

Abstract
The relationship between allergic airway inflammation and pneumococcal pneumonia is not well understood. We assessed susceptibility to experimental pneumococcal pneumonia in mice with and without allergic airway inflammation. Susceptibility to pneumococcal pneumonia was evaluated by challenging mice with a bioluminescent Streptococcus pneumoniae strain after sensitization with ovalbumin (OVA), with subsequent monitoring of pneumococcal infection using real-time photonic imaging. Of 46 OVA-sensitized mice challenged with pneumococci, 13 (28%) developed imaging findings consistent with pneumococcal pneumonia. In comparison, 28 (57%) of 49 non-sensitized control mice developed pneumococcal pneumonia (P = 0·005). While none of the control group developed meningitis (0%, none of 28), two mice in the OVA-sensitized group developed meningitis (15·4%, two of 13) (P = 0·09). The mean bacterial count in the lung was significantly lower in the OVA-sensitized than the non-sensitized group (8·26 ± 0·69 versus 9·21 ± 0·67 log10 colony-forming units (CFU)/g, P = 0·002). There was a trend towards the mean bacterial count in the spleen being higher in the OVA-sensitized versus the non-sensitized group (8·14 ± 0·89 versus 7·45 ± 1·07 log10 CFU/g, P = 0·071). A high level of interleukin (IL)-4 in lung homogenates was associated with risk of pneumococcal infection independent of sensitization with OVA (odds ratio: 49·7, 95% confidence interval 2·92-846·5, per increment of 1·0 pg/ml). In the murine model studied, acute allergic airway inflammation reduced susceptibility to pneumococcal pneumonia. IL-4 may increase the risk of pneumococcal pneumonia independently of allergic airway inflammation.
Keywords: Allergy, asthma, pneumonia, Streptococcus pneumoniae
Introduction
Despite decreasing rates of invasive pneumococcal disease (IPD) caused by vaccine-serotypes in the United States [1–3], Streptococcus pneumoniae continues to present a global threat associated with substantial morbidity and mortality. Neither natural disease nor vaccination provide complete or life-long immunity against IPD. One million children younger than 5 years of age die of IPD (including pneumonia) globally each year [4]. In the United States, the annual number of fatal pneumococcal infections is 40 000 [5]. Nasopharyngeal colonization (present in 20–50% of the population) is a prelude to IPD; 100 000 cases of pneumococcal pneumonia, 60 000 cases of sepsis and 3300 cases of meningitis occur each year in the United States. The reported case-fatality rate of IPD is 10% [6]. Talbot et al. reported recently that asthma is associated with an increased risk of IPD among Medicaid recipients of the state of Tennessee [7]. This study finding was confirmed independently in our recent study in Rochester, Minnesota [8]. Higher rates of pneumococcal nasopharyngeal colonization as well as sinusitis and otitis media have been reported among individuals with asthma [9,10]. The Advisory Committee on Immunization Practices has recommended recently that adults with asthma receive the pneumococcal vaccine [11]. The mechanisms underlying the above observations are unknown. T helper type 2 (Th2)-predominant immune responses to various environmental stimuli play a role in the development and exacerbation of atopic diseases, including asthma [12,13]. Numerous studies demonstrate that Th2 cytokines [e.g. interleukin (IL)-4)] down-regulate Th1 functions; this has been suggested to be associated with susceptibility to and severity of protozoal [14,15], mycoplasmal [16,17], bacterial [18–21], viral [22–24] and candidal infections [25]. Despite evidence that Th2 cytokines increase susceptibility to these infections, whether Th2 immune responses (i.e. increased Th2 cytokines or reciprocal counter-regulation of Th1 cytokines) influence susceptibility to IPD is unknown.
We hypothesized that mice sensitized and challenged with ovalbumin (OVA) would be more likely to develop pneumococcal pneumonia, compared with control mice, and that Th2 cytokines would increase susceptibility to pneumococcal pneumonia. To test these hypotheses, we assessed susceptibility to pneumococcal pneumonia by challenging mice with a bioluminescent S. pneumoniae strain after sensitization with OVA and monitored pneumococcal infection using real-time photonic imaging [26,27]. We also examined the roles of Th1 and Th2 cytokines in lung homogenates in relation to the risk of pneumococcal pneumonia.
Materials and methods
Experimental animals
Pathogen-free 6–8-week-old female BALB/c mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). All animal experiments were performed in accordance with guidelines of the Institutional Animal Care and Use Committee at Mayo Clinic (Rochester, MN, USA). After bacterial challenge, each group of animals was kept isolated from the others in a biohazard containment facility.
Allergen sensitization and challenge
The sensitization and challenge procedures with OVA were modified from the method described by Zhang et al.[28], as reported in our previous study [29]. Briefly, experimental mice were sensitized by intraperitoneal (i.p.) injection of 20 µg OVA adsorbed to 1 mg aluminium hydroxide gel (in a volume of 100 µl) on days 0 and 7. Experimental mice were challenged intranasally with 100 µg OVA in 50 µl phosphate-buffered saline (PBS) under light tribromoethanol anaesthesia on days 15, 16 and 17. Control mice received i.p. injection of PBS with aluminium hydroxide gel but no intranasal challenge.
Bacterial concentrate
The S. pneumoniae A 66·1 serotype 3, made bioluminescent by integration of a modified lux operon into its chromosome (Xen 10, Caliper Life Sciences, Hopkinton, MA, USA), was studied [27]. The strain was incubated in Todd-Hewitt broth at 37°C in 5% CO2 until an optical density similar to a McFarland turbidity standard of 1 was reached. Bacterial concentrations were estimated spectrophotometrically by absorbance at 620 nm and diluted using PBS. The exact number of colony-forming units (CFU) of inoculum was determined retrospectively by culture of serial dilutions of inoculum on blood agar plates.
