

Abstract
Recent reports show that 5-amino-4-imidazole carboxamide riboside (AICAR), a pharmacological activator of AMP-activated protein kinase (AMPK), inhibits the lipopolysaccharide (LPS)-induced production of proinflammatory cytokines. MRL/MPJ-Faslpr (MRL/lpr) mice show an intrinsic decreased threshold for the production of inflammatory mediators when stimulated. In our current studies, we sought to determine if AMPK activation would inhibit inflammatory mediator production in stimulated kidney mesangial cells. Cultured mesangial cells from MRL/lpr mice were treated with AICAR and stimulated with LPS/interferon (IFN)-γ. AICAR decreased dose-dependently inducible nitric oxide synthase (iNOS), cyclooxygenase-2 and interleukin-6 production in LPS/IFN-γ-stimulated mesangial cells. Mechanistically, AICAR inhibited the LPS/IFN-γ-stimulated PI3K/Akt signalling inflammatory cascade but did not affect LPS/IFN-γ-mediated inhibitory kappa B phosphorylation or nuclear factor (NF)-κB (p65) nuclear translocation. Treatment with the adenosine kinase inhibitor 5′-iodotubercidin blocked the ability of AICAR to activate AMPK and prevented AICAR from inhibiting the LPS/IFN-γ-stimulated PI3K/Akt pathway and attenuating iNOS expression. Taken together, these observations suggest that AICAR inhibits LPS/IFN-γ-induced Akt phosphorylation through AMPK activation and may serve as a potential therapeutic target in inflammatory diseases.
Keywords: AMPK, inflammation, lupus, metabolism, MRL/lpr
Introduction
MRL/lpr mice develop a lupus-like disease similar to human systemic lupus erythematosus (SLE). In both species, the pathophysiology includes immune abnormalities affecting T cells, B cells and cells of the innate immune system. The pathogenesis of lupus nephritis involves immune complex deposition coupled with inflammatory mediator production leading to damage of the kidney glomerulus. Mesangial cells are macrophage-like cells resident in the kidney, possessing both immune and vascular functions. We and others have shown that mesangial cells, similar to macrophages, produce nitric oxide (NO), superoxide (O2−) and other inflammatory mediators in response to lipopolysaccharide (LPS), interferon (IFN)-γ and interleukin (IL)-1β[1–3]. Because of their smooth muscle properties, they also have important functions in regulating local blood flow. While low-level constitutive NO production by mesangial cells is physiological and important in regulating renal blood flow, elevated levels of NO, produced either by mesangial cells or macrophages, may induce tissue damage directly or by reacting with O2− to form highly toxic peroxynitrite (ONOO−) [2,4]. Induced NO production occurs in a number of inflammatory renal diseases, including SLE. Reports show that inducible nitric oxide synthase (iNOS) expression is elevated in renal tissue of lupus patients and there is evidence of nitrated proteins in kidneys from lupus mice [1,5,6]. Other inflammatory mediators released by mesangial cells, such as cytokines [i.e. IL-6 and tumour necrosis factor (TNF)-α] and eicosanoids, also have pathogenic roles in lupus [7]. Lupus nephritis is characterized by the activation of mesangial cells leading to the uncontrolled release of inflammatory mediators. Without a therapeutic intervention, lupus nephritis in MRL/lpr mice, as well as in humans, progresses to end-stage renal disease.
AMP-activated protein kinase (AMPK) is an enzyme that participates in the cellular response to metabolic stress. Believed to function as a ‘low fuel warning system’ of the cell, AMPK activity is elevated strongly by conditions that elevate the cellular AMP : adenosine triphosphate (ATP) ratio, such as depletion of growth factors or glucose, or treatment with the pharmacological mimetic 5-amino-4-imidazole carboxamide riboside (AICAR) [8]. Once activated, AMPK phosphorylates numerous metabolic enzymes causing a global inhibition of biosynthetic pathways, thus conserving energy, and an activation of catabolic pathways, thus generating ATP. AMPK may also be activated by pathological stresses (i.e. oxidative stress), exercise and adipose tissue-derived hormones [9,10]. It is likely that AMPK also acts to limit inflammation, as recent reports have shown that AICAR inhibits TNF-α and IL-1β–induced nuclear factor (NF)-κB reporter gene expression dose-dependently in immune cells [11–14] and iNOS and cyclooxygenase-2 (COX-2) expression in stimulated macrophages [15]. Because of these reported anti-inflammatory effects of AICAR, we hypothesized that AICAR may decrease inflammatory mediator production in lupus mice mesangial cells by attenuating the LPS/IFN-γ-mediated signal transduction cascade.
Materials and methods
Animals
Eight-week-old female MRL/lpr mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Mice were maintained under specific pathogen-free conditions and used between 6 and 8 weeks of age, prior to the onset of disease. Mesangial cells were isolated from at least five mice by renal dissection, differential sieving and collagenase digestion, as we have described previously [16], and pooled for experimental procedures. Cultures were examined to ensure that they were homogeneous for expression of mesangial cell morphology and positive for α-smooth muscle actin after three to seven passages. Cultures were maintained at 37°C and 5% CO2 in a humidified atmosphere in Dulbecco's modified Eagle's medium (DMEM)/F-12 media, supplemented with 10% fetal bovine serum (FBS) and antibiotics.
Reagents
The IFN-γ was purchased from PharMingen (San Diego, CA, USA), while FBS and DMEM/F-12 were from gibco (Gaithersburg, MD, USA). The protein assay kits were purchased from BioRad (Hercules, CA, USA). Anti-iNOS Type II antibody was purchased from Transduction Laboratories (Lexington, KY, USA), while antibodies to AMPK, phospho-(Thr172)-AMPK, Akt, phospho-Akt (Ser473), inhibitory kappa B (IκB)-α and phospho-IκB-α were from Cell Signaling (Beverly, MA, USA). Compound c was purchased from Calbiochem (EMD Biosciences, San Diego, CA, USA). All other reagents, including 5′-iodotubercidin, LPS, epigallocatechin-gallate (EGCG) and metformin (were purchased from Sigma (St Louis, MO, USA).
Experimental protocol
In each experiment, mesangial cells were plated on 100 cm2 plates, serum starved and pretreated with indicated concentrations of AICAR. Cells were then stimulated with LPS (1 µg/ml) and IFN-γ (100 ng/ml) for the indicated treatment times, during which AICAR media was maintained. In some experiments, the adenosine kinase inhibitor 5′-iodotubercidin (0·1 µM) or compound c (20 uM) was also added to the cells 30 min prior to AICAR pretreatment.
Flow cytometry
Mesangial cells were treated with increasing amounts of AICAR for 16 h, washed with phosphate-buffered saline and trypsinized. Apoptosis/necrosis was evaluated using annexin V-fluorescein isothiocyanate (FITC)/propidium iodine (PI) staining, as described previously [17]. Samples were analysed by flow cytometry using a fluorescence activated cell sorter (FACS)Calibur and CellQuest software (BD Biosciences, San Jose, CA, USA). Laser excitation wavelength was set at 488 nm. The green signal from annexin V-FITC was measured at 525 nm and the red signal from PI was measured at 620 nm.
