

Abstract
The two major primary antibody deficiency disorders are X-linked hypogammaglobulinaemia (XLA) and common variable immunodeficiency (CVID). CVID patients have an elevated risk for gastric cancer and extra-nodal marginal zone lymphoma. Both diseases are associated with Helicobacter pylori infection. We investigated whether antibody deficiency leads to defective serum bactericidal activity against H. pylori. We also investigated the correlation with immunoglobulin (Ig)M levels and observed the terminal complement complex (TCC) activity. Sera of 13 CVID patients (four H. pylori positive), one patient with hyper-IgM syndrome, one patient with Good syndrome (both H. pylori positive), five XLA patients, four H. pylori seropositive controls, four H. pylori seronegative controls and a sample of pooled human serum (PHS) were incubated in vitro with bacterial suspensions of H. pylori for 30 min. After 72 h of culture, colony-forming units were counted. TCC formation was measured by enzyme-linked immunosorbent assay. We found that normal human serum is bactericidal for H. pylori, whereas heat-inactivated serum shows hardly any killing of H. pylori. Serum (1%) of hypogammaglobulinaemia patients has a decreased bactericidal activity against H. pylori. Helicobacter pylori-positive (HP+) normal individuals show more than 90% killing of H. pylori, whereas CVID patients show 35% killing (P = 0·007) and XLA patients only 19% (P = 0·003). Serum (1%) of HP+ volunteers showed significantly better killing compared with serum of H. pylori-negative (HP–) volunteers (P = 0·034). No correlation between (substituted) IgG levels and serum bactericidal activity was found, but a weak correlation between total serum IgM and serum bactericidal activity was found. In conclusion, serum bactericidal activity against H. pylori is decreased in patients with hypogammaglobulinaemia. Heat treatment of the serum abolished the bactericidal capacity, indicating that complement activity is essential for the bactericidal effect.
Keywords: CVID, Helicobacter pylori, hypogammaglobulinaemia, serum bactericidal activity, XLA
Introduction
The two main types of primary antibody deficiency disorder are X-linked hypogammaglobulinaemia (XLA) and common variable immunodeficiency (CVID). XLA, caused by mutations in the gene encoding for Bruton tyrosine kinase (BTK), is characterized by the absence of mature B cells, very low serum concentrations of all immunoglobulin isotypes and lack of specific antibody production [1,2]. The diagnosis of CVID is based on markedly reduced serum levels for immunoglobulin (Ig)G and IgA or IgM, an impaired ability to specific antibody production after vaccination or exposure, and exclusion of secondary causes for antibody deficiency [3]. The pathogenesis of this disorder is heterogeneous, but usually not within the B cell lineage [4]. Abnormal T lymphocyte regulation of B lymphocyte differentiation may play a role; several T cell and cytokine abnormalities have been described [3,5–9].
As a consequence of the deficient host defence, XLA and CVID patients suffer from a variety of infections. Most prominent are infections of the respiratory tract and ear/nose/throat region. Due to deficient opsonization, these infections are caused especially by encapsulated micro-organisms such as Haemophilus influenzae and pneumococci. IgG supplementation by the parenteral route is the mainstay of therapy to prevent these types of infection.
Other prominent infections are those within the intestinal tract, such as those caused by Giardia lamblia. In addition, prolonged intestinal carriage of Campylobacter jejuni, complicated by recurrent bacteraemia, occurs. These bacteraemic events are attributed to the lack of serum bactericidal activity against C. jejuni[10,11].
CVID and XLA are associated not only with an increased risk of infection but also with an increased risk of cancer. In particular, there is an elevated risk in CVID patients for gastric cancer and lymphomas, such as extra-nodal marginal zone lymphomas [described formerly as mucosa-associated lymphoid tissue (MALT) lymphomas][3,12–15]. In XLA the risk for colorectal cancer is elevated [16].
In the pathogenesis of gastric cancer, as well as extra-nodal marginal zone lymphoma, the Gram-negative bacterium Helicobacter pylori is a major factor [17–19]. There is increasing support for the role of H. pylori infection in the gastric pathology of patients with CVID [20–24].