Pneumococcal pneumonia model
Pneumococcal infection was defined as the presence of bioluminescence in and positive pneumococcal culture from normally sterile body tissues. Three days after OVA challenge, all mice (both the OVA-sensitized/challenged group and the non-sensitized/challenged group) were inoculated intranasally with a predetermined dose (1·5 × 104 – 3 × 104 CFU) of S. pneumoniae in 30 µl of PBS under anaesthesia [i.p. injection of ketamine (100 mg/kg) plus xylazine (10 mg/kg)]. In preliminary experiments, it was determined that this was the lowest dose of S. pneumoniae that established pneumococcal pneumonia in approximately 50% of challenged mice. After inoculation, we measured luminescence daily for 7 days using a Lumazone Imaging System (1002FE series; Roper Scientific, Tucson, AZ, USA). Animals were sedated with ketamine plus xylazine, placed in an imaging box without restraint, and imaged for a maximum of 5 min at 4 × 4 binning resolution. Initial development of pneumococcal infection was determined by detection of bioluminescence (Fig. 1). Animals that developed bioluminescence (i.e. evidence of pneumococcal infection) were killed and luminescent tissues (i.e. lung or spleen) were cultured for S. pneumoniae. For animals that died before detection of bioluminescence, lung and spleen tissues were harvested and cultured for S. pneumoniae.
Fig. 1.
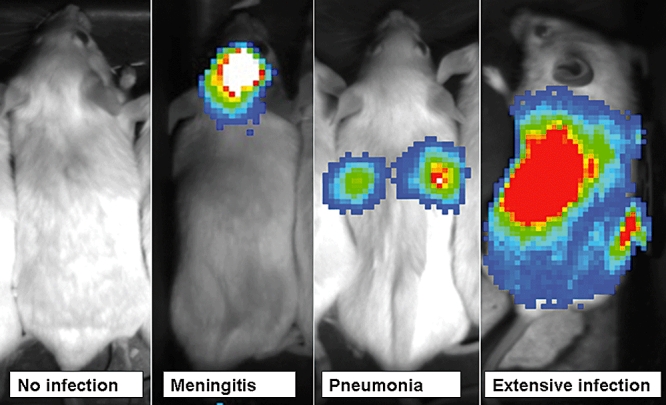
Development of pneumococcal infection after intranasal challenge with Streptococcus pneumoniae.
Animals that did not develop bioluminescence by day 7 after intranasal challenge were killed at 7 days, and then nasal lavage fluid and lung tissue were harvested and cultured for S. pneumoniae.
Killing and tissue processing
Mice were killed by i.p. injection of a lethal dose of pentobarbital (100 mg/kg). The chest and abdomen were opened and the lung and spleen were harvested aseptically. The lung and spleen were weighed and homogenized in a Stomacher 80 with 2 ml PBS; the homogenate was cultured quantitatively. Serial 10-fold dilutions in PBS were placed on blood agar plates (0·1 ml aliquot per plate) and incubated for 24 h at 37°C in 5% CO2. Tissue culture results were expressed as CFU of bacteria per gram of lung or spleen.
Nasal lavage culture
For animals that did not develop bioluminescence by day 7 after intranasal challenge, nasal lavage fluid was harvested and cultured for S. pneumoniae. With the mice sedated, nasal lavage with 100 µl of PBS was performed. Fifty µl of lavage fluid was streaked across a blood agar plate. The S. pneumoniae colony count was measured and classified as follows; <10 represented few colonies, 10–100 moderate colonies and >100 many colonies.
Histopathological evaluation of lung tissues
To ascertain the presence of allergic airway inflammation in the OVA-sensitized/challenged mice, histopathological evaluation of lung tissues was performed in eight mice. Lung sections stained with haematoxylin and eosin to assess inflammatory cell infiltrate or with periodic acid Schiff to examine hypersecretion of mucin were examined by light microscopy.
Cytokine analysis for lung homogenates
The lung tissues of mice at the time of death or pneumococcal infection, or those of mice without pneumococcal infection at day 7, were homogenized in a Stomacher 80 with 2 ml of PBS and frozen at −20°C for subsequent cytokine analysis. Cytokine levels in lung homogenates were determined by Bio-Plex assay (Bio-Rad Laboratories, Hercules, CA, USA), according to the manufacturer's instructions. IL-2, IL-4 and IL-5 concentrations below the described detection limit of the assay were extrapolated beyond the standard range.
Outcome measures and statistical analysis
We compared the proportions of mice that developed pneumococcal pneumonia after intranasal challenge with S. pneumoniae between OVA-sensitized/challenged versus non-sensitized/challenged mice. In addition, we compared the quantitative culture results (expressed as log10 CFU per gram of lung and spleen) between the same groups of mice. To assess the roles of Th2 cytokines in the risk of pneumococcal pneumonia, independent of sensitization, we compared Th2 cytokine levels between mice with and without pneumococcal pneumonia. To examine whether the impact of sensitization and challenge with OVA on the risk of pneumococcal infection is mediated through Th2-predominant immune responses (i.e. Th2 cytokines), data were fitted to a logistic regression. We calculated odds ratios (ORs) and corresponding 95% confidence intervals for sensitization status, Th2 cytokine data (continuous variable) and the interaction term between sensitization and cytokine data in predicting the risk of pneumococcal pneumonia. The Mann–Whitney U-test or Student's t-test was used to compare continuous variables depending on the distribution, and the χ2 or Fisher's exact test was used to compare categorical variables. The Kaplan–Meier method was used for analysis of development of pneumococcal infection. All statistical significance was tested at a two-tailed alpha error of 0·05.
Results
Nasal colonization with S. pneumoniae
Mice that had no bioluminescent evidence of pneumococcal infection by day 7 after intranasal challenge with pneumococci did not become ill and bacterial cultures of their lung tissues revealed fewer than 5 × 103 CFU/g. Nasal lavage fluid of mice that did not develop bioluminescent evidence of pneumococcal infection was cultured for S. pneumoniae. A higher percentage of nasal lavages yielded growth of ‘moderate’ or ‘many’ pneumococci in the non-sensitized control mice than in the OVA-sensitized/challenged mice [81·0% (17 of 21) versus 53·1% (17 of 32), P = 0·046].
