

Abstract
The hydrophobic hydration of fullerenes in water is of significant interest as the most common Buckminsterfullerene (C60) is a mesoscale sphere; C60 also has potential in pharmaceutical and nanomaterial applications. We use an all-atom molecular dynamics simulation lasting hundreds of nanoseconds to determine the behavior of a single molecule of C60 in a periodic box of water, and compare this to methane. A C60 molecule does not induce drying at the surface; however, unlike a hard sphere methane, a hard sphere C60 solute does. This is due to a larger number of attractive Lennard-Jones interactions between the carbon atom centers in C60 and the surrounding waters. In these simulations, water is not uniformly arranged but rather adopts a range of orientations in the first hydration shell despite the spherical symmetry of both solutes. There is a clear effect of solute size on the orientation of the first hydration shell waters. There is a large increase in hydrogen-bonding contacts between waters in the C60 first hydration shell. There is also a disruption of hydrogen bonds between waters in the first and second hydration shells. Water molecules in the first hydration shell preferentially create triangular structures that minimize the net water dipole near the surface near both the methane and C60 surface, reducing the total energy of the system. Additionally, in the first and second hydration shells, the water dipoles are ordered to a distance of 8 Å from the solute surface. We conclude that, with a diameter of approximately 1 nm, C60 behaves as a large hydrophobic solute.
1. Introduction
Buckminsterfullerene (C60), discovered in 1985 by Kroto et al.,1 has created a great deal of interest in many fields of biology and chemistry including polymer science, nanotechnology, and pharmaceuticals. A very active field of research regards the solubility of fullerenes, particularly simulation and experiment of fullerene solvation in polar and organic solvents and the synthesis of water-soluble derivatives of C60.2–7 The solvation of C60 in water is crucial for its use in therapeutic applications. Promising assays have shown that fullerenes can bind to the HIV protease with high affinity, acting as an HIV virus inhibitor.8 C60 is also being examined for cancer treatment; it has been shown to be cytotoxic, cleaving DNA when it is exposed to visible light.9 We will address the following questions using molecular dynamics (MD) simulations: (1) What is the atomic arrangement of water surrounding a single C60 molecule, both in terms of solute–water and water–water structure? (2) What is the diffusive behavior of the solute? (3) How is the hydrophobic solvation of a large solute, C60, different from that of a small solute, methane (CH4)? Previously, Raschke et al.10 reported on the hydration of single molecules of benzene and cyclohexane in water. We consider this a continuation of that work, in that C60 can be considered a three-dimensional analogue of benzene that is almost perfectly spherical. As such, it lends itself to rotationally averaged analysis, which is very sensitive, as it clearly enhances signal while canceling noise through averaging.
A hydrophobic solute placed in water can behave very differently depending on the size of the solute,11 and C60 (diameter ≈ 1 nm) represents a boundary case between “small” and “large” hydrophobic solutes. Small solutes are easily accommodated in the water structure, while large solutes lead to a rearrangement of the hydrogen-bonding network.12–14 The resulting reduction of possible hydrogen-bonding configurations leads to an unfavorable change in entropy. Recent theories link the likelihood of opening a cavity large enough to accommodate the solute to solute size and hydrophobicity.15,16 It has been suggested that the crossover from small-scale to large-scale behavior occurs on the nanometer scale,11,17 and solutes as small as neopentane (diameter ≈ 5.2 Å) can behave in the same way as large hydrophobic solutes18 when treated as hard spheres. What is the effect of size on solute–solvent nonbonded interactions? Theoretical studies11,12,19 have shown that these interactions contribute to the structure of water around large solutes, and that a large hard sphere solute will dry, while a van der Waals (vdW) solute of comparable size will not. However, small hard sphere solutes will not dry. Previously, fully atomistic MD simulations of a pair of C60 molecules and 2 and 16 carbon nanotubes in water found that with an initial spacing of less than 12 Å the C60 molecules and the nanotubes exhibit drying, resulting in direct contact between the solutes.20