In a number of patients with CVID we experienced great difficulty in elimination of H. pylori[24]. Therefore, we raised the question of whether the lack of immunoglobulin and of a bactericidal effect was responsible for this problem, similar to the situation with C. jejuni[10,11].
We hypothesized that the serum bactericidal activity against H. pylori may be reduced in patients with hypogammaglobulinaemia. To investigate this hypothesis, we measured the serum bactericidal activity against H. pylori in both CVID and XLA patients and compared this with the serum bactericidal activity in H. pylori-seropositive (HP+) and -seronegative (HP–) healthy volunteers. In addition, we hypothesized that a lack of serum bactericidal activity in hypogammaglobulinaemic patients could be due to a lack of specific IgG or IgM, which we explored by comparing HP+ and HP− individuals and by measuring IgM levels and terminal complement complex activity.
Methods
Patients
Thirteen patients with CVID, one patient with hyper-IgM syndrome, one patient with Good syndrome and five patients with XLA were tested. When available, H. pylori status was based on histological biopsies. If H. pylori status was unknown, a C14-urea breath test (HeliprobeTM; Noster Systems AB, Stockholm, Sweden) [25] was performed. All patients were supplemented appropriately with intravenous IgG. As controls, four healthy HP+ and four healthy HP− volunteers were investigated.
Serum
After informed consent, 10 ml of blood was drawn by venipuncture in sterile polypropylene tubes (Becton Dickinson, Franklin Lakes, NJ, USA) and kept on ice. After centrifugation, the serum was stored at –80°C. A sample of pooled human serum (PHS) was also tested.
For determination of the serum bactericidal activity, 500 µl sterile serum of each individual was diluted with Hanks' balanced salt solution (HBSS) without phenol red to obtain a 50, 25 and 5% solution. To inactivate the complement system, 250 µl of the diluted serum was heated at 56°C for 30 min.
To measure the terminal complement complex we collected a fresh blood sample from each patient in a polypropylene tube containing 50 µl lepirudin (Refludan; Pharmion, Tiel, the Netherlands) on ice. The latter is a recombinant hirudin, which is a highly specific thrombin inhibitor which does not influence the complement activation [26]. The blood was centrifuged at 4200 g for 10 min at 4°C and thereafter the sterile plasma was transferred to –80°C until assay.
Bacterial strains
Strain H. pylori ATCC-43504 is a cytotoxin-associated gene A-positive strain producing a vacuolating cytotoxin and able to induce interleukin-8 production. Bacterial cells were cultured on Dent (Biotrading, Mijdrecht, The Netherlands) plates and microaerophilic incubated at 37°C for 48 h. After this, they were cultured for a second time on Dent plates in the same conditions. For use in serum bactericidal assays the bacteria were taken from the plates and washed in sterile CaCl2. The solution was centrifuged at 4200 gfor 4 min. Next, the pellet was resuspended with CaCl2 and adjusted spectrophotometrically to an optical density value of 0·75 at 660 nm wavelength. Dilutions were made in HBSS in two steps to 1:100 [5 × 106 colony-forming units (CFU)/ml].
Serum bactericidal assay
In a first experiment, 50 µl bacterial suspension were mixed with 150 µl HBSS and 50 µl fresh serum solution in 96-well flat-bottomed tissue-culture plates. In a second experiment, heat-inactivated serum was used. Final concentrations were 1 × 106 CFU/l of bacteria and either 1, 5 or 10% serum. As a control, 50 µl bacterial suspension without serum was incubated with 200 µl HBSS. To measure the number of bacteria inoculated, a specimen of 100 µl was taken from this mixture before incubation. The 96-well plate was incubated at 37°C. After 30 min, a 100 µl sample from each well was diluted with saline to 1:10 or 1:100. Six 10-µl drops of each sample were pipetted onto Dent plates and incubated at 37°C under microaerophilic conditions for 72 h. All tests were conducted in duplicate. After incubation, CFU were counted. When fewer than 10 CFU in 12 drops of the same solution were counted, serum bactericidal activity was considered to be maximal.