Nitrite production
Supernatants were collected 24 h after stimulation and analysed for nitrite concentration (a stable reaction product of NO with oxygen) as described after conversion of nitrate to nitrite using nitrate reductase, G6P, nicotinamide adenine dinucleotide reduced (NADPH) and glucose-6-phosphate dehydrogenase (G6PDH) (Roche-Boehringer Mannheim, Indianapolis, IN, USA) [18]. Briefly, supernatants were analysed by mixing an equal volume of sample with Griess reagents (1% sulphanilamide and 0·1% naphthylethylenediamene in 2·5% H3PO4) in a 96-well plate and the absorbance determined at 550 nm. The concentration of nitrite was calculated from a standard curve produced by the reaction of NaNO2 in the assay.
Real-time reverse transcription–polymerase chain reaction
Cellular RNA was extracted by lysing cells with RNeasy kit reagents (Biorad, Hercules, CA, USA) and mRNA was converted to cDNA, as described previously [19]. Real-time polymerase chain reaction was performed with the Sybr green method on a BIO-RAD IQ5 thermocycler using specific primers targeting iNOS (forward: CAGCTGGGCTGTACAAACCTT, reverse: CATTGGAAGTGAAGCGTTTCG), IL-6 (forward: ATCCAGTTGCCTTCTTGGGACTGA, reverse: TAAGCCTCCGACTTGTGAAGTGGT), COX-2 (forward: AAAGGTTCTTCTACGGAGAGAGTTCA, reverse: CTGGGCAAAGAATGCAAACA) and glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) (forward: AACTTTGGCATTGTGGAAGGGCTC, reverse: TGGAAGAGTGGGAGTTGCTGTTGA) (Integrated DNA Technologies, Coralville, IA, USA). The relative fold induction of corresponding target genes was calculated using the delta-delta-Ct method.
Western blotting
Cellular protein was collected by lysing pelleted cells with cell-lytic M cell lysis buffer supplemented with proteinase inhibitor and sodium orthovanadate. Cell lysate protein concentration was determined using the Bradford protein assay and equal amounts of protein were loaded onto a polyacrylamide gel and electrophoresed prior to transfer to a polyvinylidene difluoride membrane. Membranes were blocked in 5% non-fat milk/Tris-buffered saline-Tween 20 and then incubated overnight at 4°C with the indicated primary antibodies and visualized after secondary antibody incubation using the enhanced chemiluminescence + chemiluminescence kit (Amersham, GE Healthcare, Piscataway, NJ, USA).
Preparation of nuclear extract
Nuclear extracts were prepared utilizing nuclear and cytoplasmic extraction reagents nuclear extraction and cytoplasmic extraction reagents and protocol (Pierce Biotechnology, Rockford, IL, USA). Briefly, extracts were prepared by lysing pelleted cells in 200 ul of ice-cold hypotonic lysis solution (10 mM HEPES, pH 7·9, 10 mM KCl, 1·5 mM MgCl2) and vortexing vigorously for 15 s. Subsequently, 11 ul of Nonidet P-40 was added to the lysate, vortexed for 15 s and centrifuged at 16 000 g for 5 min. The resulting pellet was washed, resuspended in 50 µl of hypertonic lysis solution [20 mM HEPES, pH 7·9, 420 mM NaCl, 1·5 mM MgCl2, 0·2 mM ethylenediamine tetraacetic acid, 25% glycerol], incubated for 5 min at 4°C, and centrifuged at 16 000 g for 10 min. Supernatant containing the nuclear extract was removed, quantified with the dichloroacetate protein assay kit and stored at −80°C.
Enzyme-linked immunosorbent assay
The IL-6 and TNF-α levels in supernatants were quantified by enzyme-linked immunosorbent assay as per the manufacturer's instructions (R&D Systems, Minneapolis, MN, USA).
Statistics
Results shown represent means ± standard error of the mean, with n = 3 unless stated otherwise. Statistical analysis was performed by anova with post hoc analyses or Student's t-test, where appropriate, using GraphPad Prism version 4.0 for Windows (GraphPad Software, San Diego, CA, USA).
Results
The AICAR decreases iNOS, COX-2 and IL-6 production in LPS/IFN-γ-stimulated mesangial cells in a concentration-dependent manner
Mesangial cells from MRL/lpr mice were chosen to investigate whether AICAR prevented inflammatory mediator production, given their role in the regulation of kidney homeostasis. It has been reported widely that increased IL-6, COX-2 and iNOS gene activation are associated with inflammatory lupus [18,20–22]. Mesangial cell cultures established from 8-week-old female mice were treated with various concentrations of AICAR prior to the addition of LPS/IFN-γ. After 12–24 h of LPS/IFN-γ stimulation, cell mRNA, protein and supernatant were assayed for inflammatory mediator production. Our results show that AICAR blocked iNOS production in LPS/IFN-γ-stimulated mesangial cells in a concentration-dependent manner. Although message levels of iNOS did not reach statistical significance, there was a trend for a decrease with AICAR administration (P = 0·07, Fig. 1a) and protein levels were reduced by 1·0 mM AICAR (P < 0·05, Fig. 1b). Concordantly, NO production in the supernatants as measured by Griess reaction was also reduced (P < 0·05, data not shown). The anti-inflammatory effect of AICAR was also apparent when cells were pretreated with AICAR for only 1 h and then stimulated for 24 h with LPS/IFN-γ, as evident by dose-dependent reductions in cellular protein levels of iNOS and supernatant nitrite, as assessed by Griess reaction (Fig. 1c). Of interest, the effect of AICAR was intensified during a serum starved state compared with when the cells were cultured in 10% FBS. However, the same pattern of iNOS attenuation was evident in both cases (Fig. 1d). AICAR also reduced the levels of COX-2 mRNA (P < 0·05, Fig. 1a) and tended to reduce cellular protein (Fig. 1b). In addition, AICAR reduced IL-6 mRNA (P = 0·05, Fig. 1a) and there was a trend for a reduction in IL-6 protein in the supernatants of LPS/IFN-γ-stimulated cells treated with 1·0 mM AICAR compared with non-treated cells (P = 0·10). TNF-α levels were unaffected by AICAR administration (data not shown).
Fig. 1.