Development of pneumococcal pneumonia
Lumazone imaging showed pneumonia or meningitis by real-time in vivo imaging in affected mice. Of the 46 OVA-sensitized/challenged mice, 13 (28·3%) developed pneumococcal pneumonia after challenge with pneumococci whereas 28 (57·1%) of 49 control mice developed pneumococcal pneumonia (P = 0·005) (Fig. 2). While none of the control group developed meningitis (0%, none of 28), two mice in the OVA-sensitized/challenged group developed meningitis (15·4%, two of 13) (P = 0·09). The mice with meningitis also had lung and spleen cultures positive for S. pneumoniae. The mean bacterial count in the lung was significantly lower in the OVA-sensitized/challenged group than in the control group (8·26 ± 0·69 versus 9·21 ± 0·67 log10 CFU/g, P = 0·002). There was a trend towards the mean bacterial count in the spleen being higher in the OVA-sensitized/challenged group than in the control group (8·14 ± 0·89 versus 7·45 ± 1·07 log10 CFU/g, P = 0·071).
Fig. 2.
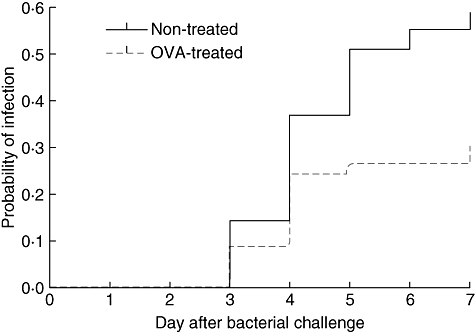
Kaplan–Meier analysis of development of pneumococcal pneumonia in ovalbumin (OVA)-treated versus non-treated control mice challenged with pneumococci (P = 0·022). The OVA-treated and control mice were challenged intranasally with pneumococci and followed for the development of pneumococcal infection for 7 days.
Histopathological assessment of lung tissue
By light microscopy, marked cellular infiltration around the airways and pulmonary blood vessels was seen in the lung interstitium of the OVA-sensitized/challenged mice compared with non-sensitized control mice (Fig. 3a and b respectively). In addition, hyperplasia of airway goblet cells and hypersecretion of mucin were observed in the OVA-sensitized/challenged but not in the control mice (Fig. 3c and d respectively). Airway mucin hypersecretion was prominent in the OVA-sensitized/challenged mice with pneumonia, whereas there was scant mucin production in the airway of the control mice with pneumonia (Fig. 3e and f respectively).
Fig. 3.
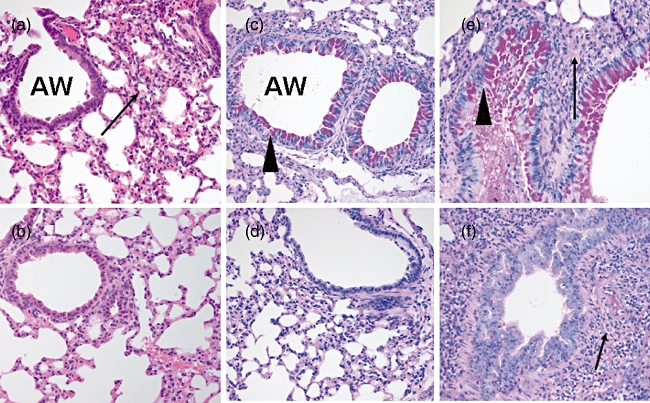
Lung tissues stained with haematoxylin and eosin and examined by light microscopy (a,b). (a) The ovalbumin (OVA)-sensitized/challenged mice exhibited marked infiltration of eosinophils and other inflammatory cells (arrows) in the interstitium of the lungs. The airways were surrounded by dense cellular infiltrate. (b) The control mice had airways of normal appearance. Inflammatory cell infiltration was absent in the lung interstitium. The lung tissues were stained with periodic acid Schiff and examined by light microscopy (c–f). (c) Airway goblet cell hyperplasia and mucin hypersecretion (arrowheads) were observed in OVA-sensitized/challenged mice. (d) Airway mucin was scant in the control mice. (e) Alveolar consolidation (arrow) and mucin hypersecretion (arrowhead) were observed in the OVA-sensitized/challenged mice with pneumococcal pneumonia. (f) Only alveolar consolidation (arrow) was observed and airway mucin was scant in the control mice with pneumococcal pneumonia.
Cytokine analysis of lung homogenates
Cytokine and chemokine analyses of the lung homogenates of the OVA-sensitized/challenged mice and the control mice are summarized in Table 1. The results suggest no significant Th2-predominant immune responses in the OVA-sensitized/challenged mice compared with the control group. The relationship between cytokines/chemokines and pneumococcal pneumonia was assessed (Table 1). Mice with pneumococcal pneumonia had higher Th2 cytokines (IL-4, IL-10 and IL-13) and Th1 cytokines [IL-12, interferon (IFN)-γ and tumour necrosis factor (TNF)-α] except IL-2, which was lower, compared with mice without pneumococcal pneumonia. Because of the inconsistent relationship between sensitization status and cytokine patterns and the association between Th2 cytokines and pneumococcal pneumonia, we assessed whether the impact of sensitization status on pneumococcal pneumonia was independent of Th2 cytokines or mediated through Th2 cytokines by fitting data to a logistic regression model (Table 2). The results suggest that an elevated IL-4 (a Th2 cytokine) level is associated with risk of pneumococcal pneumonia, independent of sensitization and its interaction with sensitization. Sensitization with OVA became insignificant in association with the risk of pneumococcal pneumonia when controlled for IL-4 levels and the interaction between IL-4 and sensitization. Similar results were observed controlling for IL-10. An elevated IFN-γ (a Th1 cytokine) level was associated with risk of pneumococcal pneumonia, but the impact was quantitatively insignificant. Sensitization status was no longer significant in predicting the risk of pneumococcal pneumonia and similar results emerged when controlled for other Th1 cytokines (i.e. IL-12, TNF-α) and chemokines [i.e. macrophage inflammatory protein (MIP)-1α, MIP-1β, eotaxin and regulated upon activation normal T cell expressed and secreted (RANTES) (data not shown)].