Terminal complement complex assay
Per well of a polypropylene 96-well flat-bottomed tissue culture plate, 20 µl of the bacterial suspension were mixed with 180 µl lepirudin plasma. As a control, 20 µl phosphate-buffered saline (PBS) was used instead of bacterial suspension. All tests were performed in duplicate. After incubation at 37°C for 30 min, samples were placed on ice and 25 mmol ethylenediamine tetraacetic acid (EDTA) was added to each well to stop complement activation. The enzyme-linked immunosorbent assay (ELISA) for measuring Terminal complement complex (TCC) used the monoclonal antibody aE11 95 B as capture antibody, which is specific for C9 incorporated in the sC5b-9 complex. The antibody aE11 95B was purified from ascites using protein A affinity chromatography and Mono Q high performance liquid chromatography (Pharmacia LKB Biotechnology, Uppsala, Sweden, obtained from Professor Mollnes, Oslo, Norway), diluted 1:10 000 in coating buffer (PBS with 0·02% azide) and incubated for 1 h at 4°C in Nunc maxisorp ELISA plates (eBiosciences, San Diego, CA, USA). Hereafter, ELISA plates were washed five times and samples diluted in dilution buffer (PBS with 0·02% azide, 0·2% Tween 20 and 0·02 M EDTA) were incubated for 1 h at 4°C. Zymosan-activated serum (kindly provided by Professor Mollnes, Oslo, Norway) was used as a standard, defining 1000 arbitrary units (AU)/ml. After washing the plates five times, the second antibody 9C4-RQ-B (also provided by Professor Mollnes) specific for C6 was added in a 1:5000 dilution. After 45 min incubation at 37°C, the plates were washed five times. Streptavidin–horseradish peroxidase (Sanquin, Amsterdam, the Netherlands) was added for 30 min at 37°C. After a final five washes, 0·003% H2O2 in 0·018% 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) (ABTS) in a 0·1 M Na-acetate buffer at pH 4·0 was added to the ELISA plates to start the colour reaction. The colour reaction was stopped by adding 50 µl H2SO4 to each well. The extinction was read in an ELISA reader (Victor Wallac Multilabel Counter, PerkinElmer, Groningen, The Netherlands). The method used for TCC analysis has been described in detail previously [27].
Statistical analysis
We used one-way anova and the Levene test for homogeneity of variance. For control, we also used the non-parametric Kruskal–Wallis test. For post hoc tests we used Bonferroni and least significance difference tests. For more specific questions, for example to compare CVID and XLA or patients and controls, we also used the t-test and Mann–Whitney U-test.
Results
In 19 of 20 hypogammaglobulinaemic patients we were able to determine whether or not they were infected with H. pylori; one patient refused the C14-urea breath test. Four CVID patients were positive for H. pylori infection. The patient with hyper-IgM syndrome and the patient with Good syndrome were also positive for H. pylori infection. None of the XLA patients tested positive for H. pylori infection. The patient characteristics are given in Table 1.
Table 1.
Patient characteristics at time of sample collection.
| Patient | Age years | Type | IgA g/l | IgG† g/l | IgM g/l | Helicobacter pylori | Clinical information |
|---|---|---|---|---|---|---|---|
| 1 | 28 | CVID | < 0·05 | 9·00 | 0·96 | Negative | |
| 2 | 47 | CVID | 0·07 | 5·55 | 0·08 | Negative | |
| 3 | 28 | CVID | < 0·25 | 2·45 | 0·15 | Negative | |
| 4 | 66 | CVID | < 0·06 | 1·40 | 0·11 | Positive | Extra-nodal marginal zone lymphoma of the stomach |
| 5 | 47 | CVID | < 0·05 | 5·01 | 0·32 | – | |
| 6 | 42 | CVID | < 0·04 | 5·56 | 0·05 | Negative | |
| 7 | 68 | CVID | 0·069 | 5·60 | 0·40 | Negative | |
| 8 | 35 | CVID | < 0·07 | 3·56 | 0·14 | Negative | |
| 9 | 33 | CVID | < 0·05 | 0·20 | Negative | Extra-nodal marginal zone lymphoma of the parotid gland | |
| 10 | 89 | CVID | < 0·08 | 5·60 | 0·52 | Positive | |
| 11 | 41 | CVID | < 0·05 | 9·27 | 0·29 | Positive | |
| 12 | 65 | CVID | < 0·07 | 12·40 | 0·95 | Positive | Gastric cancer |
| 13 | 24 | CVID | 0·11 | 4·77 | 0·57 | Negative | |
| 14 | 46 | Hyper-IgM syndrome | 0·68 | 4·30 | 6·89 | Positive | Hyper-IgM syndrome |
| 15 | 64 | Good syndrome | 0·90 | 6·70 | 0·17 | Positive | Good syndrome |
| 16 | 41 | XLA | < 0·07 | 6·26 | 0·17 | Negative | |
| 17 | 46 | XLA | < 0·06 | 6·55 | 0·09 | Negative | |
| 18 | 47 | XLA | < 0·07 | 7·42 | < 0·05 | Negative | |
| 19 | 22 | XLA | < 0·07 | 8·17 | 0·03 | Negative | |
| 20 | 41 | XLA | < 0·04 | 7·20 | < 0·04 | Negative |
These concentrations are a consequence of intravenous gammaglobulin supplementation. CVID, common variable immunodeficiency; XLA, X-linked agammaglobulinaemia; Ig, immunoglobulin.