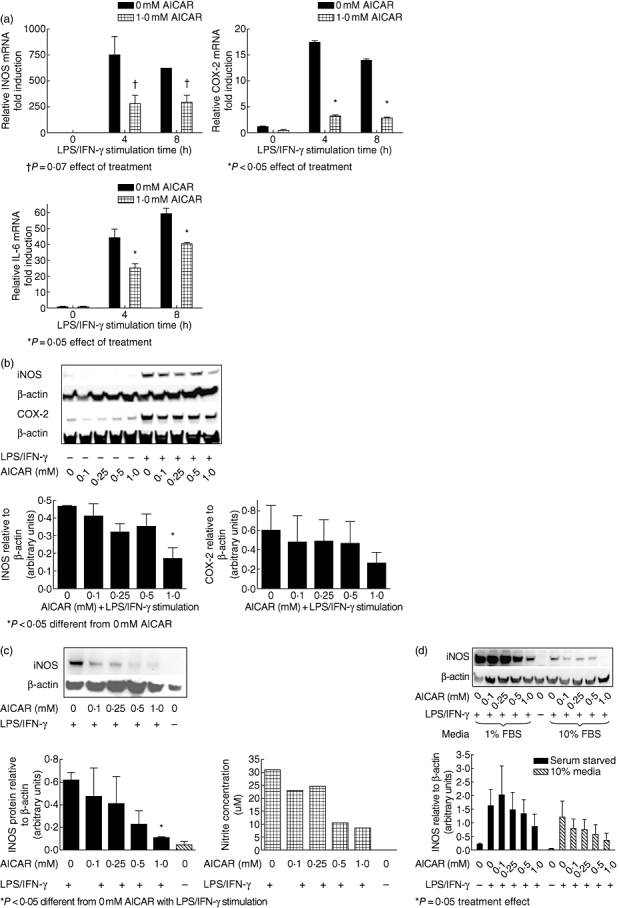
5-amino-4-imidazole carboxamide riboside (AICAR) inhibits lipopolysaccharide (LPS)/interferon (IFN)-γ-induced increases in inflammatory mediator production in mesangial cells in a concentration-dependent manner. Mesangial cell cultures from MRL/lpr mice were serum starved at confluency and pretreated with AICAR at the concentrations indicated for 16 h. Next, cells were stimulated with LPS/IFN-γ for the indicated times. (a) AICAR administration reduced inflammation dose-dependently, as evident by cellular mRNA of inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2) and interleukin (IL)-6 analysed by real-time reverse transcription–polymerase chain reaction and normalized to glyceraldehyde-3-phosphate-dehydrogenase (GAPDH). (b) Equal amounts of cellular protein were measured for iNOS and COX-2 by Western blot after 12 h. (c) AICAR also reduced iNOS levels dose-dependently with only a 1-h pretreatment prior to LPS/IFN-γ stimulation, as evident by cellular protein and supernatant nitrite levels. (d) AICAR attenuated iNOS levels whether cells were serum starved [1% fetal bovine serum (FBS)] or given regular (10% FBS) media during AICAR pretreatment and LPS/IFN-γ stimulation. Blots are representative of three experiments and densitometry analysis was conducted relative to β-actin. *P < 0·05; †P = 0·07 for effect of treatment.
The AICAR did not induce apoptosis
We sought to determine whether exposure of AICAR decreased cell viability. We used annexin V and PI to stain the cells after 16 h of AICAR exposure and cell viability was determined by flow cytometry. We found that there was no difference in cell viability at 0·05, 0·1 and 0·5 mM concentrations of AICAR; however, at 1·0 mM AICAR there was a slight increase in necrosis and apoptosis of the mesangial cells, indicating that elevated levels of AICAR may be toxic to serum starved cells (Table 1). While it is true that overnight serum starving may induce stress and decrease viability in the cells, the activation of AMPK also decreased inflammatory mediator production in the non-serum starved state (Fig. 1d), indicating that AICAR's anti-inflammatory effects are not due to stress.
Table 1.
5-amino-4-imidazole carboxamide riboside (AICAR) administration and cell viability.
| 0 mM (control) | 0·05 mM AICAR | 0·1 mM AICAR | 0·5 mM AICAR | 1·0 mM AICAR | |
|---|---|---|---|---|---|
| Necrotic | 8·71 ± 2·38 | 9·17 ± 1·55 | 6·64 ± 2·43 | 6·92 ± 1·94 | 13·76 ± 9·46 |
| Necrotic/apoptotic | 19·60 ± 4·21 | 18·24 ± 3·71 | 20·16 ± 3·23 | 28·48 ± 4·35 | 37·37 ± 3·91 |
| Live | 63·96 ± 6·30 | 64·60 ± 4·82 | 64·76 ± 5·22 | 55·40 ± 5·30 | 39·48 ± 8·35 |
| Apoptotic | 7·89 ± 1·01 | 7·97 ± 1·52 | 8·46 ± 0·76 | 9·17 ± 1·82 | 9·37 ± 1·05 |
Mesangial cells were treated with AICAR at the concentrations indicated for 16 h and apoptosis and necrosis were determined using annexin V/propidium iodine staining and analysed by flow cytometry. Data are shown as percentage of total cells.
The AICAR activates AMPK
Previous studies have shown that AMPK is activated by treatment with AICAR after conversion by adenosine kinase to the AMP analogue, AICA riboside monophosphate (ZMP). To explore the mechanism of inflammatory mediator inhibition in AICAR-treated mesangial cells, we measured phosphorylation of AMPK by Western blot with and without LPS/IFN-γ challenge in AICAR (1 mM) pretreated cells (Fig. 2a). Without the addition of AICAR, AMPK-P levels were low. The addition of AICAR increased AMPK-P, whereas addition of LPS/IFN-γ alone had no effect on the phosphorylation state of AMPK. When cells were pretreated with AICAR and then stimulated with LPS/IFN-γ, AMPK-P levels were elevated similarly. These studies indicate that AMPK is activated strongly by AICAR, whereas LPS/IFN-γ alone does not affect activation of AMPK.
Fig. 2.
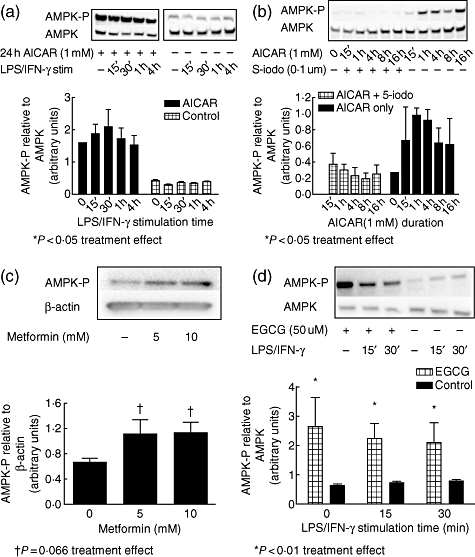
5-amino-4-imidazole carboxamide riboside (AICAR) activates AMP-activated protein kinase (AMPK) in mesangial cells, which is blocked by pretreatment with 5′-iodotubercidin, and AMPK activation is not affected by lipopolysaccharide (LPS)/interferon (IFN)-γ stimulation. (a) Mesangial cells from 8-week-old MRL/lpr mice were pretreated with or without AICAR (1 mM) for 24 h and stimulated with LPS/IFN-γ for 15 min to 4 h. Equal amounts of cellular protein loaded onto gels were measured for AMPK activation by Western blot, using antibodies for AMPK and phospho-(Thr172)-AMPK. (b) When mesangial cells were pretreated with 5′-iodotubercidin (0·1 uM) 30 min prior to the addition of AICAR (1 mM) for 15 min to 16 h, AICAR was unable to activate AMPK. (c) Metformin treatment for 1 h also activated AMPK in mesangial cells, as did epigallocatechin-gallate (EGCG) (50 uM), which was not affected by LPS/IFN-γ stimulation (d). Blots are representative of three experiments and densitometric data are expressed as AMPK-P relative to total AMPK levels. *P < 0·05; †P = 0·066 for effect of treatment.