Table 1.
Comparison of cytokines and chemokines between the ovalbumin-treated mice and non-treated mice, and the mice with and without pneumococcal pneumonia.
| Ovalbumin-treated mice | Non-treated mice | Mice with pneumococcal pneumonia | Mice without pneumococcal pneumonia | P | ||
|---|---|---|---|---|---|---|
| IL-1α | 23·7 ± 8·5 | 111·0 ± 19·4 | <0·001 | 135·0 ± 19·7 | 7·2 ± 0·6 | <0·001 |
| IL-1β | 83·1 ± 34·1 | 331·3 ± 61·1 | <0·001 | 414·0 ± 65·9 | 23·7 ± 2·1 | <0·001 |
| IL-2 | 0·6 ± 0·1 | 0·2 ± 0·1 | <0·001 | 0·3 ± 0·1 | 0·5 ± 0·1 | 0·025 |
| IL-4 | 0·8 ± 0·1 | 0·9 ± 0·1 | 0·801 | 1·1 ± 0·1 | 0·7 ± 0·1 | 0·002 |
| IL-5 | 0·9 ± 0·3 | 0·9 ± 0·3 | 0·913 | 1·1 ± 0·2 | 0·8 ± 0·3 | 0·424 |
| IL-6 | 56·6 ± 43·9 | 289·9 ± 62·8 | 0·003 | 363·1 ± 76·0 | 4·2 ± 0·8 | <0·001 |
| IL-10 | 8·6 ± 3·4 | 111·6 ± 29·7 | <0·001 | 124·3 ± 30·5 | 2·1 ± 0·4 | <0·001 |
| IFN-γ | 11·5 ± 1·2 | 23·6 ± 5·2 | 0·022 | 27·3 ± 5·2 | 8·9 ± 1·1 | 0·001 |
| TNF-α | 2·6 ± 0·7 | 11·6 ± 3·3 | 0·008 | 12·5 ± 3·4 | 2·2 ± 0·7 | 0·002 |
| GM-CSF | 5·9 ± 0·9 | 11·6 ± 1·9 | 0·006 | 14·7 ± 1·8 | 3·5 ± 0·5 | <0·001 |
| IL-12 | 21·4 ± 1·9 | 59·8 ± 12·1 | 0·002 | 61·6 ± 12·4 | 21·5 ± 2·5 | 0·001 |
| IL-13 | 16·4 ± 1·8 | 23·1 ± 3·2 | 0·064 | 29·5 ± 3·0 | 11·2 ± 1·3 | <0·001 |
| MIP-1α | 123·2 ± 38·3 | 803·8 ± 147·4 | <0·001 | 930·9 ± 146·2 | 44 ± 5·4 | <0·001 |
| MIP-1β | 53·6 ± 22·3 | 741·1 ± 134·1 | <0·001 | 823·3 ± 134·0 | 12·8 ± 2·5 | <0·001 |
| Eotaxin | 97·0 ± 17·7 | 158·5 ± 26·7 | 0·055 | 199·3 ± 27·6 | 65·0 ± 12·9 | <0·001 |
| RANTES | 456·5 ± 42·9 | 1474·7 ± 230·5 | <0·001 | 1613·2 ± 229·6 | 381·9 ± 35·0 | <0·001 |
Data are presented as mean ± standard error (pg/ml). The lung tissues of mice at the time of death or pneumococcal pneumonia or those of mice without pneumococcal pneumonia at day 7 were homogenized with 2 ml of phosphate-buffered saline and frozen at −20°C for subsequent cytokine analysis. Cytokine levels in lung homogenates were determined by Bio-Plex assay. IL, interleukin; IFN, interferon; TNF, tumour necrosis factor; GM-CSF, granulocyte–macrophage colony-stimulating factor; MIP, macrophage inflammatory protein; RANTES, regulated upon activation normal T cell expressed and secreted.
Table 2.
Logistic regression models assessing the impact of each variable on the risk of pneumococcal pneumonia.
| Variables | OR (95% CI) | P-value |
|---|---|---|
| Th2 cytokine and sensitization status | ||
| IL-4 (per an increment of 1·0 pg/ml) | 49·7 (2·92–846·5) | 0·007 |
| Sensitization with OVA | 1·12 (0·18–7·12) | 0·903 |
| Interaction term between IL-4 and sensitization status | 0·03 (0·00–0·65) | 0·025 |
| Th1 cytokine and sensitization status | ||
| IFN-γ (per an increment of 1·0 pg/ml) | 1·14 (1·04–1·26) | 0·005 |
| Sensitization with OVA | 0·48 (0·09–2·62) | 0·396 |
| Interaction term between IFN-γ and sensitization | 0·93 (0·82–1·05) | 0·243 |
OR, odds ratio; CI, confidence interval; IL, interleukin; OVA, ovalbumin; Th1/2, T helper types 1/2; IFN, interferon.
There was a significant elevation of Th2 cytokines, IL-4, IL-5, IL-13 and eotaxin, in the lung homogenates of OVA-sensitized/challenged mice compared with non-treated control mice without pneumococcal pneumonia (Fig. 4), but these anticipated results were not observed in mice with pneumococcal pneumonia. When we evaluated the concentrations of proinflammatory cytokines among the mice which developed pneumococcal pneumonia, OVA-sensitized/challenged mice showed decreased concentrations of the proinflammatory cytokines, IL-1β, IL-6, IFN-γ and TNF-α, compared with non-treated control mice (Fig. 5). MIP-1α, MIP-1β, monocyte chemotactic protein-1 and granulocyte–macrophage colony-stimulating factor levels were also significantly lower in OVA-sensitized/challenged mice with pneumococcal pneumonia compared with the control mice. Similarly, in mice without pneumococcal pneumonia, these results were inconsistent.
Fig. 4.