Figure 1 shows the mean serum bactericidal activity of pooled human serum, and of serum from healthy HP− and HP+ volunteers, CVID patients and patients with XLA. Serum bactericidal activity was determined for both fresh serum and heat-inactivated serum. In all groups, heat-inactivated serum is poorly bactericidal. With fresh serum, at only a 1% concentration, differences between the groups became apparent. At this concentration, HP+ normal individuals showed more than 90% killing of H. pylori, whereas patients with CVID showed 35% killing and XLA patients only 19% killing. The patient with Good syndrome showed 17% killing and the patient with hyper-IgM syndrome 100% killing (these two patients are not presented in Fig. 1). As expected, there was a significantly larger bactericidal effect in HP+ than in HP− volunteers (P = 0·034). Bactericidal activity of the non-XLA agammaglobulinaemic sera was significantly less than that of HP+ controls (P = 0·007); sera from XLA patients were even less bactericidal (differences with HP+ controls significant, P = 0·003). Using 5% and 10% serum, these differences were no longer apparent, except for the comparison between XLA and both control groups in 5% serum (P = 0·036, compared with HP− and P = 0·031, compared with HP+).
Fig. 1.
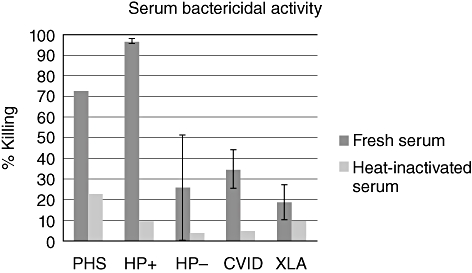
Mean bactericidal activity with standard of the mean. CVID, common variable immunodeficiency; HP+, H. pylori-positive; HP−, H. pylori-negative; PHS, pooled human serum; XLA, X-linked agammaglobulinaemia.
There were no significant differences between HP+ and HP− CVID patients in any of the serum groups. Serum IgG concentrations (presented in Table 1) in these patients were all a consequence of the gammaglobulin substitution. We did not find a correlation between serum IgG levels and serum bactericidal activity.
Because total serum IgM was crucial for bactericidal activity against C. jejuni[28], we investigated whether there was a correlation between endogenous total serum IgM concentrations and serum bactericidal activity in these hypogammaglobulinaemic patients. A poor correlation (r = 0·3, P = 0·11) was found (data not shown).
In Fig. 2, the mean TCC value for each group is shown. Helicobacter pylori is known to be a good activator of TCC formation. There were no significant differences between the groups. We did not find a correlation between serum bactericidal activity and TCC formation.
Fig. 2.
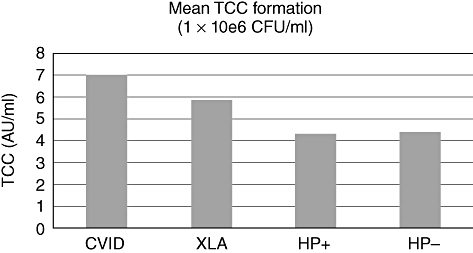
Mean terminal complement complex (TCC) formation in lepirudin samples of all groups. AU, arbitrary units; CFU, colony-forming units; CVID, common variable immunodeficiency; HP+, H. pylori-positive; HP−, H. pylori-negative; XLA, X-linked agammaglobulinaemia.