We also sought to determine if other AMPK activators would have similar effects on AMPK activation in MRL/lpr mesangial cells. We used metformin because of its ability to stimulate AMPK specifically [23]. Our results show that metformin activated AMPK dose-dependently (Fig. 2c). EGCG, a green tea extract reported widely to activate AMPK [24], also effectively activated AMPK, and was not affected by LPS/IFN-γ treatment (Fig. 2d).
5′-Iodotubercidin blocks AMPK activation
To investigate whether the anti-inflammatory effect of AICAR on mesangial cells is mediated via AMPK activation, we used the adenosine kinase inhibitor 5′-iodotubercidin. This compound blocks AMPK activation by preventing the intracellular conversion of AICAR to its active metabolic form, ZMP, by adenosine kinase. Mesangial cells were pretreated with 5′-iodotubericidin (0·1 µM) for 30 min prior to the addition of AICAR (1 mM), and AMPK activation was measured from 15 min to 16 h after AICAR addition (Fig. 2b). Pretreatment with 5′-iodotubercidin resulted in abrogation of AMPK phosphorylation at all times measured.
The AICAR attenuates LPS/IFN-γ-induced iNOS expression through AMPK activation
Because of the fact that AICAR has been shown to inhibit AMP deaminase, leading to the accumulation and subsequent release of adenosine into the extracellular space [25–27], it was essential to determine whether the anti-inflammatory effects of AICAR were attributed to AMPK activation or enhanced adenosine levels. Adenosine plays a major role in the control of renal function by regulating processes such as renal blood flow, glomerular filtration by vasoconstriction of the afferent arteriole, renin secretion and tubuloglomerular feedback, but it can also block inflammation [28,29]. Adenosine is released by the cell either directly through a transporter or as a result of cell damage. Enzymatic-mediated hydrolysis of extracellular adenine nucleotides, such as ATP, adenosine diphosphate (ADP) and AMP, increases extracellular adenosine levels. Just as intracellular adenosine is metabolized rapidly by adenosine kinase to AMP [30], AICAR is metabolized to ZMP. As an AMP analogue, ZMP can mimic the AMPK-activating effects of AMP.
We therefore treated mesangial cells with the adenosine kinase inhibitor 5′-iodotubercidin to prevent AICAR conversion to ZMP and subsequent AMPK activation. It was reasoned that if the anti-inflammatory effects of AICAR were due solely to the activation of AMPK, then the addition of 5′-iodotubercidin would prevent AICAR from blocking inflammatory mediator production. Cells were pretreated with 5′-iodotubericidin (0·1 µM) and different concentrations of AICAR for 16 h prior to 24 h stimulation with LPS/IFN-γ. Inducible-NOS protein levels were measured by Western blot. Stimulation of the cells with LPS/IFN-γ resulted in iNOS expression, and the addition of 5′-iodotubercidin prevented AICAR from attenuating the increase in iNOS protein levels (Fig. 3a), indicating that AICAR-induced reductions in iNOS expression are dependent upon AMPK activation. As an additional confirmation that AMPK activation was responsible for the reduction in iNOS, we also pretreated mesangial cells with compound c (AMPK inhibitor [31]) for 30 min prior to AICAR addition and LPS/IFN-γ stimulation (Fig. 3b). Compound c (20 µM) also prevented AICAR from attenuating iNOS expression, confirming that AMPK activation was responsible for the anti-inflammatory effect of AICAR.
Fig. 3.
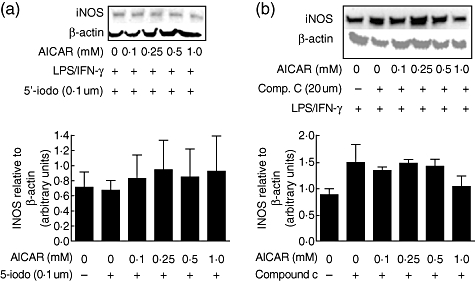
5-amino-4-imidazole carboxamide riboside (AICAR) attenuation of inducible nitric oxide synthase (iNOS) in lipopolysaccharide (LPS)/interferon (IFN)-γ-stimulated mesangial cells is abrogated by inhibition of adenosine kinase using 5′-iodotubercidin. Mesangial cells from 8-week-old MRL/lpr mice were serum starved and pretreated with either 5′-iodotubercidin (0·1 uM) or compound c (20 uM) for 30 min prior to the addition of AICAR for 16 h, then stimulated with LPS/IFN-γ for 24 h (a and b respectively). Cellular protein was collected and equal amounts loaded onto gels for determination of iNOS protein levels by Western blot using anti-iNOS/NOS Type II. Blots are representative of three experiments and densitometric data are expressed relative to β-actin.
The AICAR does not inhibit IκB/NF-κB activation in stimulated mesangial cells
The NF-κB transcription factor complex plays a central role in regulating the inflammatory, immune and anti-apoptotic responses in mammals [32–34]. Of note, LPS and IFN-γ act synergistically to activate the iNOS promoter by stimulating the binding of several transcription factors, including NF-κB, IFN regulatory factor-1 and C/EBP to their respective cognitive sites [35–37]. LPS/IFN-γ induces phosphorylation of IκB and, once phosphorylated, IκB dissociates from the NF-κB complex and is subsequently degraded. The liberated NF-κB translocates rapidly into the nucleus where it engages κB enhancer elements and activates gene expression of inflammatory proteins such as IL-6.
Considering the critical role of NF-κB in regulating iNOS transcription and inflammatory protein production [38], we tested whether AICAR treatment regulated IκB or NF-κB in mesangial cells. Mesangial cell cultures were pretreated with AICAR (1 mM) for 24 h and then stimulated with LPS/IFN-γ for 15 min to 4 h. Cytosolic and nuclear fractions were isolated and IκB, IκB-p and NF-κB protein levels were assessed by Western blot. Twenty-four-hour exposure to AICAR alone did not affect the IκB phosphorylation or nuclear translocation of NF-κB. Subsequent addition of LPS/IFN-γ resulted in increased phosphorylation of IκB (Fig. 4a) and translocation of NF-κB (p-65 subunit) to the nucleus (Fig. 4b) within 30 min of challenge. These results show that AICAR did not mediate LPS/IFN-γ-induced phosphorylation of IκB or NF-κB translocation to the nucleus.
