

Abstract
Porphyromonas gingivalis lipopolysaccharide (LPS) (strain W50) interacts with Toll-like receptor 2 (TLR-2) leading to cytokine expression and inflammation, and thereby plays a key role in the pathogenesis of periodontal disease. The aims of this study were to investigate gene expression of key regulatory mediators of innate immune responses in a human monocytic cell line (THP-1) to P. gingivalis LPS and to compare these results with those obtained using the TLR-4 ligand, Escherichia coli LPS. Custom-made Taqman low-density arrays were used for expression profiling of 45 different cytokine-related genes. Both types of LPS highly up-regulated interleukin (IL)-1α and IL-1β, IL-18 receptor (IL-18R), IL-18R accessory protein and IL-1 family (IL-1F)9. Expression levels of IL-1F6, IL-1F7 and caspase-1 were unaltered by either LPS. Genes for tumour necrosis factor-α, IL-6, leukaemia inhibitory factor and IL-32 were also highly induced by both LPS. For a subset of genes, including CXC chemokine ligand 5 (CXCL5), expression was induced only by E. coli LPS or was up-regulated more highly by E. coli compared with P. gingivalis LPS in THP-1 monocytes. A similar expression pattern was also observed in dendritic cells. Analysis of signalling pathways which lead to CXCL5 expression indicated that the mechanisms underpinning the differential responses did not involve the recruitment of different adaptor proteins by TLR-2 and TLR-4, and therefore occur downstream of the receptor–adaptor complex. We conclude that differences in signalling pathways activated by TLR-2 and TLR-4 ligands lead to differential innate immune responses which may be important in polymicrobial diseases such as periodontal disease.
Keywords: chemokines, cytokines, monocytes, PAMPS, P. gingivalis
Introduction
Porphyromonas gingivalis is a key periodontal pathogen associated with the aetiology of periodontal disease [1]. The host inflammatory response in the periodontal tissues induced by bacteria and their components present in dental plaque leads to breakdown of the attachment apparatus between the tooth and the supporting bone and may result ultimately in tooth loss [1]. In addition, epidemiological evidence is now emerging which shows that periodontal disease may be linked to systemic disorders such atherosclerosis and type 2 diabetes mellitus [2,3].
The innate immune system constitutes the first line of defence against periodontal pathogens. Monocytes play a critical part in this response, migrating to sites of infection and undergoing differentiation into macrophages and dendritic cells which are involved in phagocytosis and the recruitment of the adaptive immune system through antigen presentation. Toll-like receptors (TLRs) are a major group of pattern recognition receptors involved in the detection of bacterial pathogen-associated molecular patterns (PAMPS). Lipopolysaccharide (LPS) derived from enteric bacteria such as Escherichia coli binds to TLR-4 leading to activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and mitogen-activated protein kinase (MAPK) pathways and the production of proinflammatory cytokines [4]. LPS from P. gingivalis is structurally distinct from other types of LPS, such as E. coli, and has been shown to interact with both TLR-2 or TLR-4 [5–7]. Highly purified P. gingivalis LPS was found to contain two related but distinct isoforms which differed in the lipid A moieties [5,7]. Therefore, distinct molecular forms of P. gingivalis LPS may activate TLR-2 and TLR-4 [5]. LPS from P. gingivalis strain W50 has been well characterized [8,9] and we have confirmed that it is a TLR-2-specific ligand (unpublished data). Interaction between TLRs and their ligands leads to the recruitment of adaptor proteins to the cytoplasmic domains of the TLRs [10]. The adaptors MyD88 and MyD88 adaptor-like mediate downstream activation of the NF-κB, c-Jun N-terminal kinase (JNK) and p38 MAPK intracellular signalling pathways [11,12]. The MyD88-dependent pathway can also activate several members of the interferon regulatory factor (IRF) family of transcription factors, including IRF5 [10]. MyD88 is utilized by all the TLRs except TLR-3 to activate intracellular signalling cascades [13]. TLR-4 can also signal via a MyD88-independent pathway using the Toll/interleukin (IL)-1 receptor domain-containing adaptor inducing IFN-γ (TRIF) which is recruited via the TRIF-related adaptor molecule (TRAM) leading to NF-κB, MAPK and IRF3 activation [14].
Host immune responses mediated through TLRs are dependent not only upon specific receptors binding but also on the nature of the receptor–ligand interaction. For example, fimbriae and LPS from P. gingivalis both interact with TLR-2; however, human macrophages display differential gene expression profiles in response to these stimuli [6,15,16]. Other studies have shown that different structural components of PAMPS interact with various accessory proteins modulating the TLR-mediated response [17]. This upstream mechanism has been demonstrated in monocytes/macrophages treated with fimbrial peptides. Thus, distinct fimbral epitopes interact with CD14 or the β-integrin CD11b/CD18 which results in distinct cytokine expression profiles [17].
Differential activation of TLRs by PAMPS such as LPS leads to the activation of different intracellular signalling pathways, recruitment of transcription factors and the production of different repertoires of cytokines and other inflammatory mediators [15]. In a mixed infection such as periodontal disease, differential regulation of genes encoding inflammatory cytokines and associated molecules may have an impact on disease pathogenesis. The aims of this study were to investigate the temporal regulation of immunoregulatory genes in response to P. gingivalis LPS (a TLR-2 ligand), to compare gene expression induced by P. gingivalis and E. coli LPS (a TLR-4 ligand) and to identify genes which were regulated differentially in response to these PAMPS.
Materials and methods
Reagents
Unless stated otherwise, all laboratory reagents were purchased from Sigma (Poole, UK). P. gingivalis LPS (strain W50) was a gift from Dr M. Rangarajan, Barts and the London School of Medicine and Dentistry, London, UK. E. coli LPS (strain 0111:B4) and monophosphoryl lipid A (MPLA) were purchased from Invivogen (San Diego, CA, USA). Experiments using HEK-293 cells stably transfected with either TLR-2 or TLR-4 confirmed that P. gingivalis (strain W50) was a specific TLR-2 agonist (Jaedicke, unpublished data).