Discussion
In this paper we demonstrate that serum of patients with hypogammaglobulinaemia had a decreased bactericidal activity against H. pylori. This defect was only apparent when 1% serum was used. At this concentration, HP+ normal individuals showed more than 90% killing of H. pylori, whereas patients with CVID showed 35% killing and XLA patients only 19% killing. The patient with Good syndrome showed 17% killing and the patient with hyper-IgM syndrome showed 100% killing. Heat treatment of the serum abolished the bactericidal capacity, indicating that complement activity is essential for the bactericidal effect. This is in line with the observation by Berstad et al., that H. pylori is complement-sensitive and activates the classic pathway even in the absence of specific antibodies [29].
Gonzalez-Valencia et al.[30] investigated the susceptibility of H. pylori to the bactericidal activity of human serum. They also concluded that H. pylori is susceptible to complement, C3 binding being a major locus of variability. With the serum of the one patient with CVID tested they found a significantly lower bactericidal capacity, in agreement with our results in CVID patients.
In essence, we investigated different kinds of sera: (1) human pooled serum, containing all Ig classes and antibody activity against H. pylori; (2) serum from HP+ individuals with similar properties as the human pooled serum; (3) serum from HP− individuals, containing all Ig classes and no activity against H. pylori; (4) serum from CVID patients containing low but variable concentrations of endogenous immunoglobulin from the various classes and near-normal concentrations of IgG from many donors and presumably antibody activity against H. pylori– in these patients, virtually no immunoglobulin other than IgG is being substituted; and (5) serum from XLA patients, containing negligible concentrations of endogenous immunoglobulin and near-normal concentrations of IgG derived from intravenous (i.v.) Ig supplementation, containing normal activity against H. pylori. The observation that sera from CVID and XLA patients were less bactericidal than HP+ sera (1 and 2) suggests that immunoglobulins play a role in the serum bactericidal effect. That the defect remains in the hypogammaglobulinaemic patients despite substitution with pooled IgG (which contains H. pylori-specific IgG, data not published) to near-physiological normal levels of IgG suggests that immunoglobulins other than IgG are responsible for the serum bactericidal effect. This could be IgM, as was found in earlier studies for C. jejuni[10,11]. The greater bactericidal activity found with 5% HP− serum compared with 5% XLA serum is compatible with this notion. Also, the observation of near-normal serum bactericidal activity in the hyper-IgM patient and in patient 1, a CVID patient with normal IgM levels, supports this (Table 1, patients 1 and 14). It is most likely that complement is activated by immunoglobulin, especially IgM, directed against H. pylori. Within the IgG class, IgG1 and IgG3 are the two subclasses that are able to induce complement activation. It should be noted, however, that substitution with IgG does not lead to a normal distribution of IgG concentrations. Large amounts of IgG1 and only low amounts of IgG3 (mean 60–81% IgG1 and 3–8% IgG3 [31]) (http://www.sanquin.nl) are being substituted. It is therefore unlikely that IgG3 would be the main activator of complement in the interaction with H. pylori. However, it is possible that IgG1-mediated complement activation accounts for the serum bactericidal activity, when more concentrated serum samples are tested.
The discrepancy we found between serum bactericidal activity and TCC formation is confirmed by earlier observations. Drogari-Aparanthitou et al. found that C3b and MAC formation on meningococci was independent of the presence of specific IgG, while serum bactericidal activity was dependent upon classical pathway activation [32]. In addition, we found earlier that TCC formation by meningococci in whole blood was dependent upon alternative and lectin pathway activation, but independent of classical pathway activation [26]. These observations indicated that IgG is essential for proper MAC insertion into the membrane of meningococci and subsequent bacterial lysis, but that TCC formation by meningococci is dependent upon other complement pathways. Our present observation that hypogammaglobulinaemia affected only H. pylori serum bactericidal activity at low (1%) concentrations of serum suggests that, under such conditions, classical pathway activation is crucial for effective lysis. At higher concentrations of complement components, alternative – and possibly lectin – pathway activation also contributes to lysis of H. pylori.