In this paper, we report on the atomic structure of water surrounding one molecule of C60. Water forms a structured configuration surrounding C60, with a dense and structured first hydration shell (S1), a pronounced area of low water density, and a distinctly well-formed second hydration shell (S2). Water surrounding CH4 shows less structure, with a more diffuse S1 and very little structure in S2. Water adopts a distinct pattern of orientations around both hydrophobic solutes. Specifically, we observe two populations of water molecules, one with the water dipole pointing toward the solute center and another with the water dipole pointing away from the solute center. In the first population, an OH bond may point toward the solute center, whereas, in the second population, at least one OH bond is tangential to the solute surface. We find a small but significant ordering in the water dipoles surrounding the solutes that extends to 8 Å from the solute surface, in agreement with the finding that the protein surface affects interdomain water to a distance of 8 Å.21 This water dipole ordering may be essential to hydrophobic long-range interactions and therefore to protein folding.22,23
We use a simple distance cutoff to classify water molecules into spherical shells surrounding the solutes. The hydrogen bonding within and between shells is explored by looking at the distance and angle distribution of water–water contacts, rather than by defining a strict hydrogen-bonding geometry.24,25 The hydrogen-bonding behavior we find is similar for both solutes, specifically an increase in short distance, near-linear hydrogen-bonding contacts in S1 compared to bulk. However, C60 shows a much larger increase compared to CH4. We further expand our analysis to look at three-water interactions in triangles in S1. In the first hydration shell, we find an increase in the frequency of water triangles that minimize the net water dipole near the solute surface compared to bulk water, and a subsequent decrease in S1 in the frequency of water triangles that strengthen the net water dipole near the solute surface.
To examine drying, we “turn off” the attractive part of the Lennard-Jones (LJ) interaction between the solute C atom and water O atom to study the effect of nonbonded attractive solute–solvent interactions on hydration shell structure. Our simulations confirm that attractive LJ interactions make important contributions to the water structure surrounding the larger solute, with the modified LJ C60 showing considerably less structure in S1 and no water structure beyond S1. We find that the large solute C60 does dry when treated as a hard sphere but that the modified LJ CH4 does not dry, and there is little difference between the hard sphere and the original solute in the structure of the hydration shells. The behavior of the hard sphere solutes is the largest difference we find between C60 and CH4.
We also report on the rotational and translational diffusion rates of C60 and compare to calculation by the Debye–Stokes–Einstein (DSE) equation, which regards the solute as a perfect sphere. The rotational relaxation time26 of C60 in our simulation allows us to measure the rotational diffusion constant of C60 in water, which is unavailable through experiment.
2. Methods
2.1. Molecular Dynamics (MD) Simulations
Simulations were run using the GROMACS software package. We simulate a single molecule of solute, CH4 or C60, in a (4 nm)3 simulation box containing over 2000 explicit water molecules using the rigid SPC water model. We have previously compared a variety of water models in the hydration of benzene and cyclohexane.10 The geometry of the solvating water molecules was not affected by water model type in the MD simulations. We therefore chose SPC, as it is the simplest (three-point) model, is commonly used, and reduced our computation time. Neither solute is constrained in our simulations. However, due to the highly interconnected geometry of the C60, it is very rigid. In our simulations, the root mean square deviation (rmsd) of C60 from frame to frame is between 0.154 and 0.168 Å (mean rmsd is 0.158 Å). CH4 does have flexibility, especially around the angle of the H atoms in the tetrahedron. The rmsd of CH4 from frame to frame is between 0.008 and 0.138 Å (mean rmsd is 0.053 Å); however, CH4 is of course much smaller than C60.