Fig. 4.
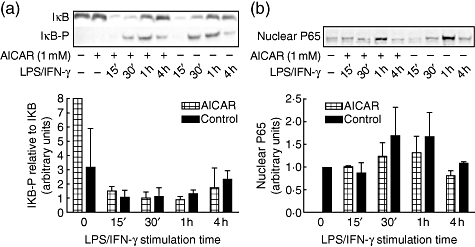
5-amino-4-imidazole carboxamide riboside (AICAR) does not inhibit inhibitory kappa B (IκB) phosphorylation or nuclear factor (NF)-κB translocation in lipopolysaccharide (LPS)/interferon (IFN)-γ-stimulated mesangial cells. Mesangial cells from 8-week-old MRL/lpr mice were pretreated with or without AICAR (1 mM) for 24 h and stimulated with LPS/ IFN-γ for 15 min to 4 h. (a) Cellular protein was assessed for IκB protein and phosphorylated IκB protein expression by Western blot using anti-IκB-α and anti-IκB-α-p. (b) Nuclear protein was collected and assessed for p65 protein expression by Western blot. Blots are representative of three experiments.
The AMPK activation blocks LPS/IFN-γ-induced phosphorylation of Akt
The LPS-induced PI3-kinase/Akt signalling pathway has been shown to play an important role in inflammatory responses. We therefore sought to determine whether AICAR decreased the PI3K/Akt inflammatory pathway triggered by LPS/IFN-γ. Mesangial cells were serum starved, pretreated with AICAR and stimulated with LPS/IFN-γ for 24 h. Akt phosphorylation was measured from 15 min to 4 h after stimulation. Our results show that LPS/IFN-γ treatment induced Akt phosphorylation at ser473 within 15 min of stimulation, while pretreatment with AICAR reduced significantly the phosphorylation of Akt (Fig. 5a). To determine whether AICAR-mediated inhibition of the LPS-induced-PI3K/Akt signalling pathway was through AMPK, the cells were given 5′-iodotubercidin (0·1 uM) 30 min prior to the addition of AICAR (Fig. 5b). Treatment with 5′-iodotubercidin blocked AICAR from inhibiting Akt-P, indicating the importance of AMPK in AICAR regulation of the LPS-induced-PI3K/Akt pathway in mesangial cells.
Fig. 5.
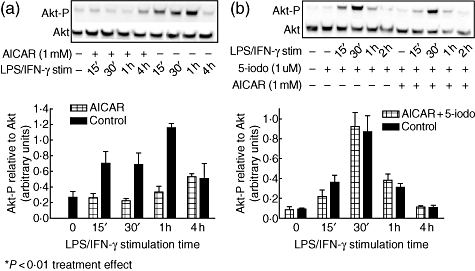
5-amino-4-imidazole carboxamide riboside (AICAR) attenuates Akt-P in lipopolysaccharide (LPS)/interferon (IFN)-γ-stimulated mesangial cells, which is dependent upon AMP-activated protein kinase (AMPK) activation. Serum starved mesangial cells from 8-week-old MRL/lpr mice were pretreated with AICAR (1 mM) for 24 h and then stimulated with LPS/IFN-γ. Cellular protein was assessed for Akt activation by Western blot using Akt and Akt-p (ser473). (a) LPS/IFN-γ stimulation alone induced Akt-p, while AICAR pretreatment attenuated the rise in Akt-P as determined by Western blot (*P < 0·01); (b) when cells were pretreated with 5′-iodotubercidin, AICAR was unable to attenuate the rise in Akt-P. Blots are representative of n = 3 and densitometric data are expressed as Akt-P relative to total Akt.
Next, we sought to determine if other AMPK activators (metformin and EGCG) would have similar effects on blocking Akt activation. Mesangial cells were pretreated with either EGCG or metformin and stimulated with LPS/IFN-γ for various times, and Akt phosphorylation was determined. While EGCG effectively inhibited Akt phosphorylation (Fig. 6a), metformin pretreatment did not decrease significantly LPS/IFN-γ-induced Akt activation (Fig. 6b).
Fig. 6.
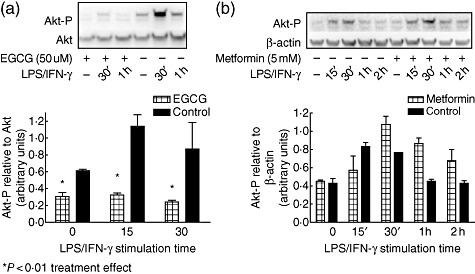
Epigallocatechin-gallate (EGCG) and metformin affect differentially lipopolysaccharide (LPS)/interferon (IFN)-γ-induced activation of Akt. (a) Mesangial cells from 8-week-old MRL/lpr mice were pretreated with EGCG (50 uM) for 1 h and then stimulated for the indicated times with LPS/IFN-γ for assessment of Akt activation. (b) Mesangial cells were pretreated with metformin (5 mM) and stimulated with LPS/IFN-γ for the indicated times and assessed for Akt activation. Blots are representative of three experiments and densitometric data are expressed as Akt-P relative to β-actin. *P < 0·01 for effect of treatment.
Discussion
Several studies have examined the role of AMPK activation in cells using AICAR as an AMPK agonist [39,40]. However, the study of how AMPK activation can reduce the production of inflammatory mediators has been less explored. Recent studies provide evidence that AICAR reduces inflammatory mediators [12,13]. The extent to which AICAR is successful and the mechanism by which it acts appear to be cell type-dependent. For example, Giri et al. reported that AICAR pretreatment abrogated LPS-induced expression of proinflammatory cytokines IL-1β, IL-6 and TNF-α in rat primary astrocytes, microglia and peritoneal macrophages, with AMPK suggested to play an important role [12]. However, in RAW 264·7 macrophages, AICAR's anti-inflammatory effects appeared to be independent of AMPK [13]. These studies led us to test the effects of AICAR on inflammation in cells instrumental in kidney inflammation and disease progression in a mouse model of SLE.
The AICAR is a cell-permeable compound whose phosphorylated metabolite activates AMPK in a variety of cell types [8]. Incubation of rat hepatocytes with the non-phosphorylated AICAR results in accumulation of the monophosphorylated derivative, ZMP [41]. Within the cell, ZMP mimics the activating effects of AMP on AMPK, i.e. direct allosteric activation of phosphotransferase activity of AMPK [42]. AMPK is a heterotrimer consisting of a catalytic α-subunit and non-catalytic β- and α-subunits [43]. Several isoforms of each subunit, termed α1 or α2, β1 or β2 and γ1, γ2 and γ3 are known [43]. AMPK is a multi-substrate protein kinase, which appears to play a central role in response to metabolic stress and regulates numerous functions within the cell [44]. It is thought that the AMPK system evolved to protect cells against ATP depletion by inhibiting biosynthetic pathways while stimulating energy-generating pathways [40]. Unlike other methods for activating AMPK in intact cells, AICAR does not perturb the cellular concentrations of ATP, ADP or AMP [10,45]. We investigated the effects of AICAR-induced AMPK activation on inhibition of the inflammatory signalling cascade in mesangial cells isolated from MRL/lpr mice. We provide evidence that phosphorylation/activation of AMPK by AICAR profoundly inhibits LPS/IFN-γ-stimulated inflammatory mediator production.