The variation that we observed may have been due to the variation in complement activity in the sera used. We took great care to keep complement intact but there is, of course, individual variation. The TCC values varied from 1·21 AU/ml to 12·25 AU/ml in the lepirudin samples and did not correlate with serum bactericidal activity.
Two of our CVID patients had a marginal zone lymphoma and one had gastric cancer. The clinical details of the two lymphoma patients have been described elsewhere [24,33]. The patient with gastric cancer (Table 1, patient 12) and one with extra-nodal marginal zone lymphoma (Table 1, patient 4), both with a history of H. pylori infection, had a bactericidal activity below the mean of the total CVID group. In the patient with extra-nodal marginal zone lymphoma of the stomach, treatment of H. pylori was extremely difficult. The serum of the third patient, suffering from HP− extra-nodal marginal zone lymphoma of the parotid gland (Table 1, patient 9), showed considerably better bactericidal activity than the mean of the total CVID group.
Although bactericidal activity was more severely hampered in XLA patients than in CVID, gastric pathology (antrum gastritis, gastric cancer and extra-nodal marginal zone lymphoma) is not a prominent problem in XLA, whereas it definitely is in CVID. In line with this, we did not detect H. pylori infections in our XLA patients, whereas six of our 15 CVID patients were infected with H. pylori. We may conclude that the lack of immunoglobulin or impaired bactericidal activity does not seem to explain the gastric pathology that occurs in CVID patients. It is more likely that, in the pathogenesis of HP+, gastric inflammation and cancer in CVID T cell abnormalities play a role [3–9,34–36].
In conclusion, we show that the bacterial killing of H. pylori is complement-mediated. Greater serum bactericidal activity in HP+ individuals fits with a role for immunoglobulin. The serum bactericidal activity against H. pylori is lower in patients with hypogammaglobulinaemia. Although this does not seem to explain the risk for gastric cancer and extra-nodal marginal zone lymphoma in CVID, it may contribute to the difficulty of eliminating H. pylori in these patients.
Acknowledgments
We would like to thank Professor Mollnes from Oslo, Norway, for providing the reagents.
Disclosure
None of the authors wish to declare a conflict of interest.
References
- 1.Tsukada S, Saffran DC, Rawlings DJ, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72:279–90. doi: 10.1016/0092-8674(93)90667-f. [DOI] [PubMed] [Google Scholar]
- 2.Vetrie D, Vorechovsky I, Sideras P, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein–tyrosine kinases. Nature. 1993;361:226–33. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- 3.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34–48. doi: 10.1006/clim.1999.4725. [DOI] [PubMed] [Google Scholar]
- 4.van der Meer JW, Zegers BJ. Agammaglobulinaemia. Neth J Med. 1994;45:250–6. [PubMed] [Google Scholar]
- 5.Waldmann TA, Durm M, Broder S, Blackman M, Blaese RM, Strober W. Role of suppressor T cells in pathogenesis of common variable hypogammaglobulinaemia. Lancet. 1974;2:609–13. doi: 10.1016/s0140-6736(74)91940-0. [DOI] [PubMed] [Google Scholar]
- 6.Guazzi V, Aiuti F, Mezzaroma I, et al. Assessment of thymic output in common variable immunodeficiency patients by evaluation of T cell receptor excision circles. Clin Exp Immunol. 2002;129:346–53. doi: 10.1046/j.1365-2249.2002.01893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cunningham-Rundles S, Cunningham-Rundles C, Siegal FP, et al. Defective cellular immune response in vitro in common variable immunodeficiency. J Clin Immunol. 1981;1:65–72. doi: 10.1007/BF00915478. [DOI] [PubMed] [Google Scholar]