Simulations were run in the NPT ensemble (constant number of molecules, pressure, and temperature) using the potential energy parameters based on the OPLS (optimized potentials for liquid simulations) all-atom force field for the solutes. The carbon atom type for the C60 C atom was the same as that for the C-delta atom of the aromatic tryptophan amino acid. The model for C60 included Lennard-Jones (LJ) terms, but the partial charge of each C atom was zero. The interaction potential parameters are detailed in Table 1. The smooth particle mesh Ewald (PME) summation of 27 was used for the electrostatic interactions, and a cutoff of 10 Å was used for the van der Waals interactions.
TABLE 1.
Nonbonded Energy Parameters of Solute, Water, and Solute–Water Interactions for the Standard OPLS Potential as Well as the Modified “Hard Sphere” (hs) and “No Partial Charge” (npc) Potentialsa
| parameterb | C60 | C60 hsc,d | CH4 | CH4 hs | CH4 npce |
|---|---|---|---|---|---|
| σC (nm) | 0.355 | 0.355 | 0.350 | 0.350 | 0.350 |
| εC (kJ mol−1) | 0.293 | 0.293 | 0.276 | 0.293 | 0.276 |
| σH (nm) | 0.250 | 0.250 | 0.250 | ||
| εH (kJ mol−1) | 0.126 | 0.126 | 0.126 | ||
| σOW (nm) | 0.317 | 0.317 | 0.317 | 0.317 | 0.317 |
| εOW (kJ mol−1) | 0.650 | 0.650 | 0.650 | 0.650 | 0.650 |
| σHW (nm) | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| εHW (kJ mol−1) | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| σOW–C (nm) | 0.336 | 0.336 | 0.334 | 0.334 | 0.334 |
| εOW–C (kJ mol−1) | 0.436 | 0.436 | 0.424 | 0.424 | 0.424 |
| σOW–H (nm) | 0.284 | 0.284 | 0.284 | ||
| εOW–H (kJ mol−1) | 0.286 | 0.286 | 0.286 | ||
| qC (electron) | 0 | 0 | −0.240 | −0.240 | 0 |
| qH (electron) | 0.060 | 0.060 | 0 | ||
| qOW (electron) | −0.820 | −0.820 | −0.820 | −0.820 | −0.820 |
| qHW (electron) | 0.410 | 0.410 | 0.410 | 0.410 | 0.410 |
The Lennard-Jones potential between two atoms is VLJ(rij) = 4εij((σij/rij)12 − (σij/rij)6). VLJ(rij) = 4εij(σij/rij)6 represents the attractive part of the LJ potential, and VLJ(rij) = 4εij(σij/rij)12 represents the repulsive part. The parameters σij and εij are derived from the atomic parameters as σij = (σi + σj) and εij = (εiεj)1/2.
OW, HW: SPC water oxygen and water hydrogen atoms.
hs: hard sphere.
The modified LJ hard sphere potential between the solute C atom and the water O atom has the form VLJ(rij) = 4εij(σij/rij)12. The interaction therefore has no attractive part.
npc: no partial charge.
A set of 10 simulations of 10 ns each was run for C60, for a total simulation time of 100 ns, and a set of 30 simulations of 10 ns each was run for CH4, for a total simulation time of 300 ns. More simulation time was needed to get comparable statistical accuracy by having the same number of shell 1 (S1) water molecules sampled (C60 has approximately 3 times as many water molecules in the first shell as CH4). Each system was initially minimized using steepest descent energy minimization for 250 steps, followed by a short, equilibration MD simulation of 100 000 steps with a small time step of 1 fs. All simulations were then run at 300 K, with a time step of 2 fs, storing coordinates every 1000 steps, or 2 ps. The first 500 ps of the simulations were not used in the subsequent analysis, to ensure that the system has reached equilibrium. The individual simulations differed in the random seed used to assign initial velocities.
The volume of the simulation box did not fluctuate significantly: the mean box volume was 67.1 ± 1.3 nm3, a fluctuation of 0.0002%.