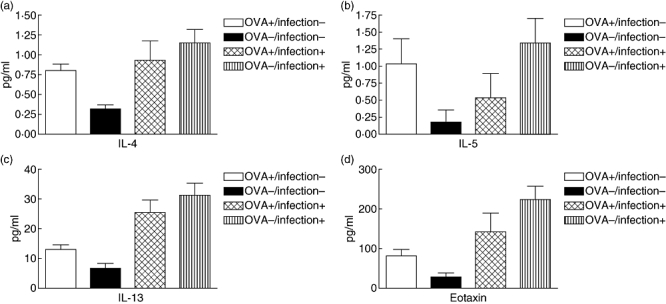
Levels of interleukin (IL)-4, IL-5, IL-13 and eotaxin in lung homogenates of the ovalbumin (OVA)-treated versus control mice. Data are presented as mean ± standard error (pg/ml). The mean levels were significantly higher in the OVA-treated mice without pneumococcal pneumonia than in the control mice (P < 0·05). OVA+/infection-, OVA-sensitized/challenged mice without pneumococcal pneumonia; OVA-/infection-, non-sensitized/challenged mice without pneumococcal pneumonia; OVA+/infection+, OVA-sensitized/challenged mice with pneumococcal pneumonia; OVA-/infection+, non-sensitized/challenged mice with pneumococcal pneumonia.
Fig. 5.
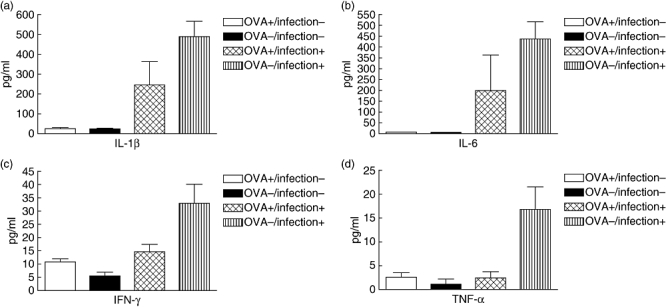
Levels of proinflammatory cytokines, interleukin (IL)-1β, IL-6, interferon (IFN)-γ and tumour necrosis factor (TNF)-α in lung homogenates of ovalbumin (OVA)-treated versus control mice with pneumococcal pneumonia. Data are presented as mean ± standard error (pg/ml). The mean levels were significantly lower in the OVA-treated mice with pneumococcal pneumonia than the control mice with pneumococcal pneumonia (P < 0·05). OVA+/infection-, OVA-sensitized/challenged mice without pneumococcal pneumonia; OVA-/infection-, non-sensitized/challenged mice without pneumococcal pneumonia; OVA+/infection+, OVA-sensitized/challenged mice with pneumococcal pneumonia; OVA-/infection+, non-sensitized/challenged mice with pneumococcal pneumonia.
Discussion
Paradoxically, in the mouse model studied, sensitization/challenge with OVA reduced the risk of pneumococcal pneumonia (28% versus 57%) and the number of pneumococci in the lung homogenates of animals with pneumonia. These are unexpected findings given the literature, which suggests an increased risk, severity and delayed clearance of microbial infections in both humans and mice in a Th2-predominant milieu [16–18,20,21,24,25]. Blair et al. reported that BALB/c mice sensitized with OVA had an increased rate of sinus infection with S. pneumoniae after intranasal challenge with pneumococci, compared with non-sensitized mice [30]. Potential mechanisms for increased risk of microbial infections in asthmatics have been suggested [16,19,20,22,31,32].
In interpreting the discrepant results between our study and the literature, a few explanations can be considered. First, although control mice in our study received i.p. aluminium hydroxide gel (instead of OVA) they had no intranasal challenge, whereas test mice had intranasal challenge with OVA. The lack of intranasal challenge in control mice might not have controlled fully the impact of intranasal challenge itself on airway inflammation. However, in our previous study using PBS–airway-challenged control mice, OVA-sensitized/challenged mice showed airway changes compatible with allergic inflammation (e.g. cellular infiltration and mucin hyperproduction), while PBS-challenged control mice did not show any inflammatory change in airway and lung interstitial tissues [33]. Therefore, we believe the lack of intranasal challenge in this study is unlikely to account for the results reported. Intranasal challenge with OVA induces marked infiltration of eosinophils, other granulocytes, especially neutrophils, and mononuclear cells in the airways [34,35].
During the acute phase of airway inflammation, infiltration of inflammatory cells into the airways and the presence of their inflammatory mediators might create a hostile environment for pneumococci, and may even deplete granulocytes or mononuclear cells in the bloodstream or spleen, resulting in reduced susceptibility to pneumonia but increased susceptibility to extrapulmonary infection. In support of this possibility, cigarette smoke-induced airway inflammation in a mouse model reduced the number of non-typeable Haemophilus influenzae in lung [36]. Thus, it can be postulated that airway inflammation, whether allergic or otherwise, may reduce the risk of bacterial pneumonia during the acute phase. Acute allergic airway inflammation induced by OVA and host defence against pneumococci in the mouse model studied are likely to differ from the chronic airway inflammation and remodelling of the airway present in humans with asthma [37]. Immunogenetic factors contributing to both asthma and host defence against microbial infection in humans are not accounted for in the mouse model [38–40]. In addition, humans are exposed to various pneumococcal serotypes with different levels of virulence for a longer duration, whereas our mice experienced a single challenge with a single strain. Also noted was a trend towards more extrapulmonary infection in OVA-sensitized/challenged mice, although this finding was not statistically significant. Further study on the association between allergic inflammation and susceptibility to pneumococcal bacteraemia or extrapulmonary infection (i.e. IPD) in a mouse model is needed, which may help us to understand possible mechanisms underlying the association.