- 8.Kondratenko I, Amlot PL, Webster AD, Farrant J. Lack of specific antibody response in common variable immunodeficiency (CVID) associated with failure in production of antigen-specific memory T cells. MRC Immunodeficiency Group. Clin Exp Immunol. 1997;108:9–13. doi: 10.1046/j.1365-2249.1997.d01-993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stagg AJ, Funauchi M, Knight SC, Webster AD, Farrant J. Failure in antigen responses by T cells from patients with common variable immunodeficiency (CVID) Clin Exp Immunol. 1994;96:48–53. doi: 10.1111/j.1365-2249.1994.tb06228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van der Meer JW, Mouton RP, Daha MR, Schuurman RK. Campylobacter jejuni bacteraemia as a cause of recurrent fever in a patient with hypogammaglobulinaemia. J Infect. 1986;12:235–9. doi: 10.1016/s0163-4453(86)94190-3. [DOI] [PubMed] [Google Scholar]
- 11.Kerstens PJ, Endtz HP, Meis JF, et al. Erysipelas-like skin lesions associated with Campylobacter jejuni septicemia in patients with hypogammaglobulinemia. Eur J Clin Microbiol Infect Dis. 1992;11:842–7. doi: 10.1007/BF01960888. [DOI] [PubMed] [Google Scholar]
- 12.Kinlen LJ, Webster AD, Bird AG, et al. Prospective study of cancer in patients with hypogammaglobulinaemia. Lancet. 1985;1:263–6. doi: 10.1016/s0140-6736(85)91037-2. [DOI] [PubMed] [Google Scholar]
- 13.Mellemkjaer L, Hammarstrom L, Andersen V, et al. Cancer risk among patients with IgA deficiency or common variable immunodeficiency and their relatives: a combined Danish and Swedish study. Clin Exp Immunol. 2002;130:495–500. doi: 10.1046/j.1365-2249.2002.02004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cunningham-Rundles C, Cooper DL, Duffy TP, Strauchen J. Lymphomas of mucosal-associated lymphoid tissue in common variable immunodeficiency. Am J Hematol. 2002;69:171–8. doi: 10.1002/ajh.10050. [DOI] [PubMed] [Google Scholar]
- 15.Park MA, Li JT, Hagan JB, Maddox DE, Abraham RS. Common variable immunodeficiency: a new look at an old disease. Lancet. 2008;372:489–502. doi: 10.1016/S0140-6736(08)61199-X. [DOI] [PubMed] [Google Scholar]
- 16.van der Meer JW, Weening RS, Schellekens PT, van Munster IP, Nagengast FM. Colorectal cancer in patients with X-linked agammaglobulinaemia. Lancet. 1993;341:1439–40. doi: 10.1016/0140-6736(93)90883-i. [DOI] [PubMed] [Google Scholar]
- 17.IARC Working group on the evaluation of carcinogenic risks to Humans. Schistosomes, liver flukes and Helicobacter pylori. Lyon, 7–14 June 1994. IARC Monogr Eval Carcinog Risks Hum. 1994;61:1–241. [PMC free article] [PubMed] [Google Scholar]
- 18.The EUROGAST Study Group. An international association between Helicobacter pylori infection and gastric cancer. Lancet. 1993;341:1359–62. [PubMed] [Google Scholar]
- 19.Parsonnet J, Hansen S, Rodriguez L, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med. 1994;330:1267–71. doi: 10.1056/NEJM199405053301803. [DOI] [PubMed] [Google Scholar]
- 20.Zullo A, Romiti A, Rinaldi V, et al. Gastric pathology in patients with common variable immunodeficiency. Gut. 1999;45:77–81. doi: 10.1136/gut.45.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van der Hilst JC, Smits BW, van der Meer JW. Hypogammaglobulinaemia: cumulative experience in 49 patients in a tertiary care institution. Neth J Med. 2002;60:140–7. [PubMed] [Google Scholar]