The mechanism of AICAR's anti-inflammatory effects has not been elucidated completely, and may depend upon the cell type studied. AICAR is known to increase AMPK activation, but it also increases the pool of intracellular adenosine, an effect that can be enhanced further with administration of the adenosine kinase inhibitor 5′-iodotubercidin. Adenosine mediates a diverse array of biological functions, including heart rate, blood pressure, pain and nerve conduction, in addition to inflammation [29]. In the kidney, adenosine plays a major role in homeostasis and anti-inflammatory effects mediated through its receptor. The adenosine A2A receptor has emerged as a critical regulator of immune function and inflammation and is expressed on cells of haematopoietic origin, including T cells, neutrophils, platelets, macrophages, monocytes, dendritic cells and mesangial cells [46–48].
To distinguish between the anti-inflammatory mechanisms of AICAR mediated through AMPK from those of adenosine, we used the adenosine kinase inhibitor 5′-iodutubercidin. We demonstrated that the activation of AMPK, rather than increased adenosine, is involved in LPS/IFN-γ-mediated iNOS expression in mesangial cells. Similarly, we showed that AICAR treatment and AMPK activation corresponded with an abrogation of the LPS-IFN-γ-induced phosphorylation of Akt. While EGCG, another AMPK activator, effectively inhibited Akt phosphorylation, surprisingly, metformin (another well-characterized activator of AMPK) did not block the LPS/IFN-γ-induced Akt phosphorylation. These results are intriguing, and several possible explanations exist: first, the fact that AMPK can be phosphorylated at different sites, including α-Thr172, α1-Ser 485, α2-Ser491, Ser497 and Ser1179 [49,50]. Differences in sites of phosphorylation of AMPK can affect its ability to regulate other intracellular enzymes [49]. Current studies in our laboratory are aimed at detailing where AMPK is phosphorylated with different activators in order to clarify this issue. It is also possible that the level of AMPK activation following metformin was not sufficient to inhibit LPS/IFN-γ-induced Akt phosphorylation, as Thr172 phosphorylation following metformin did not appear as strong as with EGCG or AICAR. One additional explanation could be that both EGCG and AICAR act not only to induce AMPK activation, but also act on other intracellular proteins to inhibit LPS/IFN-γ-induced Akt phosphorylation. Although this possibility exists, treatment with 5′-iodotubercidin prevented AMPK activation and subsequent LPS/IFN-γ-induced increases in Akt-p were not attenuated by AICAR. These studies suggest that the inhibition of the LPS/IFN-γ-induced PI3K/Akt pathway in MRL/lpr mesangial cells was mediated through AMPK.
The iNOS promoter exhibits homologies to binding sites for numerous transcription factors known to be involved in the LPS-cytokine-mediated induction of transcription. Co-activators that are involved in iNOS promoter activation have been reported to involve the binding of p300 to the iNOS promoter and activation of Akt [51,52]. IL-6 is a multi-functional cytokine that plays a central role in both innate and acquired immune responses, and is the predominant mediator of the acute phase response, an innate immune mechanism which is triggered by infection and inflammation. IL-6 plays multiple roles in the development of acquired immunity against incoming pathogens, including regulation of cytokine and chemokine expression, stimulation of antibody production by B cells and regulation of macrophage and dendritic cell differentiation. IL-6 levels have been shown to be elevated in chronic inflammatory conditions, such as SLE [53,54]. Our studies show that LPS/IFN-γ-induced IL-6 production and iNOS expression are attenuated by the activation of AMPK.
Activation of the IκB/NF-κB pathway has been demonstrated to be involved in LPS-induced iNOS expression and NO release in RAW 264·7 macrophages. In our experiments using AICAR, we did not observe a decrease in LPS-induced IκB phosphorylation or NF-κB translocation. However, it is possible that AMPK affects the ability of NF-κB to induce nuclear transcription. Recent studies indicate that post-translational modification of NF-κB is critical for its transcriptional competence. It is possible that in mesangial cells, AICAR interferes directly with the binding of NF-κB to DNA, as observed recently by Kuo et al. in macrophages and microglial cells [15].
In conclusion, through AMPK activation, AICAR inhibits the LPS/IFN-γ-stimulated PI3K/Akt-mediated pathway, but does not inhibit significantly NF-κB translocation to the nucleus in MRL/lpr mesangial cells. These studies delineate, in part, the signal transduction pathways modulated by AMPK activation and show that AMPK activation can inhibit inflammatory mediator production in stimulated cells. Taken together, these studies show that AICAR acts as an anti-inflammatory agent by activating AMPK and suggest that targeting AMPK specifically may reduce chronic inflammation in diseases such as SLE.
Acknowledgments
This work was supported by the Arthritis Foundation.
Disclosure
None.