2.2. Modified LJ Solute MD Simulations
In order to explore how the nonbonded interactions between solute and solvent influence the hydration shell structure, we simulate two modified LJ solute models. We use models where the attractive part of the LJ interaction between the solute C atom and water O atom has been removed (Table 1). This creates a solute that is even more repulsive than the WCA repulsive solute–solvent interaction.28 The modified LJ solutes were run in the NPT ensemble. Again, we found very little fluctuation in the volume of the simulation box (mean box volume 67.8 ± 1.4 nm3), and therefore very little fluctuation in water density that would affect our results. All other run parameters were the same. Second, we neutralize the partial charges on the CH4 molecule run under the standard MD conditions in the NPT ensemble.
2.3. Radial Distribution Functions (RDF)
We calculate radial distribution functions in one dimension for the solute center to water O atom gsol–O(r) and water H atom gsol–H(r) in the usual fashion,29 using a bin width of 0.05 Å. In order to obtain orientational information, a two-dimensional RDF, g(r,cos(ω)), is also computed, as previously described.30 The tilt angle ω is defined as the angle between the water dipole μ (bisecting the water HOH angle) and the radial vector r, connecting the water O to the solute center of mass. The distance bin width was 0.05 Å, and the angle bin width was 1°. For uniformly sampled orientations, cos(ω) is expected to be a uniform distribution.
2.4. Water Dipole Distribution
We measure the spherically averaged water dipole 〈cos(ω)〉 defined as 〈μ̂·r̂〉, where μ̂ and r̂ are the unit vectors along the water dipole moment (from O to H) and along the line connecting the solute center to the water oxygen, respectively. This has a value of −1 for a water molecule with its dipole oriented toward the solute center and 1 for a water molecule with its dipole pointing away from the solute center. For an isotropic distribution, this value averages to 0. The value of 〈μ̂·r̂〉 is small with a maximum absolute value of 0.04 and sensitive to noise. We smooth the data to allow for the very small number of counts, n, for water oxygen atoms close to the solute surface. This is done by starting at the first bin, i, and iteratively setting , where α taken as 0.1 and ni is the number of counts in bin i. As has already been smoothed whereas νi+1 has not, the loop over all bins is repeated four times.
2.5. Water Classification into Hydration Shells with Distance Cutoffs
We divide the simulation box volume into spherical shells surrounding the solute using a distance cutoff (see Table 2). The first cutoff distance reflects the location of the first minimum in the gsol–O(r) surrounding the solute, and the second cutoff reflects the location of the second minimum (Figure 1). Surrounding CH4 (radius = 1.8 Å), water molecules with an O atom <5.4 Å from the C center were defined as shell 1 (S1), with a thickness of 3.6 Å, water molecules with an O atom between 5.4 and 8.9 Å from the C center were defined as shell 2 (S2), with a thickness of 3.5 Å, and all water molecules >8.9 Å were defined as bulk (B). Surrounding C60 (radius = 5.0 Å), water molecules with an O center <8.6 Å from the solute center of mass were defined as S1, with a thickness of 3.6 Å, water molecules with an O center between 8.6 and 12.1 Å were defined as S2, with a thickness of 3.5 Å, and all water molecules >12.1 Å were defined as B. We further classify water molecules in S1 according to their orientation relative to the solute center. Water molecules with a tilt angle of ω < 90°(cos(ω) > 0) point the water dipole toward the solute and are classified as “1a”, and water molecules with a tilt angle of ω > 90° (cos(ω) < 0) point the water dipole away from the solute and are classified as “1b”.
TABLE 2.
Solute Parameters
| C60 | CH4 | |
|---|---|---|
| radius (Å) | 5.0 | 1.8 |
| shell 1 (S1) cutoff (Å) | 8.6 | 5.4 |
| shell 2 (S2) cutoff (Å) | 12.1 | 8.9 |
| mean number of water molecules in S1 per MD frame | 209.0 ± 5.0 | 28.2 ± 2.3 |
| mean number of water molecules in S2 per MD frame | 266.8 ± 7.0 | 91.9 ± 4.5 |
| D simulation (10−9 m2 s−1) (predicted) | 1.03 ± 0.92(0.773) | 2.67 ± 1.86(2.15) |
| τ1 simulation (ps) (predicted) | 625(323) | 21.6(15.1) |
| τ2 simulation (ps) (predicted) | 128(108) | 7.07(5.03) |
Figure 1.