Human monocyte (THP-1) cell culture
Human pro-monocytic THP-1 cells were purchased from the European Collection of Cell Cultures (Salisbury, Wilts, UK). Cells were cultured in RPMI-1640 medium, supplemented with fetal calf serum (FCS) (10% v/v), L-glutamine (2 mM), penicillin (100 U/ml) and maintained at 37°C and 5% CO2. Prior to use, 4 × 106 THP-1 cells per well were differentiated into monocytes in six-well plates using 0·1 µM 1,25 dihydroxyvitamin D3 (Merck Biosciences, Nottingham, UK) for 24 h [18]. After 24 h the ability of the cells to adhere to the plastic tissue culture dish (indicative of differentiation from pro-monocytes to monocytes) was assessed by microscopy. Cell viability was assessed by Trypan blue exclusion.
Isolation and culture of peripheral blood monocytes
Buffy coats obtained from the National Blood Service (Newcastle upon Tyne, UK) were mixed in a 1:1 ratio with phosphate-buffered saline (PBS)/ethylenediamine tetraacetic acid (EDTA) (5 mM), then layered on top of an equal volume of Histopaque (Sigma). Following centrifugation (300 g for 20 min), the buffy coat layer was removed and mixed with an equal volume of PBS/EDTA (5 mM). The cells were pelleted at 300 g (5 min) and the supernatant was discarded. To remove erythrocyte contamination, cells were resuspended in erythrocyte lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0·1 mM EDTA, pH 7·3) for 3 min before quenching in 10 ml PBS for 10 min. Platelets were removed by resuspending the cells in PBS/EDTA (2 mM) and centrifuged at 300 g for 10 min. Peripheral blood mononuclear cells within the buffy coat were counted prior to incubation in six-well plates at 1 × 106 cells/well. Cells were incubated for 12 h in RPMI-1640 medium, the non-adherent cells (lymphocytes) were removed and the adherent cells were gently washed twice in media. Fluorescence-activated cell sorting (FACS), using a CD14/fluorescein isothiocyanate-conjugated antibody, confirmed that the isolated cells were monocytes (data not shown). The purity of the monocyte populations was between 75 and 82% in all experiments.
Differentiation of peripheral blood monocytes into dendritic cells
Monocyte-derived dendritic cells (MDDCs) were produced by plating peripheral blood monocytes at 5 × 105 cells/ml in six-well plates in RPMI-1640 medium supplemented with IL-4 (50 ng/ml) and granulocyte–macrophage colony-stimulating factor (GM-CSF) (10 ng/ml) for 7 days [19]. Differentiation was confirmed by observing morphological changes using light microscopy. Changes in expression of CD1a, CD11C, CD14, human leucocyte antigen D-related and CD83 were measured by FACS to determine the phenotype of MDDCs prior to experimentation (data not shown).
Cell stimulation experiments
All assays measuring gene expression or secreted protein levels were performed following stimulation of cells with LPS or MPLA. Previous studies have demonstrated that monocytes are highly responsive to P. gingivalis LPS (strain W50), and maximal induction of tumour necrosis factor (TNF)-α expression was observed following treatment with 100 ng/ml P. gingivalis LPS [18,20]. Relevant controls were also measured in parallel. E. coli, P. gingivalis LPS (100 ng/ml) or MPLA (1 µg/ml) were used to stimulate 1 × 106 cells/ml or 5 × 105 cells/ml (MDDCs) in six-well plates for 0·5 to 48 h.
RNA extraction and reverse transcription
Total RNA was isolated from myeloid cells using a RNeasy Mini Kit (Qiagen, Crawley, UK). The cells were lysed directly in situ using RLT buffer (Qiagen). RNA was isolated according to the manufacturer's instructions and was eluted in RNase-free water (30 µl). Total RNA (1 µg) was then reverse transcribed using random hexamers in 20 µl reactions using a high capacity cDNA kit (Applied Biosystems, Warrington, UK) (ABI) according to the manufacturer's instructions.
Gene expression assays
Custom-made Taqman low-density arrays (TLDAs) (ABI) were used for expression profiling based on real-time quantitative reverse transcription–polymerase chain reaction (RT–PCR) to compare the gene expression induced by P. gingivalis or E. coli LPS with unstimulated THP-1 monocytes. These arrays contained predesigned primers and Taqman probes (FAM™ reporter dye at the 5′ end of each Taqman® MGB probe and a non-fluorescent quencher at the 3′ end). Each array contained eight sample loading ports and 2·5 µl of each 20 µl cDNA reaction was loaded into each port. These arrays contained primers and probes for 45 different cytokine-related genes which regulate inflammatory processes (Table 1). RNA polymerase was used as endogenous control. Amplification and real-time analysis of cDNA samples loaded onto the TLDAs were performed using an ABI 7900HT real-time PCR machine. The results were analysed using sds version 2.2 software (ABI). Single-assay real-time RT–PCR using Taqman primers and probes (ABI) were performed for CXC chemokine ligand 5 (CXCL5) (Hs00171085) to confirm the results obtained from the arrays. RNA polymerase II (Hs00172187_ml) was used as an endogenous control.
Table 1.
Primers and probes for the quantification of gene expression using Taqman low-density arrays (TLDAs).