References
- 1.Kashem A, Endoh M, Yano N, Yamauchi F, Nomoto Y, Sakai H. Expression of inducible-NOS in human glomerulonephritis: the possible source is infiltrating monocytes/macrophages. Kidney Int. 1996;50:392–9. doi: 10.1038/ki.1996.328. [DOI] [PubMed] [Google Scholar]
- 2.Cook HT, Smith J, Salmon JA, Cattell V. Functional characteristics of macrophages in glomerulonephritis in the rat. O2- generation, MHC class II expression, and eicosanoid synthesis. Am J Pathol. 1989;134:431–7. [PMC free article] [PubMed] [Google Scholar]
- 3.Cattell V, Jansen A. Inducible nitric oxide synthase in inflammation. Histochem J. 1995;27:777–84. [PubMed] [Google Scholar]
- 4.Farrell AJ, Blake DR, Palmer RM, Moncada S. Increased concentrations of nitrite in synovial fluid and serum samples suggest increased nitric oxide synthesis in rheumatic diseases. Ann Rheum Dis. 1992;51:1219–22. doi: 10.1136/ard.51.11.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cattell V. Nitric oxide – potential mediator in glomerulonephritis? Nephrol Dial Transplant. 1995;10:759–61. Editorial. [PubMed] [Google Scholar]
- 6.Cattell V. Nitric oxide and glomerulonephritis. Semin Nephrol. 1999;19:277–87. [PubMed] [Google Scholar]
- 7.Suryaprabha P, Das UN, Ramesh G, Kumar KV, Kumar GS. Reactive oxygen species, lipid peroxides and essential fatty acids in patients with rheumatoid arthritis and systemic lupus erythematosus. Prostaglandins Leukot Essent Fatty Acids. 1991;43:251–5. doi: 10.1016/0952-3278(91)90038-7. [DOI] [PubMed] [Google Scholar]
- 8.Ruderman NB, Park H, Kaushik VK, et al. AMPK as a metabolic switch in rat muscle, liver and adipose tissue after exercise. Acta Physiol Scand. 2003;178:435–42. doi: 10.1046/j.1365-201X.2003.01164.x. [DOI] [PubMed] [Google Scholar]
- 9.Whitehead JP, Richards AA, Hickman IJ, Macdonald GA, Prins JB. Adiponectin – a key adipokine in the metabolic syndrome. Diabetes Obes Metab. 2006;8:264–80. doi: 10.1111/j.1463-1326.2005.00510.x. [DOI] [PubMed] [Google Scholar]
- 10.Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J Biol Chem. 2006;281:32207–16. doi: 10.1074/jbc.M606357200. [DOI] [PubMed] [Google Scholar]
- 11.Levine YC, Li GK, Michel T. Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells. Evidence for an AMPK -> Rac1 -> Akt -> endothelial nitric-oxide synthase pathway. J Biol Chem. 2007;282:20351–64. doi: 10.1074/jbc.M702182200. [DOI] [PubMed] [Google Scholar]
- 12.Giri S, Nath N, Smith B, Viollet B, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci. 2004;24:479–87. doi: 10.1523/JNEUROSCI.4288-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jhun BS, Jin Q, Oh YT, et al. 5-Aminoimidazole-4-carboxamide riboside suppresses lipopolysaccharide-induced TNF-alpha production through inhibition of phosphatidylinositol 3-kinase/Akt activation in RAW 264·7 murine macrophages. Biochem Biophys Res Commun. 2004;318:372–80. doi: 10.1016/j.bbrc.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 14.Jin Q, Jhun BS, Lee SH, et al. Differential regulation of phosphatidylinositol 3-kinase/Akt, mitogen-activated protein kinase, and AMP-activated protein kinase pathways during menadione-induced oxidative stress in the kidney of young and old rats. Biochem Biophys Res Commun. 2004;315:555–61. doi: 10.1016/j.bbrc.2004.01.093. [DOI] [PubMed] [Google Scholar]
- 15.Kuo C-L, Ho F-M, Chang MY, Prakash E, Lin W-W. Inhibition of lipopolysaccharide-induced inducible nitric oxide synthase and cyclooxygenase-2 gene expression by 5-aminoimidazole-4-carboxamide riboside is independent of AMP-activated protein kinase. J Cell Biochem. 2008;103:931–40. doi: 10.1002/jcb.21466. [DOI] [PubMed] [Google Scholar]
- 16.Reilly CM, Oates JC, Sudian J, Crosby MB, Halushka PV, Gilkeson GS. Prostaglandin J(2) inhibition of mesangial cell iNOS expression. Clin Immunol. 2001;98:337–45. doi: 10.1006/clim.2000.4985. [DOI] [PubMed] [Google Scholar]
- 17.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 18.Gilkeson G, Cannon C, Oates J, Reilly C, Goldman D, Petri M. Correlation of serum measures of nitric oxide production with lupus disease activity. J Rheumatol. 1999;26:318–24. [PubMed] [Google Scholar]
- 19.Huang Y, Li T, Sane DC, Li L. IRAK1 serves as a novel regulator essential for lipopolysaccharide-induced interleukin-10 gene expression. J Biol Chem. 2004;279:51697–703. doi: 10.1074/jbc.M410369200. [DOI] [PubMed] [Google Scholar]
- 20.al-Janadi M, al-Balla S, al-Dalaan A, Raziuddin S. Cytokine profile in systemic lupus erythematosus, rheumatoid arthritis, and other rheumatic diseases. J Clin Immunol. 1993;13:58–67. doi: 10.1007/BF00920636. [DOI] [PubMed] [Google Scholar]
- 21.Furusu A, Miyazaki M, Abe K, et al. Expression of endothelial and inducible nitric oxide synthase in human glomerulonephritis. Kidney Int. 1998;53:1760–8. doi: 10.1046/j.1523-1755.1998.00907.x. [DOI] [PubMed] [Google Scholar]
- 22.Tomasoni S, Noris M, Zappella S, et al. Upregulation of renal and systemic cyclooxygenase-2 in patients with active lupus nephritis. J Am Soc Nephrol. 1998;9:1202–12. doi: 10.1681/ASN.V971202. [DOI] [PubMed] [Google Scholar]
- 23.Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase – development of the energy sensor concept. J Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moon H-S, Lee H-G, Choi Y-J, Kim T-G, Cho C-S. Proposed mechanisms of (-)-epigallocatechin-3-gallate for anti-obesity. Chem Biol Interact. 2007;167:85–98. doi: 10.1016/j.cbi.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Beckers A, Organe S, Timmermans L, et al. Methotrexate enhances the antianabolic and antiproliferative effects of 5-aminoimidazole-4-carboxamide riboside. Mol Cancer Ther. 2006;5:2211–17. doi: 10.1158/1535-7163.MCT-06-0001. [DOI] [PubMed] [Google Scholar]
- 26.McGuire JJ, Haile WH, Yeh CC. 5-amino-4-imidazolecarboxamide riboside potentiates both transport of reduced folates and antifolates by the human reduced folate carrier and their subsequent metabolism. Cancer Res. 2006;66:3836–44. doi: 10.1158/0008-5472.CAN-05-3226. [DOI] [PubMed] [Google Scholar]
- 27.Cronstein BN. Low-dose methotrexate: a mainstay in the treatment of rheumatoid arthritis. Pharmacol Rev. 2005;57:163–72. doi: 10.1124/pr.57.2.3. [DOI] [PubMed] [Google Scholar]
- 28.Martinez-Salgado C, Garcia-Cenador B, Fuentes-Calvo I, Macias Nunez JF, Lopez-Novoa JM. Effect of adenosine in extracellular matrix synthesis in human and rat mesangial cells. Mol Cell Biochem. 2007;305:163–9. doi: 10.1007/s11010-007-9540-4. [DOI] [PubMed] [Google Scholar]