Radial distribution functions and averaged dipole projections for C60 and CH4. The radial distribution functions of the solute center to water oxygen atom, gsol–O(r) (in red), and both of the water hydrogen atoms, gsol–H(r) (in blue), are shown in part A for CH4 and in part B for C60. For both systems, the distribution of hydrogen atoms is broader than that of oxygen atoms. The oxygen distribution for C60 shows much clearer peaks, in particular the second peak corresponding to the second hydration shell. The positions of these peaks serve to define the radii of the water shell boundaries. For C60, S1 is defined from 5.0 Å < r < 8.6 Å and S2 is defined from 8.6 Å < r < 12.1, and for CH4, S1 is defined from 1.8 Å < r < 5.4 Å and S2 is defined from 5.4 Å < r < 8.9 Å. S1 is 3.6 Å thick, and S2 is 3.5 Å thick. (C) The spherically averaged projection 〈μ̂·r̂〉 (in green) of the unit dipole vector μ̂ on the unit vector r̂ connecting the water oxygen to the solute center at each distance r from the solute center. 〈μ̂·r̂〉 = −1 for a population of water molecules with all dipoles oriented toward the solute center, 〈μ̂·r̂〉 = 1 for a population of water molecules with all dipoles pointing away from the solute center, and 〈μ̂·r̂〉 = 0 for an isotropic distribution. There is a clear alternating pattern of 〈μ̂·r̂〉 to at least 8 Å from the solute surface. A striking difference between C60 and CH4 is the peak of inward pointing water dipoles below 3 Å; this means that water dipoles are pointing toward the methane surface. 〈μ̂·r̂〉 is shown for CH4 in part C and in part D for C60. (E) The number of water oxygen atoms (in red) and water hydrogen atoms (in blue) in shell 1 (S1) at distance r is shown for CH4 S1 and in part F for C60 S1.
2.6. Hydrogen Bonding within and between Hydration Shells
Following Raschke and Levitt,30 all water molecules with oxygen–oxygen (O···O) distances within 8 Å were considered to be in contact. The contacts were divided into classes by the occurrence of the atom pair in different hydration shells: S1···S1, S2···S2, B···B, S1···S2, and S2···B. The OOH angle θ between water molecules in contact was defined as the smallest of the four possible angles between the water pair.24 We used 0.05 Å bins for the distances and 1° bins for the angles, and divided each bin by the total number of contacts found for that class of water contacts to scale the number of contacts. Normalization of the O···O contact distances was achieved by random sampling of points31 with a finite radius equal to that of water in a (40 Å)3 box, and assignment of those points to a shell or to bulk using our predefined shell distance cutoffs. The distances between these random points were tabulated in a histogram having the same 0.05 Å bins and used to normalize the observed O···O contact distances.
2.7. Water Triangles
We define a water triangle when two water O atoms are at a contact distance between 3.6 and 5.08 Å. We term this the “long” contact. We then look for a third water molecule that is contacting and hydrogen bonded to both water molecules, which we call the apical or mediating water. Here, a hydrogen bond is defined by an O···O distance less than 3.3 Å and an O···H distance less than 2.2 Å. This corresponds to a triangle with an angle at the apex between 80 and 130°. The two far water molecules in the “long” contact are considered in contact but are not necessarily hydrogen bonded to each other; however, the apical water must be hydrogen bonded to both according to our geometric criteria (Figure 6). Not all long contacts will be counted as not all long contacts will be mediated through a water molecule that is hydrogen bonded to both water molecules; in fact, we find that only 40–50% of long contacts are part of a triangle as defined (Table 3).