| Inflammatory mediator | Primers and probe assay ID |
|---|---|
| IL-1α (interleukin-1 alpha) | Hs00174092_m1 |
| IL-1β (interleukin-1 beta) | Hs00174097_m1 |
| IL-Ra (interleukin-1 receptor antagonist) | Hs00277299_m1 |
| IL-1R1 (interleukin-1 receptor-1) | Hs00168392_m1 |
| IL-1Acp (interleukin-1 accessory protein) | Hs00370506_m1 |
| ICE (IL-1 converting enzyme) (caspase-1) | Hs00354836_m1 |
| IL-18 (interleukin-18) | Hs00155517_m1 |
| IL-18R1 (interleukin-18 receptor) | Hs00175381_m1 |
| IL-18Acp (interleukin-1 accessory protein) | Hs00187256_m1 |
| IL-1F6 (interleukin-1 family 6) | Hs00205367_m1 |
| IL-1F7 (interleukin-1 family 7) | Hs00367199_m1 |
| IL-1F8 (interleukin-1 family 8) | Hs00758166_m1 |
| IL-1F9 (interleukin-1 family 9) | Hs00219742_m1 |
| IL-1F10 (interleukin-1 family 10) | Hs00544661_m1 |
| IL-1Rrp2 (IL-1 receptor-related protein 2) | Hs00187259_m1 |
| TNF-α (tumour necrosis factor alpha) | Hs00174128_m1 |
| TNFRSF1A (TNF alpha receptor superfamily 1A) | Hs00236902_m1 |
| TNFRSF1B (TNF alpha receptor superfamily 1B) | Hs00153550_m1 |
| TACE (TNF-α converting enzyme) | Hs00234221_m1 |
| IL-4 (interleukin-4) | Hs00174122_m1 |
| IL-6 (interleukin-6) | Hs00174131_m1 |
| OSM (oncostatin M) | Hs00171165_m1 |
| LIF (leukaemia inhibitory factor) | Hs00171455_m1 |
| OSMR (oncostatin M receptor) | Hs00384278_m1 |
| LIFR (leukaemia inhibitory factor receptor) | Hs00158730_m1 |
| IL-10 (interleukin 10) | Hs00174086_m1 |
| IL-12a (interleukin-12 chain a) | Hs00168405_m1 |
| IL-12b (interleukin-12 chain b) | Hs00233688_m1 |
| IL-32 (interleukin-32) | Hs00170403_m1 |
| GM-CSF (granulocyte macrophage-colony stimulating factor) | Hs00171266_m1 |
| IFN-γ (interferon gamma) | Hs00174143_m1 |
| CCL2 (monocyte chemottractant protein 1) | Hs00234140_m1 |
| CX3CL1 (fractalkine) | Hs00171086_m1 |
| CXCL5 (epithelial-derived neutrophil activating protein 78) (ENA-78) | Hs00607029_g1 |
| CCL5 (regulation on activation normal T cell expressed and secreted) (RANTES) | Hs00174575_m1 |
| CXCL8 (IL-8) | Hs00174103_m1 |
| CXCL10 | Hs00171042_m1 |
| Leptin | Hs00174877_m1 |
| LeptinR (leptin receptor) | Hs00174497_m1 |
| ADIPOR1 (adiponectin receptor-1) | Hs00360422_m1 |
| ADIPOR2 (adiponectin receptor-2) | Hs00226105_m1 |
| Visfatin | Hs00237184_m1 |
| INSR (insulin receptor) | Hs00169631_m1 |
| RAGE (receptor for advanced glycation end products) | Hs00153957_m1 |
| CD14 | Hs00169122_g1 |
| Control genes | |
| GAPDH (glyceraldehyde-3-phosphate-dehydrogenase) | Hs99999905_m1 |
| 18S | Hs99999901_s1 |
| RNA pol II | Hs00222679_m1 |
Measurement of cytokines and chemokines secreted by myeloid cells
The concentrations of CXCL5, CXCL8, IL-1β and TNF-α in culture supernatants were measured by enzyme-linked immunosorbent assay using commercially available kits (DuoSet; R&D Systems, Abingdon, Oxford, UK). Each sample was measured in duplicate and results were obtained from three independent experiments.
Statistical analyses
Statistically significant differences between groups and over time were assessed using anova with Bonferroni post-hoc corrections. For non-parametric data, the Kruskal–Wallis test and Mann–Whitney U-test were used.
Results
Regulation of gene expression by P. gingivalis and E. coli LPS
The temporal regulation of cytokine-related genes in THP-1 monocytes following treatment with P. gingivalis or E. coli LPS for 0·5, 2, 6 and 24 h was assessed using TLDA. Fifteen of the 45 different genes represented on the arrays (Table 1) were up-regulated by both types of LPS compared with unstimulated cells. These genes included those encoding mediators with well-established roles in chronic inflammation and the pathogenesis of periodontal disease (e.g. IL-1β, IL-6 and TNF-α) (Table 2). Some of these genes were up-regulated maximally 2 h after stimulation with LPS, for example IL-1α, IL-1β, IL-18 receptor (IL-18R) and CXCL8. Other genes were up-regulated maximally after 6 h, such as IL-6, IL-10 and CCL2, while IL-32 was expressed most highly 24 h post-stimulation (Table 2). Eight of the genes investigated were found to be up-regulated significantly more highly by E. coli LPS compared with P. gingivalis LPS (Table 3). These included the IL-1 family cytokine IL-1 family (IL-1F)8, the haematopoietic growth factor GM-CSF and the chemokines CCL5, CX3CL1 and CXCL5. Some of these differentially regulated genes were induced most highly 2 h after stimulation with LPS, for example oncostatin M, while other genes were up-regulated maximally after 6 h (GM-CSF) and 24 h (visfatin) post-stimulation. Eighteen of the genes assessed were expressed but were not regulated by either type of LPS. Of the remaining genes, four were not expressed constitutively or were not induced in monocytes (IL-1F10, IL R-related protein 2, IFN-γ and leptin).
Table 2.
Immunoregulatory genes that are upregulated by both Porphyromonas gingivalis and Escherichia coli lipopolysaccharide (LPS).
