Bingle L, Judge HM, et al. Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thromb Haemost. 2005;94:831–8. [PubMed] [Google Scholar]
- 93.Tan X, Essengue S, Talreja J, Reese J, Stechschulte DJ, Dileepan KN. Histamine directly and synergistically with lipopolysaccharide stimulates cyclooxygenase-2 expression and prostaglandin I(2) and E(2) production in human coronary artery endothelial cells. J Immunol. 2007;179:7899–906. doi: 10.4049/jimmunol.179.11.7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Talreja J, Kabir MH, Filla MB, Stechschulte DJ, Dileepan KN. Histamine induces Toll-like receptor 2 and 4 expression in endothelial cells and enhances sensitivity to Gram-positive and Gram-negative bacterial cell wall components. Immunology. 2004;113:224–33. doi: 10.1111/j.1365-2567.2004.01946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ. Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation. 2002;105:1158–61. [PubMed] [Google Scholar]
- 96.Schoneveld AH, Hoefer I, Sluijter JP, Laman JD, de Kleijn DP, Pasterkamp G. Atherosclerotic lesion development and Toll like receptor 2 and 4 responsiveness. Atherosclerosis. 2008;197:95–104. doi: 10.1016/j.atherosclerosis.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 97.Xu XH, Shah PK, Faure E, et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104:3103–8. doi: 10.1161/hc5001.100631. [DOI] [PubMed] [Google Scholar]
- 98.Castrillo A, Joseph SB, Vaidya SA, et al. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell. 2003;12:805–16. doi: 10.1016/s1097-2765(03)00384-8. [DOI] [PubMed] [Google Scholar]
- 99.Michelsen KS, Wong MH, Shah PK, et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci USA. 2004;101:10679–84. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest. 2005;115:3149–56. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Naiki Y, Sorrentino R, Wong MH, et al. TLR/MyD88 and liver X receptor alpha signaling pathways reciprocally control Chlamydia pneumoniae-induced acceleration of atherosclerosis. J Immunol. 2008;181:7176–85. doi: 10.4049/jimmunol.181.10.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wiedermann CJ, Kiechl S, Dunzendorfer S, et al. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: prospective results from the Bruneck Study. J Am Coll Cardiol. 1999;34:1975–81. doi: 10.1016/s0735-1097(99)00448-9. [DOI] [PubMed] [Google Scholar]
- 103.Lehr HA, Sagban TA, Ihling C, et al. Immunopathogenesis of atherosclerosis: endotoxin accelerates atherosclerosis in rabbits on hypercholesterolemic diet. Circulation. 2001;104:914–20. doi: 10.1161/hc3401.093153. [DOI] [PubMed] [Google Scholar]
- 104.Engelmann MG, Redl CV, Nikol S. Recurrent perivascular inflammation induced by lipopolysaccharide (endotoxin) results in the formation of atheromatous lesions in vivo. Lab Invest. 2004;84:425–32. doi: 10.1038/labinvest.3700065. [DOI] [PubMed] [Google Scholar]
- 105.Vink A, Schoneveld AH, van der Meer JJ, et al. In vivo evidence for a role of toll-like receptor 4 in the development of intimal lesions. Circulation. 2002;106:1985–90. doi: 10.1161/01.cir.0000032146.75113.ee. [DOI] [PubMed] [Google Scholar]
- 106.Schoneveld AH, Oude Nijhuis MM, van Middelaar B, Laman JD, de Kleijn DP, Pasterkamp G. Toll-like receptor 2 stimulation induces intimal hyperplasia and atherosclerotic lesion development. Cardiovasc Res. 2005;66:162–9. doi: 10.1016/j.cardiores.2004.12.016. [DOI] [PubMed] [Google Scholar]