Figure 6.
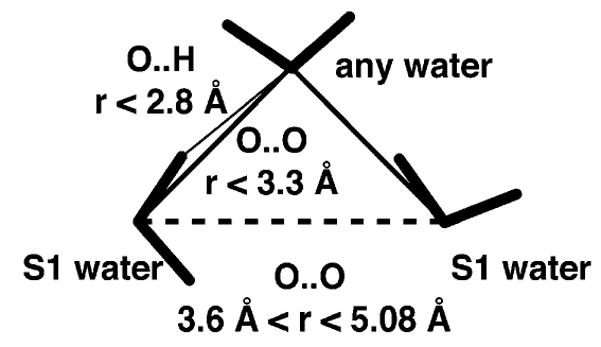
Schematic showing how the water triangular structure is defined.
TABLE 3.
Frequency of Triangular Water Structures
| CH4 shell 1 |
C60 shell 1 |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| bulk | S1 | 1a···1a | 1a···1b | 1b···1b | S1 | 1a···1a | 1a···1b | 1b···1b | |
| O···H–O···H–O | 0.45 | 0.36 | 0.39 | 0.30 | 0.37 | 0.44 | 0.46 | 0.34 | 0.36 |
| O···H–O–H···O | 0.29 | 0.35 | 0.13 | 0.34 | 0.50 | 0.30 | 0.28 | 0.34 | 0.46 |
| O–H···O···H–O | 0.22 | 0.25 | 0.44 | 0.31 | 0.10 | 0.21 | 0.22 | 0.26 | 0.14 |
| other | 0.04 | 0.04 | 0.04 | 0.05 | 0.03 | 0.04 | 0.04 | 0.06 | 0.04 |
| fraction of long contacts in triangle(total number) | 0.42(434744340) | 0.43(4202876) | 0.49(6102406) | ||||||
| fraction of long contacts not in triangle(total number) | 0.58(605443422) | 0.57(5488484) | 0.51(6294266) | ||||||
Once a triangle is found, it is classified according to the orientation of the waters in the long contact (i.e., the waters that are not necessarily hydrogen bonded) to the solute. If both waters are of type “1a” (dipole is pointing toward the solute), the triangle is classed as 1a···1a. If both waters are of type “1b” (dipole is pointing away from the solute), the triangle is classed as 1b···1b, and a 1a···1b triangle has one water of type “1a” and one water of type “1b” in the long contact.
We define six possible triangle “arrangements”; three are fully hydrogen bonded (O–H···O···H–O, O–H···O–H···O, and O···H–O–H···O), and three are not (O–H···O···O, O···H–O···O, and O···O···O). The smallest of the OOH angles at each of the three vertices define which H atoms are considered. The net dipole of a water triangle is simply the vector sum of the dipole moment of the three water molecules. The distributions of the six possible triangle arrangements were tabulated for water molecules in S1 and bulk.
2.8. Diffusion Properties
The simplest model of diffusion is that of a sphere of radius a diffusing through a liquid of viscosity η with a diffusion constant D. In this case, the linear diffusion constant is given by the Debye–Stokes–Einstein (DSE) equation:32
| (1) |
where k is the Boltzmann constant and T is the temperature in degrees Kelvin.
The diffusion constant D is related to the mean square distance (MSD) traveled by the solute by the Einstein relation:29
| (2) |
The DSE formula for the rotational diffusion constant Dr is
| (3) |
Dr is related to the experimental measurement of the τ1 and τ2 relaxation time in nuclear magnetic resonance (NMR).33 The orientational correlation function Cl can be fit to the exponential
| (4) |
The orientational correlation function Cl is related to the rotational diffusion constant Dr by
| (5) |
Note that τ1 = 3τ2. This NMR measurement cannot be carried out experimentally because of the large size of C60 and its insolubility in water; therefore, there is no experiment to directly measure the rotational diffusion of C60 in water. However, we can calculate Cl through our simulations. If μ is a normalized orientational vector that rotates with the solute, then Cl is defined as
| (6) |
where Pl is the Legendre polynomial of l. We ran an additional set of MD simulations under the same conditions outlined in section 2.1. Ten simulations of 1 ns each were run, sampled every 10 fs (every five time steps). All autocorrelation functions were calculated from these more finely sampled simulations.