| Fold change |
Fold change |
Fold change |
Fold change |
|||||
|---|---|---|---|---|---|---|---|---|
| 0·5 h |
2 h |
6 h |
24 h |
|||||
| Gene | P. gingivalis | E. coli | P. gingivalis | E. coli | P. gingivalis | E. coli | P. gingivalis | E. coli |
| IL-1 family | ||||||||
| IL-1α | 25·5 ± 9·4 | 18·6 ± 10·2 | 1904 ± 624 | 1443·2 ± 1001·1 | 78·2 ± 32·2 | 245·1 ± 49·5 | 10·6 ± 2·1 | 41·9 ± 18·5 |
| IL-1β | 26·7 ± 17·8 | 70·3 ± 29·2 | 470 ± 121 | 1125·1 ± 894·3 | 47·0 ± 13·7 | 472·2 ± 290·2 | 26·8 ± 12·3 | 389·2 ± 78·2 |
| IL-18R | 0·8 ± 0·4 | 0·8 ± 0·5 | 108 ± 90·3 | 149·2 ± 103·6 | 99·4 ± 87·7 | 44·0 ± 26·5 | 28·8 ± 22·8 | 74·0 ± 40·6 |
| IL-18Acp | 0·4 ± 0·04 | 0·7 ± 0·3 | 71·8 ± 29·0 | 159·5 ± 144·3 | 26·1 ± 10·7 | 35·3 ± 21·5 | 3·7 ± 1·5 | 7·6 ± 5·1 |
| IL-1F9 | 1·1 ± 0·3 | 1·2 ± 0·6 | 37·1 ± 4·5 | 62·1 ± 25·9 | 4·5 ± 2·1 | 3·2 ± 2·1 | 2·1 ± 1·1 | 1·7 ± 0·8 |
| IL-6 family | ||||||||
| IL-6 | 0·2 ± 0·0 | 1·0 ± 0·2 | 1325 ± 1009·3 | 52·2 ± 28·8 | 12·0 ± 7·5 | 788·1 ± 442·1 | 6·3 ± 4·2 | 135·3 ± 117·1 |
| LIF | 2·1 ± 1·0 | 0·8 ± 0·3 | 293·1 ± 218·4 | 607·4 ± 546·1 | 3·8 ± 3·0 | 12·9 ± 8·3 | 2·6 ± 1·9 | 13·1 ± 0·6 |
| TNF family | ||||||||
| TNF-α | 20·1 ± 13·4 | 41·7 ± 12·5 | 77·8 ± 30·9 | 109·3 ± 82·1 | 2·3 ± 0·9 | 6·2 ± 1·7 | 1·2 ± 0·6 | 2·6 ± 0·5 |
| TNFRSF1B | 0·7 ± 0·2 | 0·7 ± 0·3 | 0·3 ± 0·1 | 2·7 ± 2·2 | 1·1 ± 0·2 | 3·7 ± 0·8 | 2·4 ± 0·1 | 8·5 ± 1·8 |
| Chemokines | ||||||||
| CCL2 | 2·3 ± 1·1 | 1·9 ± 0·7 | 26·9 ± 14·5 | 8·6 ± 5·0 | 129·1 ± 72·3 | 56·3 ± 20·7 | 89·8 ± 47·2 | 21·0 ± 18·2 |
| CXCL8 | 7·8 ± 4·7 | 13·2 ± 4·9 | 20·9 ± 4·8 | 43·0 ± 33·0 | 4·8 ± 2·4 | 17·7 ± 6·3 | 2·3 ± 0·9 | 6·9 ± 1·1 |
| CXCL10 | 13·0±6·1 | 14·7±11·4 | 469·5±160·1 | 73·4±45·4 | 2476±1566 | 503·3±37·5 | 1086·1±736·0 | 93·5±77·9 |
| Other cytokines | ||||||||
| IL-10 | 1·3 ± 0·6 | 1·1 ± 0·32 | 7·5 ± 2·8 | 2·9 ± 0·7 | 4·4 ± 0·4 | 27·7 ± 11·9 | 3·8 ± 0·5 | 13·3 ± 1·8 |
| IL-12B | 2·6 ± 1·6 | 2·6 ± 0·9 | 794·2 ± 541·1 | 570·2 ± 288 | 137·0 ± 17·3 | 686·1 ± 157·3 | 9·1 ± 5·1 | 127·3 ± 118·0 |
| IL-32 | 1·0 ± 0·7 | 1·5 ± 0·7 | 48·5 ± 22·2 | 40·0 ± 17·7 | 67·1 ± 35·3 | 118·0 ± 55·2 | 433·5 ± 56·7 | 247·2 ± 145·1 |
THP-1 monocytes were treated with LPS (100 ng/ml) from either P. gingivalis or E. coli for the times indicated above. Gene expression was quantified using Taqman low-density arrays by real-time polymerase chain reaction and normalized to RNA polymerase II. The values shown are the mean fold change compared with untreated cells ± standard deviation. These results were obtained from three independent experiments. IL, interleukin; TNF, tumour necrosis factor; TNFRSF1B, TNF alpha receptor superfamily 1B; LIF, leukaemia inhibitory factor.
Table 3.
Escherichia coli lipopolysaccharide (LPS) upregulates differentially a subset of genes compared with Porphyromonas gingivalis LPS.
| Fold change |
Fold change |
Fold change |
Fold change |
|||||
|---|---|---|---|---|---|---|---|---|
| 0·5 h |
2 h |
6 h |
24 h |
|||||
| Gene | P. gingivalis | E. coli | P. gingivalis | E. coli | P. gingivalis | E. coli | P. gingivalis | E. coli |
| IL-1 family | ||||||||
| IL-1F8 | 0·0 ± 0·0 | 2·7 ± 1·1 | 0·0 ± 0·0 | 3·8 ± 0·8 | 0·8 ± 0·0 | 1·7 ± 0·7 | 0·0 ± 0 | 101·1 ± 62·0 |
| IL-6 family | ||||||||
| OSM | 3·0 ± 2·0 | 3·4 ± 1·5 | 0·9 ± 0·2 | 8·2 ± 5·9 | 0·1 ± 0·0 | 0·2 ± 0·1 | 0·1 ± 0·1 | 1·7 ± 0·9 |
| OSMR | 0·3 ± 0·1 | 0·5 ± 0·2 | 0·3 ± 0·0 | 6·4 ± 4·6 | 0·8 ± 0·3 | 5·5 ± 1·2 | 2·8 ± 1·4 | 8·0 ± 2·4 |
| Chemokines | ||||||||
| CCL5 | 0·6 ± 0·2 | 0·7 ± 0·1 | 1·3 ± 0·2 | 11·6 ± 8·3 | 1·2 ± 0·2 | 9·7 ± 4·3 | 2·7 ± 0·7 | 10·4 ± 3·8 |
| CXCL5 | 0·0 ± 0·0 | 0·0 ± 0·0 | 1·0 ± 0·0 | 1·0 ± 0·0 | 1·2 ± 0·2 | 6·3 ± 2·1 | 1·0 ± 0 | 27·5 ± 5·1 |
| CX3CL1 | 0·0 ± 0·0 | 0·0 ± 0·0 | 2·0 ± 0·4 | 35·6 ± 17·0 | 2·1 ± 0·3 | 3·9 ± 0·6 | 0·0 ± 0 | 0·0 ± 0·0 |
| Other cytokines | ||||||||
| GM-CSF | 0·0 ± 0·0 | 0·0 ± 0·0 | 12·9 ± 7·6 | 58·1 ± 6·5 | 2·4 ± 1·3 | 161 ± 52·1 | 3·0 ± 1·0 | 105·2 ± 68·8 |
| PBEF/visfatin | 0·6 ± 0·2 | 0·8 ± 0·1 | 1·3 ± 0·3 | 2·2 ± 0·4 | 1·6 ± 0·5 | 5·1 ± 2·3 | 2·3 ± 0·6 | 15·2 ± 4·2 |
THP-1 monocytes were treated with LPS (100 ng/ml) from either P. gingivalis or E. coli for the times indicated above. Gene expression was quantified using Taqman low-density arrays by real-time polymerase chain reaction. The values shown are the mean fold change compared with untreated cells ± standard deviation. These results were obtained from three independent experiments. IL, interleukin; OSM, oncostatin M; OSMR, oncostatin M receptor; GM-CSF, granulocyte–macrophage colony-stimulating factor; PBEF, pre-B cell colony-enhancing factor.