- 22.Teahon K, Webster AD, Price AB, Weston J, Bjarnason I. Studies on the enteropathy associated with primary hypogammaglobulinaemia. Gut. 1994;35:1244–9. doi: 10.1136/gut.35.9.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Misbah SA, Chapel HM, Johnston BJ, Ali MH, Reed PI, O'Sullivan D. Attempt to reverse atrophic gastritis associated with common variable immunodeficiency. J Clin Gastroenterol. 1992;15:354–5. [PubMed] [Google Scholar]
- 24.Desar IM, Keuter M, Raemaekers JM, Jansen JB, van Krieken JH, van der Meer JW. Extranodal marginal zone (MALT) lymphoma in common variable immunodeficiency. Neth J Med. 2006;64:136–40. [PubMed] [Google Scholar]
- 25.Hegedus O, Ryden J, Rehnberg AS, Nilsson S, Hellstrom PM. Validated accuracy of a novel urea breath test for rapid Helicobacter pylori detection and in-office analysis. Eur J Gastroenterol Hepatol. 2002;14:513–20. doi: 10.1097/00042737-200205000-00008. [DOI] [PubMed] [Google Scholar]
- 26.Sprong T, Brandtzaeg P, Fung M, et al. Inhibition of C5a-induced inflammation with preserved C5b-9-mediated bactericidal activity in a human whole blood model of meningococcal sepsis. Blood. 2003;102:3702–10. doi: 10.1182/blood-2003-03-0703. [DOI] [PubMed] [Google Scholar]
- 27.Mollnes TE. Analysis of the in vivo complement activation. In: Herzenberg LA, Weir DM, Blackwell C, editors. Weir's handbook of experimental immnunology. Boston: Blackwell Science; 1997. pp. 78.1–8. [Google Scholar]
- 28.Borleffs JC, Schellekens JF, Brouwer E, Rozenberg-Arska M. Use of an immunoglobulin M containing preparation for treatment of two hypogammaglobulinemic patients with persistent Campylobacter jejuni infection. Eur J Clin Microbiol Infect Dis. 1993;12:772–5. doi: 10.1007/BF02098467. [DOI] [PubMed] [Google Scholar]
- 29.Berstad AE, Hogasen K, Bukholm G, Moran AP, Brandtzaeg P. Complement activation directly induced by Helicobacter pylori. Gastroenterology. 2001;120:1108–16. doi: 10.1053/gast.2001.23248. [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez-Valencia G, Perez-Perez GI, Washburn RG, Blaser MJ. Susceptibility of Helicobacter pylori to the bactericidal activity of human serum. Helicobacter. 1996;1:28–33. doi: 10.1111/j.1523-5378.1996.tb00005.x. [DOI] [PubMed] [Google Scholar]
- 31.Alyanakian MA, Bernatowska E, Scherrmann JM, Aucouturier P, Poplavsky JL. Pharmacokinetics of total immunoglobulin G and immunoglobulin G subclasses in patients undergoing replacement therapy for primary immunodeficiency syndromes. Vox Sang. 2003;84:188–92. doi: 10.1046/j.1423-0410.2003.00278.x. [DOI] [PubMed] [Google Scholar]
- 32.Drogari-Apiranthitou M, Kuijper EJ, Dekker N, Dankert J. Complement activation and formation of the membrane attack complex on serogroup B Neisseria meningitidis in the presence or absence of serum bactericidal activity. Infect Immun. 2002;70:3752–8. doi: 10.1128/IAI.70.7.3752-3758.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desar IM, Weemaes CM, van DM, van der Meer JW. Reversible hypogammaglobulinaemia. Neth J Med. 2007;65:381–5. [PubMed] [Google Scholar]
- 34.Hussell T, Isaacson PG, Crabtree JE, Spencer J. The response of cells from low-grade B-cell gastric lymphomas of mucosa-associated lymphoid tissue to Helicobacter pylori. Lancet. 1993;342:571–4. doi: 10.1016/0140-6736(93)91408-e. [DOI] [PubMed] [Google Scholar]
- 35.Sneller MC, Strober W, Eisenstein E, Jaffe JS, Cunningham-Rundles C. NIH conference. New insights into common variable immunodeficiency. Ann Intern Med. 1993;118:720–30. doi: 10.7326/0003-4819-118-9-199305010-00011. [DOI] [PubMed] [Google Scholar]
- 36.Wright JJ, Wagner DK, Blaese RM, Hagengruber C, Waldmann TA, Fleisher TA. Characterization of common variable immunodeficiency: identification of a subset of patients with distinctive immunophenotypic and clinical features. Blood. 1990;76:2046–51. [PubMed] [Google Scholar]