An alternative explanation for our study findings is an independent role of Th2 cytokines from sensitization status with OVA with regard to the risk of pneumococcal pneumonia. Although the relationship between sensitization/OVA challenge and its expected cytokine concentrations in lung homogenates depended upon pneumococcal pneumonia status, as shown in Table 1, overall mice sensitized/challenged with OVA did not show clearly the expected Th2-predominant immune responses, compared with non-sensitized mice; this might be due to the Th1 effect induced by exposure to S. pneumoniae, because S. pneumoniae is a potent Th1-inducing agent [41]. Non-sensitized mice, despite the strong Th1 response elicited by exposure to S. pneumoniae, had comparable (IL-4 and IL-5) or higher levels (IL-10 and IL-13) of Th2 cytokines and chemokines associated with airway inflammation (e.g. RANTES and eotaxin) [42,43] compared with sensitized mice. These data suggest that mice sensitized with OVA might not have the expected classic Th2 immune responses and non-sensitized mice might have undiminished Th2 responses for unknown reasons. This might be a potential reason for which an association between sensitization/challenge with OVA and pneumococcal infection was not observed in our study. Because we used a previously studied dose of OVA for sensitization [28], we did not compare cytokine profiles at baseline (time zero) between mice with and without allergic sensitization. Differential effects (Th1 versus Th2) of sensitization with OVA in BALB/c mice depending on the sensitization dose of OVA have been reported [i.e. sensitization with low-dose (8 µg) versus high-dose (50 µg) OVA for Th1 versus Th2 response respectively][44]. Given this potential divergent relationship between cytokine pattern and sensitization status, we explored the impact of sensitization/challenge with OVA on the risk of pneumococcal pneumonia mediated through cytokines and chemokines. Mice with pneumococcal pneumonia had increased levels of Th2 cytokines (IL-4, IL-10 and IL-13) and chemokines associated with allergic airway inflammation (RANTES and eotaxin) compared with non-sensitized mice. We interpret that elevated Th2 cytokines are not due to exposure to S. pneumoniae because S. pneumoniae has been reported to induce typically Th1 but not Th2 responses and, thus, elevated Th1 cytokines might reflect an inflammatory process as a result of exposure to S. pneumoniae[41,45]. Thus, we adjusted the results for cytokines and interaction between cytokines and sensitization status. We found that IL-4, a Th2 cytokine, played a more important role in predicting the risk of pneumococcal pneumonia (OR: 50) than did sensitization status (OR: 1·12) (Table 2). This was true for the number of pneumococci in the spleen. As expected, IL-4 (β = 2·4, P = 0·001) was a more important factor in predicting the number of pneumococci in the spleen than sensitization status itself (β = −1·5, P = 0·21) (data not shown). In support of these findings, Beisswenger et al. reported that allergic airway inflammation inhibits anti-bacterial host defence in mice (innate immunity) [20] and demonstrated that IL-4 and IL-13 inhibit anti-microbial activity of human epithelial cells [46]. Khan et al. showed that Th2 cytokines (IL-4 or IL-10) reduced humoral immune responses to intact pneumococcal challenges, whereas Th1 cytokines (TNF-α) augmented humoral immune responses [47]. Thus, mice with attenuated production of proinflammatory cytokines might have increased further the risk of pneumococcal pneumonia in our study [20]. Controlling for IFN-γ and its interaction with sensitization status the impact of sensitization became insignificant, but IFN-γ is unlikely to be a risk factor for pneumococcal pneumonia because elevated Th1 cytokines and proinflammatory chemokines may be an effect of exposure to S. pneumoniae, instead of a cause of pneumococcal pneumonia (as discussed above). Understanding how Th2 cytokines suppress anti-microbial host defence remains to be determined.
Strengths of our study include the large sample size and use of in vivo imaging, permitting identification of mice with pneumococcal infection without killing. Limitations include lack of availability of cytokine data before pneumococcal challenge and sensitivity of detection of pneumococcal infection by bioluminescence, which may be subject to non-differential misclassification bias affecting both groups of mice. Although histopathological features in the OVA-sensitized/challenged mice were compatible with allergic airway inflammation, cellular analysis of bronchoalveolar lavage fluid, which may have been helpful to ascertain the presence of allergic airway inflammation, was not performed in this study. In addition, we studied a single pneumococcal strain; our results may not be applicable to other strains of S. pneumoniae.
In conclusion, in our mouse model, acute allergic airway inflammation reduced susceptibility to pneumococcal pneumonia and was associated with a trend towards increased extrapulmonary pneumococcal infection, suggesting that allergic airway inflammation may play a permissive role for development of invasive pneumococcal infection. Independent of allergic airway inflammation, IL-4 may play a crucial role in determining susceptibility to pneumococcal pneumonia.
Acknowledgments
We would like to thank Koji Iijima, James L. Checkel and Diane L. Squillace for technical support. The authors would like to thank Xenogen Corporation for their kind gift of Streptococcus pneumoniae serotype 3 (Xen10). This work was supported by a Bridge Award from the Mayo Foundation (Rochester, MN, USA). Cheol-In Kang was supported by funding from Samsung Medical Center (Seoul, South Korea).
Disclosure
The investigators have nothing to disclose that poses a conflict of interest.