The CXCL5 is regulated differentially by P. gingivalis and E. coli LPS
The array data showed that the chemokine CXCL5 was up-regulated more highly by LPS from E. coli than P. gingivalis (Table 3). This pattern of expression emerged 6 h post-stimulation with LPS (Table 3). Single-assay real-time PCR confirmed the data obtained from the TLDAs (Fig. 1a). Secreted protein levels of CXCL5 were also measured and showed a similar pattern of regulation by LPS (Fig. 1b). At both the mRNA and protein levels, significant differences in CXCL5 expression were detected 48 h post-stimulation (Fig. 1a and b). These data were obtained using the human THP-1 monocytic cell line. Therefore, further experiments were performed to establish whether similar expression patterns were observed for CXCL5 expression in primary cells. Primary human monocytes were treated with LPS from either P. gingivalis or E. coli for 3–48 h. The untreated cells secreted high levels of CXCL5 (Fig. 1c), and these levels were increased in monocytes treated with P. gingivalis and E. coli LPS. Following 24 h of treatment with E. coli LPS, there was a significant increase in CXCL5 levels compared with CXCL5 levels from monocytes treated with P. gingivalis LPS or the untreated control cells. MDDCs also expressed CXCL5 constitutively (Fig. 1d) and secreted levels of this cytokine were up-regulated significantly by E. coli LPS compared with P. gingivalis LPS and untreated controls (48 h).
Fig. 1.
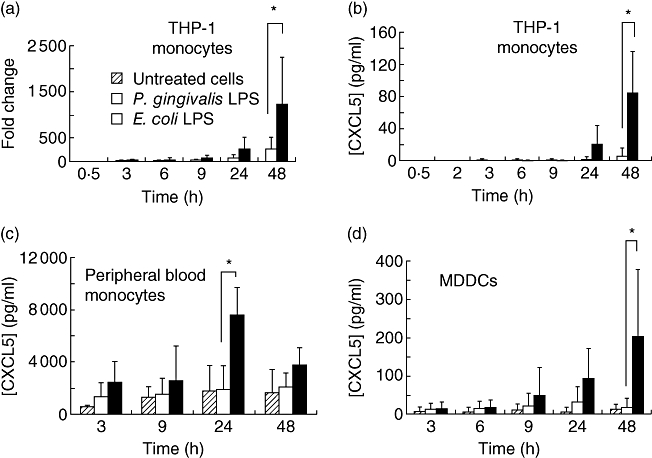
CXCL5 is differentially regulated by Porphyromonas gingivalis and Escherichia coli lipopolysaccharide (LPS) in myeloid cells. (a) Real-time reverse transcription–polymerase chain reaction analysis of CXCL5 expression using cDNA derived from THP-1 monocytes. Cells were treated with E. coli LPS (100 ng/ml) or P. gingivalis LPS (100 ng/ml) until the indicated time-points. Enzyme-linked immunosorbent assay was used to measure secreted CXCL5 in culture supernatants from (b) THP-1 monocytes, (c) peripheral blood monocytes and (d) monocyte-derived dendritic cells. The results were obtained from three independent experiments and are presented as average ± standard deviation.
The CXCL5 secretion does not occur via an IL-1β- or TNF-α-dependent mechanism
The differential regulation of CXCL5 in myeloid cells occurs relatively late (24–48 h) compared with other cytokines; for example, CCL5 is expressed maximally 2 h post-stimulation. It is possible that this effect may not be a direct result of TLR stimulation but may occur indirectly through induction of other proinflammatory cytokines. Previously, IL-1β and TNF-α have been shown to regulate CXCL5 expression in the A459 pulmonary epithelial cell line [21]. The TLDA data show that both IL-1β and TNF-α were up-regulated highly by both P. gingivalis and E. coli LPS at the mRNA level (Fig. 2a and c). Real-time PCR showed that after 24 h, IL-1β levels induced by E. coli LPS were higher than those induced by P. gingivalis LPS. Secreted IL-1β and TNF-α were both present in the culture supernatants taken from cells treated with both types of LPS (Fig. 2b and d). However, there was no significant difference in the levels of these cytokines produced in response to either P. gingivalis or E. coli LPS. When THP-1 monocytes were treated with IL-1β (250 pg/ml) or TNF-α (500 pg/ml), no secreted CXCL5 was detected. However, CXCL8 was detected in the same culture supernatants (data not shown), demonstrating that the THP-1 monocytes were responsive to both IL-1β and TNF-α and were incapable of specifically inducing CXCL5 expression.
Fig. 2.
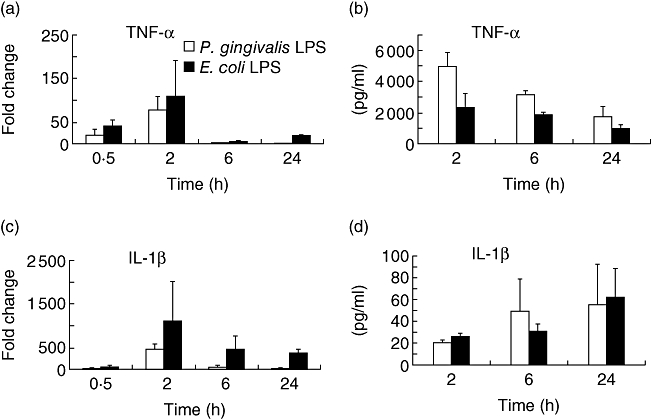
Porphyromonas gingivalis and Escherichia coli lipopolysaccharide (LPS) up-regulate interleukin (IL)-1β and tumour necrosis factor (TNF)-α (a,c) Real-time reverse transcription–polymerase chain reaction was used to quantify IL-1β and TNF-α using Taqman low-density arrays with cDNA derived from THP-1 cells treated with either P. gingivalis or E. coli LPS. The results were normalized to RNA polymerase II and calculated as a fold change compared with results obtained from unstimulated cells. (b,d) IL-1β and TNF-α levels in culture supernatant from the THP-1 cells treated with either P. gingivalis or E. coli LPS, measured by enzyme-linked immunosorbent assay. The results were obtained from three independent experiments and are presented as average ± standard deviation.