It is interesting that, for a globular protein (or other organic molecule of comparable density), the volume in eq 3 is proportional to the mass. When converted to appropriate units, the experiment viscosity of water gives a surprising and useful expression for the rotational correlation time τ1 rate (using eq 5) that is roughly the molecular weight expressed in picoseconds. Thus, for CH4, we would expect τ1 = 14 ps and for C60 τ1 = 720 ps.
3. Results and Disscussion
3.1. Water–Solute Structure
The radial distribution functions for the CH4 and C60 solute center of mass to water O atom (gsol–O(r)) and water H atom (gsol–H(r)) are shown in Figure 1A and B. The relative size and density of the hydration shells is emphasized. C60 gsol–O(r) has a sharp first hydration shell (S1) water peak with a maximum value of 2.5, though not as sharp as the first pure water peak,34 and a large second peak with a maximum value of 1.17, indicating an ordered second hydration shell (S2). The first minimum is also very pronounced with a value of 0.60. gsol–H(r) is smaller in magnitude, with broader peaks. In comparison, the CH4 gsol–O(r) is smaller, with an S1 maximum of 1.8 and a minimum of 0.76. S2 is less structured, with a maximum second peak height of 1.04. It is interesting that gsol–H(r) peaks are broadened relative to gsol–O(r), an indication of the rotational freedom of the water molecules. In the region directly adjacent to the solute surface (<1Å), we find only water H atoms, allowed by the lack of vdW radius of H atoms in the SPC model.
Canceling the LJ attractive interactions between the solute C atom and the water O atom causes a depletion of water molecules at the C60 surface (drying) as well an almost total loss of structure (Figure 2). The effect is not seen for CH4, where there is no drying and only a small loss of structure. We conclude that this is the single most marked difference between the solutes in our study.
Figure 2.

Radial distribution functions gsol–O(r) (in red) and gsol–H(r) (in blue) for the modified LJ hard sphere solutes (Table 2) modified (A) from C60 and (B) from CH4. Hard sphere C60 shows evidence of drying at the solute surface and an almost complete loss of structure, whereas the water structure surrounding hard sphere CH4 is not greatly affected.
We would like to emphasize that while the details of our results are greatly influenced by the LJ parameters of the simulation, the overriding conclusion that multiple attractive vdW interactions at the solute surface in the case of C60 are the dominant forces determining the water structure remains valid regardless of the details of LJ parametrization. The CH4 C to water O interaction has parameters of σ(C–OW) = 0.334 nm and ε(C–OW) = 0.424 kJ mol−1. The C60 C to water O interaction has parameters of σ(C–OW) = 0.336 nm and ε(C–OW) = 0.436 kJ mol−1. These agree well with the previously established parameters of Werder et al.20 (Table 1).