References
- 1.Whitney CG, Farley MM, Hadler J, et al. Decline in invasive pneumococcal disease after the introduction of protein–polysaccharide conjugate vaccine. N Engl J Med. 2003;348:1737–46. doi: 10.1056/NEJMoa022823. [DOI] [PubMed] [Google Scholar]
- 2.Huang SS, Platt R, Rifas-Shiman SL, Pelton SI, Goldmann D, Finkelstein JA. Post-PCV7 changes in colonizing pneumococcal serotypes in 16 Massachusetts communities, 2001 and 2004. Pediatrics. 2005;116:e408–13. doi: 10.1542/peds.2004-2338. [DOI] [PubMed] [Google Scholar]
- 3.Kaplan SL, Mason EO, Jr, Wald ER, et al. Decrease of invasive pneumococcal infections in children among 8 children's hospitals in the United States after the introduction of the 7-valent pneumococcal conjugate vaccine. Pediatrics. 2004;113:443–9. doi: 10.1542/peds.113.3.443. [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention (CDC) Prevention of pneumococcal disease: recommendation of the Advisory Committee on Immunization Practices (ACIP) MMWR Recomm Rep. 1997;46:1–24. [PubMed] [Google Scholar]
- 5.Obaro S, Adegbola R. The pneumococcus: carriage, disease and conjugate vaccines. J Med Microbiol. 2002;51:98–104. doi: 10.1099/0022-1317-51-2-98. [DOI] [PubMed] [Google Scholar]
- 6.Robinson KA, Baughman W, Rothrock G, et al. Epidemiology of invasive Streptococcus pneumoniae infections in the United States, 1995–1998: opportunities for prevention in the conjugate vaccine era. JAMA. 2001;285:1729–35. doi: 10.1001/jama.285.13.1729. [DOI] [PubMed] [Google Scholar]
- 7.Talbot TR, Hartert TV, Mitchel E, et al. Asthma as a risk factor for invasive pneumococcal disease. N Engl J Med. 2005;352:2082–90. doi: 10.1056/NEJMoa044113. [DOI] [PubMed] [Google Scholar]
- 8.Juhn YJ, Kita H, Yawn BP, et al. Increased risk of serious pneumococcal disease in patients with asthma. J Allergy Clin Immunol. 2008;122:719–23. doi: 10.1016/j.jaci.2008.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cernelc D, Gerbec M, Cernelc P. Comparative study of virological infections in asthmatic and nonasthmatic children. Acta Allergol. 1975;30:423–33. doi: 10.1111/j.1398-9995.1975.tb01676.x. [DOI] [PubMed] [Google Scholar]
- 10.Eldeirawi K, Persky VW. History of ear infections and prevalence of asthma in a national sample of children aged 2 to 11 years: the Third National Health and Nutrition Examination Survey, 1988 to 1994. Chest. 2004;125:1685–92. doi: 10.1378/chest.125.5.1685. [DOI] [PubMed] [Google Scholar]
- 11.Centers for Disease Control and Prevention (CDC) ACIP provisional recommendations for use of pneumococcal vaccines. [accessed 12 March 2009]. Available at: http://www.cdc.gov/vaccines/recs/provisional/downloads/pneumo-oct-2008-508.pdf.
- 12.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–93. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 13.Smart JM, Kemp AS. Increased Th1 and Th2 allergen-induced cytokine responses in children with atopic disease. Clin Exp Allergy. 2002;32:796–802. doi: 10.1046/j.1365-2222.2002.01391.x. [DOI] [PubMed] [Google Scholar]
- 14.James SL, Sher A. Cell-mediated immune response to schistosomiasis. Curr Top Microbiol Immunol. 1990;155:21–31. doi: 10.1007/978-3-642-74983-4_2. [DOI] [PubMed] [Google Scholar]
- 15.Sjolander A, Baldwin TM, Curtis JM, Handman E. Induction of a Th1 immune response and simultaneous lack of activation of a Th2 response are required for generation of immunity to leishmaniasis. J Immunol. 1998;160:3949–57. [PubMed] [Google Scholar]
- 16.Chaplin D. Clearance of Mycoplasma pneumoniae is impaired in mice with established allergic airway inflammation. J Allergy Clin Immunol. 2007;119:S132. [Google Scholar]
- 17.Chu HW, Thaikoottathil J, Rino JG, et al. Function and regulation of SPLUNC1 protein in Mycoplasma infection and allergic inflammation. J Immunol. 2007;179:3995–4002. doi: 10.4049/jimmunol.179.6.3995. [DOI] [PubMed] [Google Scholar]
- 18.Kincy-Cain T, Bost KL. Increased susceptibility of mice to Salmonella infection following in vivo treatment with the substance P antagonist, spantide II. J Immunol. 1996;157:255–64. [PubMed] [Google Scholar]
- 19.Kincy-Cain T, Clements JD, Bost KL. Endogenous and exogenous interleukin-12 augment the protective immune response in mice orally challenged with Salmonella dublin. Infect Immun. 1996;64:1437–40. doi: 10.1128/iai.64.4.1437-1440.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beisswenger C, Kandler K, Hess C, et al. Allergic airway inflammation inhibits pulmonary antibacterial host defense. J Immunol. 2006;177:1833–7. doi: 10.4049/jimmunol.177.3.1833. [DOI] [PubMed] [Google Scholar]
- 21.Mizuki D, Miura T, Sasaki S, Mizuki M, Madarame H, Nakane A. Interference between host resistance to Listeria monocytogenes infection and ovalbumin-induced allergic responses in mice. Infect Immun. 2001;69:1883–8. doi: 10.1128/IAI.69.3.1883-1888.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fischer JE, Johnson JE, Kuli-Zade RK, et al. Overexpression of interleukin-4 delays virus clearance in mice infected with respiratory syncytial virus. J Virol. 1997;71:8672–7. doi: 10.1128/jvi.71.11.8672-8677.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graham MB, Braciale VL, Braciale TJ. Influenza virus-specific CD4+ T helper type 2 T lymphocytes do not promote recovery from experimental virus infection. J Exp Med. 1994;180:1273–82. doi: 10.1084/jem.180.4.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moran TM, Isobe H, Fernandez-Sesma A, Schulman JL. Interleukin-4 causes delayed virus clearance in influenza virus-infected mice. J Virol. 1996;70:5230–5. doi: 10.1128/jvi.70.8.5230-5235.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romani L, Mocci S, Bietta C, Lanfaloni L, Puccetti P, Bistoni F. Th1 and Th2 cytokine secretion patterns in murine candidiasis: association of Th1 responses with acquired resistance. Infect Immun. 1991;59:4647–54. doi: 10.1128/iai.59.12.4647-4654.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Contag CH, Contag PR, Mullins JI, Spilman SD, Stevenson DK, Benaron DA. Photonic detection of bacterial pathogens in living hosts. Mol Microbiol. 1995;18:593–603. doi: 10.1111/j.1365-2958.1995.mmi_18040593.x. [DOI] [PubMed] [Google Scholar]