Regulation of CXCL5 by other TLR ligands
The MPLA is a TLR-4 agonist but studies in mice have demonstrated that this agonist functions with a bias towards the use of the TRIF adaptor [22] and therefore utilizing principally the TRIF–TRAM MyD88-independent pathway. Although TRIF–TRAM mediates TLR-4 signalling it is not involved in TLR-2-mediated gene expression [10]. To investigate if the differential pattern of expression of CXCL5 was mediated via the TRIF adaptor, THP-1 monocytes were treated with either MPLA (1 µg/ml) or E. coli LPS (100 ng/ml) for 48 h, as this was the time at which maximal CXCL5 expression was detected in the previous experiments and at which point differences in expression levels of CXCL5 following P. gingivalis and E. coli LPS stimulation reached significant levels. MPLA did not induce CXCL5 secretion, although it was induced by two different preparations of E. coli LPS (Fig. 3a). However, MPLA induced TNF-α secretion at a similar level to E. coli LPS (Fig. 3b) in the same tissue culture supernatants in which CXCL5 levels were measured, indicating that the TRIF–TRAM adaptors are not required for TLR-4-driven CXCL5 in THP-1 monocytes.
Fig. 3.
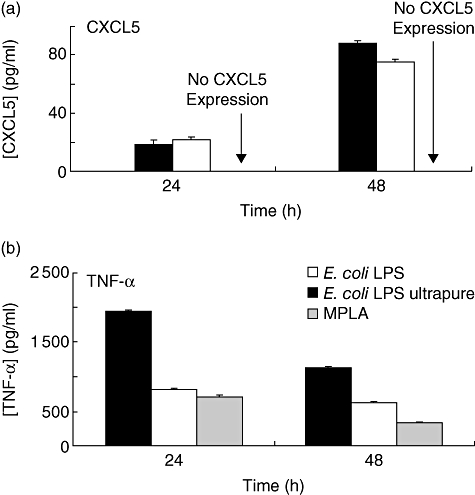
CXCL5 expression is not induced by the Toll/interleukin (IL)-1 receptor (TIR) domain-containing adaptor inducing interferon (IFN)-γ (TRIF)-biased Toll-like receptor (TLR)-4 ligand monophosphoryl lipid A (MPLA). THP-1 monocytes were treated with two different preparations of Escherichia coli lipopolysaccharide (LPS) (100 ng/ml), either strain 0111:B4 pure (black) or ultrapure LPS (white) or MPLA (1 µg/ml) (grey) for 24 or 48 h. (a) CXCL5 levels in culture supernatants following treatment with E. coli LPS or MPLA. (b) Tumour necrosis factor-α levels in the same culture supernatants following treatment with the above ligands. The results were obtained from three independent experiments and represent average ± standard deviation.
Discussion
This study was undertaken to investigate gene expression in monocytes following exposure to PAMPS which exert their effects via TLR-2 and TLR-4, and our data significantly extend current understanding of cytokine responses by monocytes following exposure to LPS. In addition, new data are presented which reveal co-ordinate and temporal gene expression of key inflammatory mediators in response to LPS from P. gingivalis, an important pathogen associated with chronic periodontitis. Furthermore, our results show that P. gingivalis and E. coli LPS have differential effects at the molecular level, leading to quantitative differences in gene expression for a subset of cytokine-related genes.
The IL-1 family cytokines have been shown to play an important role in a variety of chronic inflammatory diseases, including periodontitis and rheumatoid arthritis [23,24]. Thus, in periodontitis, IL-1β and IL-18 levels in gingival crevicular fluid correlate with severity of disease [18]. Both types of LPS up-regulated IL-1α, IL-1β, IL-18R, IL-18 R accessory protein (IL-18RAcp) and IL-1F9. Interestingly, certain members of the IL-1 cytokine family such as IL-18, IL-1R, IL-1RAcp and IL-1Ra were not regulated by LPS at the mRNA level. Previously, IL-18 mRNA has been shown to be expressed constitutively in peripheral blood mononuclear cells; however, stimuli such as LPS up-regulate IL-18 secretion [25]. Furthermore, work from our own laboratory has shown that P. gingivalis LPS stimulates IL-18 secretion in monocytes [20,26]. These data emphasize the importance of post-translational mechanisms of cytokine secretion and therefore, although analysis of mRNA expression demonstrates differential cytokine regulation by LPS from different species of bacteria, further differences may be revealed from proteomic analysis.
Both types of LPS significantly up-regulated IL-32 expression compared with the unstimulated cells. This cytokine has been detected in epithelial cells, natural killer cells and T cells and possesses proinflammatory properties such the ability to induce IL-8 and TNF-α[27]. Recently, it has also been reported that IL-32 is expressed by an oral epithelial cell line (H400) in response to LPS from the periodontal pathogens P. gingivalis and F. nucleatum[28]. These data, together with the finding that IL-32 plays a role in other inflammatory diseases [29,30] and its ability to induce monocyte differentiation [31], suggest that this cytokine may also contribute to the pathogenesis of periodontal disease. Therefore, investigation into the role of this cytokine in periodontitis could provide further understanding of how cytokine networks function in this disease.