- 29.Gallos G, Ruyle TD, Emala CW, Lee HT. A1 adenosine receptor knockout mice exhibit increased mortality, renal dysfunction, and hepatic injury in murine septic peritonitis. Am J Physiol Renal Physiol. 2005;289:F369–76. doi: 10.1152/ajprenal.00470.2004. [DOI] [PubMed] [Google Scholar]
- 30.Tostes RC, Giachini FR, Carneiro FS, Leite R, Inscho EW, Webb RC. Determination of adenosine effects and adenosine receptors in murine corpus cavernosum. J Pharmacol Exp Ther. 2007;322:678–85. doi: 10.1124/jpet.107.122705. [DOI] [PubMed] [Google Scholar]
- 31.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Vera ME, Taylor BS, Wang Q, Shapiro RA, Billiar TR, Geller DA. Dexamethasone suppresses iNOS gene expression by upregulating I-kappa B alpha and inhibiting NF-kappa B. Am J Physiol. 1997;273:G1290–6. doi: 10.1152/ajpgi.1997.273.6.G1290. [DOI] [PubMed] [Google Scholar]
- 33.Eberhardt W, Pluss C, Hummel R, Pfeilschifter J. Molecular mechanisms of inducible nitric oxide synthase gene expression by IL-1beta and cAMP in rat mesangial cells. J Immunol. 1998;160:4961–9. [PubMed] [Google Scholar]
- 34.Grassl C, Luckow B, Schlondorff D, Dendorfer U. Transcriptional regulation of the interleukin-6 gene in mesangial cells. J Am Soc Nephrol. 1999;10:1466–77. doi: 10.1681/ASN.V1071466. [DOI] [PubMed] [Google Scholar]
- 35.Kinugawa K, Shimizu T, Yao A, Kohmoto O, Serizawa T, Takahashi T. Transcriptional regulation of inducible nitric oxide synthase in cultured neonatal rat cardiac myocytes. Circ Res. 1997;81:911–21. doi: 10.1161/01.res.81.6.911. [DOI] [PubMed] [Google Scholar]
- 36.Futaki M, Inokuchi K, Hanawa H, Tanosaki S, Dan K, Nomura T. Possible transforming activity of interferon regulatory factor 2 in tumorigenicity assay of NIH3T3 cells transfected with DNA from chronic myelogenous leukemia patients. Leuk Res. 1996;20:601–5. doi: 10.1016/0145-2126(96)00013-6. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Z, Qian Y, Wald D, Xia YF, Geng JG, Li X. IFN regulatory factor-1 is required for the up-regulation of the CD40-NF-kappa B activator 1 axis during airway inflammation. J Immunol. 2003;170:5674–80. doi: 10.4049/jimmunol.170.11.5674. [DOI] [PubMed] [Google Scholar]
- 38.Fu Y, Xie C, Yan M, et al. The lipopolysaccharide-triggered mesangial transcriptome: evaluating the role of interferon regulatory factor-1. Kidney Int. 2005;67:1350–61. doi: 10.1111/j.1523-1755.2005.00212.x. [DOI] [PubMed] [Google Scholar]
- 39.Baumann P, Mandl-Weber S, Emmerich B, Straka C, Schmidmaier R. Activation of adenosine monophosphate activated protein kinase inhibits growth of multiple myeloma cells. Exp Cell Res. 2007;313:3592–603. doi: 10.1016/j.yexcr.2007.06.020. [DOI] [PubMed] [Google Scholar]
- 40.Kim J, Yoon MY, Choi SL, et al. Effects of stimulation of AMP-activated protein kinase on insulin-like growth factor 1- and epidermal growth factor-dependent extracellular signal-regulated kinase pathway. J Biol Chem. 2001;276:19102–10. doi: 10.1074/jbc.M011579200. [DOI] [PubMed] [Google Scholar]
- 41.Sabina RL, Patterson D, Holmes EW. 5-Amino-4-imidazolecarboxamide riboside (Z-riboside) metabolism in eukaryotic cells. J Biol Chem. 1985;260:6107–14. [PubMed] [Google Scholar]
- 42.Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-Aminoimidazole-4-carboxamide ribonucleoside, a specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–65. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- 43.Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- 44.Baron SJ, Li J, Russell RR, III, et al. Dual mechanisms regulating AMPK kinase action in the ischemic heart. Circ Res. 2005;96:337–45. doi: 10.1161/01.RES.0000155723.53868.d2. [DOI] [PubMed] [Google Scholar]
- 45.Winder WW. AMP-activated protein kinase: possible target for treatment of type 2 diabetes. Diabetes Technol Ther. 2000;2:441–8. doi: 10.1089/15209150050194305. [DOI] [PubMed] [Google Scholar]
- 46.Lange-Sperandio B, Forbes MS, Thornhill B, Okusa MD, Linden J, Chevalier RL. A2A adenosine receptor agonist and PDE4 inhibition delays inflammation but fails to reduce injury in experimental obstructive nephropathy. Nephron Exp Nephrol. 2005;100:e113–23. doi: 10.1159/000085057. [DOI] [PubMed] [Google Scholar]
- 47.Awad AS, Huang L, Ye H, et al. A2A receptor activation attenuates inflammation and injury in diabetic nephropathy. Am J Physiol Renal Physiol. 2006;290:F828–37. doi: 10.1152/ajprenal.00310.2005. [DOI] [PubMed] [Google Scholar]
- 48.Dubey RK, Gillespie DG, Mi Z, Jackson EK. Adenosine inhibits PDGF-induced growth of human glomerular mesangial cells via A(2B) receptors. Hypertension. 2005;46:628–34. doi: 10.1161/01.HYP.0000178464.63393.88. [DOI] [PubMed] [Google Scholar]
- 49.Hurley RL, Barre LK, Wood SD, et al. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J Biol Chem. 2006;281:36662–72. doi: 10.1074/jbc.M606676200. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y, Lee T-S, Kolb EM, et al. AMP-activated protein kinase is involved in endothelial NO synthase activation in response to shear stress. Arterioscler Thromb Vasc Biol. 2006;26:1281–7. doi: 10.1161/01.ATV.0000221230.08596.98. [DOI] [PubMed] [Google Scholar]
- 51.Torchia J, Rose DW, Inostroza J, et al. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature. 1997;387:677–84. doi: 10.1038/42652. see Comments. [DOI] [PubMed] [Google Scholar]
- 52.Giri S, Rattan R, Singh AK, Singh I. The 15-deoxy-delta12,14-prostaglandin J2 inhibits the inflammatory response in primary rat astrocytes via down-regulating multiple steps in phosphatidylinositol 3-kinase-Akt-NF-kappaB-p300 pathway independent of peroxisome proliferator-activated receptor gamma. J Immunol. 2004;173:5196–208. doi: 10.4049/jimmunol.173.8.5196. [DOI] [PubMed] [Google Scholar]
- 53.Akaogi J, Yamada H, Kuroda Y, Nacionales DC, Reeves WH, Satoh M. Prostaglandin E2 receptors EP2 and EP4 are up-regulated in peritoneal macrophages and joints of pristane-treated mice and modulate TNF-alpha and IL-6 production. J Leukoc Biol. 2004;76:227–36. doi: 10.1189/jlb.1203627. [DOI] [PubMed] [Google Scholar]
- 54.Sawada T, Falk LA, Rao P, Murphy WJ, Pluznik DH. IL-6 induction of protein-DNA complexes via a novel regulatory region of the inducible nitric oxide synthase gene promoter: role of octamer binding proteins. J Immunol. 1997;158:5267–76. [PubMed] [Google Scholar]