- 27.Francis KP, Yu J, Bellinger-Kawahara C, et al. Visualizing pneumococcal infections in the lungs of live mice using bioluminescent Streptococcus pneumoniae transformed with a novel Gram-positive lux transposon. Infect Immun. 2001;69:3350–8. doi: 10.1128/IAI.69.5.3350-3358.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Lamm WJ, Albert RK, et al. Influence of the route of allergen administration and genetic background on the murine allergic pulmonary response. Am J Respir Crit Care Med. 1997;155:661–9. doi: 10.1164/ajrccm.155.2.9032210. [DOI] [PubMed] [Google Scholar]
- 29.Radhakrishnan S, Iijima K, Kobayashi T, Rodriguez M, Kita H, Pease LR. Blockade of allergic airway inflammation following systemic treatment with a B7-dendritic cell (PD-L2) cross-linking human antibody. J Immunol. 2004;173:1360–5. doi: 10.4049/jimmunol.173.2.1360. [DOI] [PubMed] [Google Scholar]
- 30.Blair C, Nelson M, Thompson K, et al. Allergic inflammation enhances bacterial sinusitis in mice. J Allergy Clin Immunol. 2001;108:424–9. doi: 10.1067/mai.2001.117793. [DOI] [PubMed] [Google Scholar]
- 31.Contoli M, Message SD, Laza-Stanca V, et al. Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med. 2006;12:1023–6. doi: 10.1038/nm1462. [DOI] [PubMed] [Google Scholar]
- 32.Wark PA, Johnston SL, Bucchieri F, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201:937–47. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kobayashi T, Iijima K, Kita H. Marked airway eosinophilia prevents development of airway hyper-responsiveness during an allergic response in IL-5 transgenic mice. J Immunol. 2003;170:5756–63. doi: 10.4049/jimmunol.170.11.5756. [DOI] [PubMed] [Google Scholar]
- 34.Schroder NW, Maurer M. The role of innate immunity in asthma: leads and lessons from mouse models. Allergy. 2007;62:579–90. doi: 10.1111/j.1398-9995.2007.01386.x. [DOI] [PubMed] [Google Scholar]
- 35.Taube C, Dakhama A, Rha YH, et al. Transient neutrophil infiltration after allergen challenge is dependent on specific antibodies and Fc gamma III receptors. J Immunol. 2003;170:4301–9. doi: 10.4049/jimmunol.170.8.4301. [DOI] [PubMed] [Google Scholar]
- 36.Stampfli M, Skrtic M, Gaschler G. Cellular and cytokine profile following in vivo challenge with nontypeable Haemophilus influenzae in cigarette smoke exposed mice. European Respiratory Society Annual Meeting 2008; Berlin, Germany.
- 37.Henderson WR, Jr, Tang LO, Chu SJ, et al. A role for cysteinyl leukotrienes in airway remodeling in a mouse asthma model. Am J Respir Crit Care Med. 2002;165:108–16. doi: 10.1164/ajrccm.165.1.2105051. [DOI] [PubMed] [Google Scholar]
- 38.Juhn YJ, Kita H, Lee LA, et al. Childhood asthma and human leukocyte antigen type. Tissue Antigens. 2007;69:38–46. doi: 10.1111/j.1399-0039.2006.00719.x. [DOI] [PubMed] [Google Scholar]
- 39.Jacobson RM, Poland GA, Vierkant RA, et al. The association of class I HLA alleles and antibody levels after a single dose of measles vaccine. Hum Immunol. 2003;64:103–9. doi: 10.1016/s0198-8859(02)00741-3. [DOI] [PubMed] [Google Scholar]
- 40.Kinsman OS, McKenna R, Noble WC. Association between histocompatability antigens (HLA) and nasal carriage of Staphylococcus aureus. J Med Microbiol. 1983;16:215–20. doi: 10.1099/00222615-16-2-215. [DOI] [PubMed] [Google Scholar]
- 41.Arva E, Andersson B. Induction of phagocyte-stimulating and Th1-promoting cytokines by in vitro stimulation of human peripheral blood mononuclear cells with Streptococcus pneumoniae. Scand J Immunol. 1999;49:417–23. doi: 10.1046/j.1365-3083.1999.00481.x. [DOI] [PubMed] [Google Scholar]
- 42.Park SJ, Lee KS, Kim SR, et al. Change of connexin 37 in allergen-induced airway inflammation. Exp Mol Med. 2007;39:629–40. doi: 10.1038/emm.2007.69. [DOI] [PubMed] [Google Scholar]
- 43.Mori A, Ogawa K, Kajiyama Y, Suko M, Kaminuma O. Th2-cell-mediated chemokine synthesis is involved in allergic airway inflammation in mice. Int Arch Allergy Immunol. 2006;140(Suppl. 1):55–8. doi: 10.1159/000092712. [DOI] [PubMed] [Google Scholar]
- 44.Morokata T, Ishikawa J, Yamada T. Antigen dose defines T helper 1 and T helper 2 responses in the lungs of C57BL/6 and BALB/c mice independently of splenic responses. Immunol Lett. 2000;72:119–26. doi: 10.1016/s0165-2478(00)00188-7. [DOI] [PubMed] [Google Scholar]
- 45.Koh YY, Park Y, Lee HJ, Kim CK. Levels of interleukin-2, interferon-gamma, and interleukin-4 in bronchoalveolar lavage fluid from patients with Mycoplasma pneumonia: implication of tendency toward increased immunoglobulin E production. Pediatrics. 2001;107:E39. doi: 10.1542/peds.107.3.e39. [DOI] [PubMed] [Google Scholar]
- 46.Klescz F, Beisswenger C, Kaendler K, et al. Allergic airway inflammatory inhibits pulmonary antibacterial host defense. Am J Respir Crit Care Med. 2007;175:A209. [Google Scholar]
- 47.Khan AQ, Shen Y, Wu ZQ, Wynn TA, Snapper CM. Endogenous pro- and anti-inflammatory cytokines differentially regulate an in vivo humoral response to Streptococcus pneumoniae. Infect Immun. 2002;70:749–61. doi: 10.1128/iai.70.2.749-761.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]