Previous research has shown that different types of E. coli LPS, acting through TLR-4, induce distinct but overlapping gene expression profiles [32]. Consistent with our findings, chemokines were among the genes which were regulated differentially by LPS [32]. CXCL5 and CXCL8 are both neutrophil chemoattractants and potent angiogenic factors [33,34] These chemokines act through the same receptor (CXCR2) [35] and are produced concomitantly in response to IL-1β and TNF-α in pulmonary epithelial cells (A459) [21]. The data from our study show that CXCL8 was up-regulated by both types of LPS, while CXCL5 was up-regulated significantly only by E. coli LPS at the mRNA and protein level in both monocytes and dendritic cells. Secretion of CXCL5 following LPS stimulation is a relatively late event (24+ h) compared with other cytokines and therefore may be mediated by other proinflammatory cytokines. In the A459 cell line, which are pulmonary-derived epithelial cells, both IL-1β and TNF-α were shown to regulate CXCL5 transcription [21]. In our study, both IL-1β and TNF-α were up-regulated by LPS in monocytes at the mRNA level and were detected in culture supernatants from LPS-treated cells. However, when THP-1 monocytes were treated with either IL-1β or TNF-α at comparable concentrations to those that were recorded in the supernatants no secreted CXCL5 was detected, although CXCL8 levels were up-regulated by both cytokines. This discrepancy, with respect to previous data obtained in pulmonary epithelial cells, may be due to the differences in cytokine concentrations used. In our study, cells were stimulated with either 250 pg/ml or 500 pg/ml of IL-1β or TNF-α, whereas the pulmonary epithelial cells were treated with 10–20 ng/ml of IL-1β and TNF-α[21]. The differences may be due to differential responses to these cytokines by monocytes and pulmonary epithelial cells. Although IL-1β and TNF-α are not responsible for TLR-mediated CXCL5 expression, we cannot rule out the possibility that this effect is mediated by other proinflammatory cytokines. Studies carried out in fibroblasts with an inactive transforming growth factor-β kinase 1 (TAK1) revealed that CXCL5 belonged to a group of chemokine genes whose expression was suppressed most strongly by this inactive kinase [36]. Furthermore, in fibroblasts CXCL5 expression was shown to be predominantly mediated by the NF-κB and JNK pathways [36].
The MPLA is a derivative of LPS and is a TLR-4 agonist which is associated with a bias for TRIF signalling [22]. TLR-4 activation occurs via the adaptors MyD88 and TRIF, whereas TLR-2 utilizes only MyD88 [14,16]. Therefore, THP-1 monocytes were treated with MPLA to investigate whether the differential expression induced by E. coli LPS compared with P. gingivalis LPS is due to the recruitment of TRIF to TLR-4 by E. coli LPS. The results showed that CXCL5 was not expressed in response to MPLA. However, TNF-α was produced at similar levels to those induced by E. coli LPS, indicating that the selective use of TRIF adaptor by TLR-4 was not the mechanism by which E. coli LPS up-regulated CXCL5.
Other cytokines were also expressed differentially in response to P. gingivalis and E. coli LPS. For some of these genes, differential regulation occurred at an earlier time-point compared with CXCL5. For example, regarding the oncostatin M receptor and GM-CSF, differential regulation was observed 2 h post-stimulation with LPS. Therefore, this points to multiple mechanisms contributing to differential expression in response to these different PAMPS. The data presented here confirm and extend previous information on the effects of different PAMPS on monocytes. Understanding the mechanisms involved in differential responses will be important in deciphering pathogen–host interactions and how these can be manipulated to prevent tissue destruction associated with chronic inflammatory disease.
Acknowledgments
We would like to thank Dr M. Rangarajan, Barts and the London School of Medicine and Dentistry, London, UK for the gift of P. gingivalis LPS. This study was supported by UK Department of Health Clinician Scientist Fellowship DHCS/03/G121/46 awarded to Philip Preshaw.
Disclosure
None.
References
- 1.Darveau RP, Tanner A, Page RC. The microbial challenge in periodontitis. Periodontol 2000. 1997;14:12–32. doi: 10.1111/j.1600-0757.1997.tb00190.x. [DOI] [PubMed] [Google Scholar]
- 2.Gibson FC, III, Genco CA. Porphyromonas gingivalis mediated periodontal disease and atherosclerosis: disparate diseases with commonalities in pathogenesis through TLRs. Curr Pharm Des. 2007;13:3665–75. doi: 10.2174/138161207783018554. [DOI] [PubMed] [Google Scholar]
- 3.Preshaw PM, Foster N, Taylor JJ. Cross-susceptibility between periodontal disease and type 2 diabetes mellitus: an immunobiological perspective. Periodontol 2000. 2007;45:138–57. doi: 10.1111/j.1600-0757.2007.00221.x. [DOI] [PubMed] [Google Scholar]
- 4.Takeuchi O, Hoshino K, Kawai T, et al. Differential roles of TLR2 and TLR4 in recognition of Gram-negative and Gram-positive bacterial cell wall components. Immunity. 1999;11:443–51. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 5.Darveau RP, Pham TT, Lemley K, et al. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both Toll-like receptors 2 and 4. Infect Immun. 2004;72:5041–51. doi: 10.1128/IAI.72.9.5041-5051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hajishengallis G, Tapping RI, Harokopakis E, et al. Differential interactions of fimbriae and lipopolysaccharide from Porphyromonas gingivalis with the Toll-like receptor 2-centred pattern recognition apparatus. Cell Microbiol. 2006;8:1557–70. doi: 10.1111/j.1462-5822.2006.00730.x. [DOI] [PubMed] [Google Scholar]
- 7.Dixon DR, Darveau RP. Lipopolysaccharide heterogeneity: innate host responses to bacterial modification of lipid a structure. J Dent Res. 2005;84:584–95. doi: 10.1177/154405910508400702. [DOI] [PubMed] [Google Scholar]
- 8.Bramanti TE, Wong GG, Weintraub ST, Holt SC. Chemical characterization and biologic properties of lipopolysaccharide from Bacteroides gingivalis strains W50, W83, and ATCC 33277. Oral Microbiol Immunol. 1989;4:183–92. doi: 10.1111/j.1399-302x.1989.tb00250.x. [DOI] [PubMed] [Google Scholar]
- 9.Shapira L, Champagne C, Van Dyke TE, Amar S. Strain-dependent activation of monocytes and inflammatory macrophages by lipopolysaccharide of Porphyromonas gingivalis. Infect Immun. 1998;66:2736–42. doi: 10.1128/iai.66.6.2736-2742.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–64. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 11.Fitzgerald KA, Palsson-McDermott EM, Bowie AG, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413:78–83. doi: 10.1038/35092578. [DOI] [PubMed] [Google Scholar]
- 12.Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol. 2001;2:835–41. doi: 10.1038/ni0901-835. [DOI] [PubMed] [Google Scholar]
- 13.Jiang Z, Zamanian-Daryoush M, Nie H, Silva AM, Williams BR, Li X. Poly(I-C)-induced Toll-like receptor 3 (TLR3)-mediated activation of NFkappa B and MAP kinase is through an interleukin-1 receptor-associated kinase (IRAK)-independent pathway employing the signaling components TLR3-TRAF6-TAK1-TAB2-PKR. J Biol Chem. 2003;278:16713–19. doi: 10.1074/jbc.M300562200. [DOI] [PubMed] [Google Scholar]
- 14.Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science. 2003;301:640–3. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 15.Zhou Q, Amar S. Identification of signaling pathways in macrophage exposed to Porphyromonas gingivalis or to its purified cell wall components. J Immunol. 2007;179:7777–90. doi: 10.4049/jimmunol.179.11.7777. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Q, Desta T, Fenton M, Graves DT, Amar S. Cytokine profiling of macrophages exposed to Porphyromonas gingivalis, its lipopolysaccharide, or its FimA protein. Infect Immun. 2005;73:935–43. doi: 10.1128/IAI.73.2.935-943.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hajishengallis G, Ratti P, Harokopakis E. Peptide mapping of bacterial fimbrial epitopes interacting with pattern recognition receptors. J Biol Chem. 2005;280:38902–13. doi: 10.1074/jbc.M507326200. [DOI] [PubMed] [Google Scholar]
- 18.Foster N, Cheetham J, Taylor JJ, Preshaw PM. VIP inhibits Porphyromonas gingivalis LPS-induced immune responses in human monocytes. J Dent Res. 2005;84:999–1004. doi: 10.1177/154405910508401106. [DOI] [PubMed] [Google Scholar]
- 19.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179:1109–18. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foster N, Lea SR, Preshaw PM, Taylor JJ. Pivotal advance: vasoactive intestinal peptide inhibits up-regulation of human monocyte TLR2 and TLR4 by LPS and differentiation of monocytes to macrophages. J Leukoc Biol. 2007;81:893–903. doi: 10.1189/jlb.0206086. [DOI] [PubMed] [Google Scholar]
- 21.Chang MS, McNinch J, Basu R, Simonet S. Cloning and characterization of the human neutrophil-activating peptide (ENA-78) gene. J Biol Chem. 1994;269:25277–82. [PubMed] [Google Scholar]
- 22.Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316:1628–32. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- 23.Barksby HE, Lea SR, Preshaw PM, Taylor JJ. The expanding family of interleukin-1 cytokines and their role in destructive inflammatory disorders. Clin Exp Immunol. 2007;149:217–25. doi: 10.1111/j.1365-2249.2007.03441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dinarello CA. Interleukin-18 and the pathogenesis of inflammatory diseases. Semin Nephrol. 2007;27:98–114. doi: 10.1016/j.semnephrol.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 25.Puren AJ, Fantuzzi G, Dinarello CA. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1beta are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc Natl Acad Sci USA. 1999;96:2256–61. doi: 10.1073/pnas.96.5.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foster N, Andreadou K, Jamieson L, Preshaw PM, Taylor JJ. VIP inhibits P. gingivalis LPS-induced IL-18 and IL-18BPa in monocytes. J Dent Res. 2007;86:883–7. doi: 10.1177/154405910708600915. [DOI] [PubMed] [Google Scholar]
- 27.Dinarello CA, Kim SH. IL-32, a novel cytokine with a possible role in disease. Ann Rheum Dis. 2006;65(Suppl. 3):iii61–4. doi: 10.1136/ard.2006.058511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milward MR, Chapple IL, Wright HJ, Millard JL, Matthews JB, Cooper PR. Differential activation of NF-kappaB and gene expression in oral epithelial cells by periodontal pathogens. Clin Exp Immunol. 2007;148:307–24. doi: 10.1111/j.1365-2249.2007.03342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shioya M, Nishida A, Yagi Y, et al. Epithelial overexpression of interleukin-32alpha in inflammatory bowel disease. Clin Exp Immunol. 2007;149:480–6. doi: 10.1111/j.1365-2249.2007.03439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shoda H, Fujio K, Yamaguchi Y, et al. Interactions between IL-32 and tumor necrosis factor alpha contribute to the exacerbation of immune-inflammatory diseases. Arthritis Res Ther. 2006;8:R166. doi: 10.1186/ar2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Netea MG, Lewis EC, Azam T, et al. Interleukin-32 induces the differentiation of monocytes into macrophage-like cells. Proc Natl Acad Sci USA. 2008;105:3515–20. doi: 10.1073/pnas.0712381105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen C, Coats SR, Bumgarner RE, Darveau RP. Hierarchical gene expression profiles of HUVEC stimulated by different lipid A structures obtained from Porphyromonas gingivalis and Escherichia coli. Cell Microbiol. 2007;9:1028–38. doi: 10.1111/j.1462-5822.2006.00849.x. [DOI] [PubMed] [Google Scholar]
- 33.Koch AE, Polverini PJ, Kunkel SL, et al. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. 1992;258:1798–801. doi: 10.1126/science.1281554. [DOI] [PubMed] [Google Scholar]
- 34.Koch AE, Volin MV, Woods JM, et al. Regulation of angiogenesis by the C-X-C chemokines interleukin-8 and epithelial neutrophil activating peptide 78 in the rheumatoid joint. Arthritis Rheum. 2001;44:31–40. doi: 10.1002/1529-0131(200101)44:1<31::AID-ANR5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 35.Wuyts A, Proost P, Lenaerts JP, Ben-Baruch A, Van Damme J, Wang JM. Differential usage of the CXC chemokine receptors 1 and 2 by interleukin-8, granulocyte chemotactic protein-2 and epithelial-cell-derived neutrophil attractant-78. Eur J Biochem. 1998;255:67–73. doi: 10.1046/j.1432-1327.1998.2550067.x. [DOI] [PubMed] [Google Scholar]
- 36.Thiefes A, Wolter S, Mushinski JF, et al. Simultaneous blockade of NFkappaB, JNK, and p38 MAPK by a kinase-inactive mutant of the protein kinase TAK1 sensitizes cells to apoptosis and affects a distinct spectrum of tumor necrosis factor [corrected] target genes. J Biol Chem. 2005;280:27728–41. doi: 10.1074/jbc.M411657200. [DOI] [PubMed] [Google Scholar]