The coupling of water dipoles at hydrophobic surfaces may create initial long-range attractions between hydrophobic solutes and may also be a factor in protein folding.22,23 In Figure 1C and D, we show the spherically averaged water dipole defined as 〈μ̂·r̂〉 = 〈cos(ω)〉. A value of 〈μ̂·r̂〉 = 1 indicates water molecule dipoles pointing directly toward the solute center. A value of 〈μ̂·r̂〉 = 0 indicates an isotropic distribution of water dipoles, and a value of 〈μ̂·r̂〉 = −1 indicates water molecule dipoles pointing directly away from the solute center. The value of 〈μ̂·r̂〉 is negative through S1, is positive through S2, and continues to oscillate to a distance of over 8 Å from the hydrophobic surface. It has been shown that interdomain water dynamics and hydrogen bonding are affected by the protein surface to a distance of 8 Å,21 in agreement with our finding. The oscillation is very clear for both C60 and CH4. After the first peak, the magnitude of 〈μ̂·r̂〉 is twice as large for C60, showing that larger hydrophobic solutes induce more ordering of the dipole in surrounding water molecules. At the solute surface, the first peak is positive, indicating a preference for the water dipole to turn toward the solute. This first peak is significantly higher for CH4; however, it is important to remember that this represents only a small number of waters at the solute surface (Figure 1E and F shows the number of water molecules at distance r in S1). The large first peak right at the CH4 surface may be due to the partial charge of −0.24 electrons on the CH4 carbon atom, which attracts the water dipole. In addition, the smaller size and high curvature of CH4 allows waters to interact around the solute, creating a softer solute.
C60–C60 interactions have been attributed to a strong favorable interaction between the solutes as a consequence of the high atomic density on the surface of C60, rather than to a hydrophobic, water driven clustering.5 It has been shown that two carbon nanotubes in a water slab under a purely repulsive potential do not cluster together, as they do under a LJ potential, yet they do exhibit drying.20 Here, as in our work, the van der Waals forces are shown to dominate C60 and nanotube hydration. In our classical MD calculation, we do not simulate the effects of surface electrostatics and cannot estimate their effect on water dipole ordering.
The two-dimensional gsol–water(r,cos(ω)) of C60 and CH4 are shown in Figure 3. Water adopts a well-defined distribution of orientations around both solutes, with a large plateau at (−0.6 < cos(ω) < 0.4). Waters in S1 surrounding C60 show a broader distribution than those in S1 surrounding CH4. C60 has two almost equally preferred populations. The first population, centered at cos(ω) = 0.19 (ω = 79°), points the dipole toward the solute, and no OH bond in this population will be more than ~80° from the solute surface considering all possible rotations around the μ vector. This population can therefore be seen as pointing inward. The second population, centered at cos(ω) = −0.45 (ω = 117°), points the dipole away from the solute, and at least one OH bond is tangential to the solute in this case considering all rotations. Tangentially arranged water molecules have been experimentally demonstrated to surround small hydrophobic solutes.35 Water in S1 surrounding CH4 shows a narrower distribution, with more waters favoring the tangential orientation than the inward orientation. There is a clear effect of surface size on the orientation of water near a solute surface. A small, highly curved surface with the width of approximately one carbon atom will induce the narrow distribution we see near the CH4 surface, with a preference for a tangential orientation. The orientation of waters surrounding CH4 is similar to that of the equatorial waters surrounding cyclohexane,30 i.e., those near the edge of cyclohexane, with a width of ~1.5 Å or one carbon atom. The orientation of waters surrounding C60 is similar to that of the axial waters surrounding cyclohexane, with a broad distribution and equal preference for inward and tangential orientations. The axial waters are near the hexagonal face of the cyclohexane chair. It is interesting that the crossover from one behavior to the other occurs when going from a width of one carbon atom to six hexagonally arranged carbon atoms.
Figure 3.

Two-dimensional radial distribution function, gsol–O(r,cos(ω)), in part A for CH4 and in part B for C60. Water molecules in shell 1 (S1) tend to arrange tangentially around both solutes at a distance from the surface of about 1 Å and centered at a tilt angle of ω = 100° (cos(ω) = −0.43) and peak height of 0.39 for CH4 and centered at a tilt angle of ω = 117° (cos(ω) = −0.45) and peak height 0.28 for C60. Both solutes have a second population of water molecules in S1 with an inward orientation. This orientation is more populated in C60, centered at ω = 79° (cos(ω) = 0.19) and a peak height of 0.37. We observe that waters near a small highly curved surface adopt the narrower distribution with a preference for the tangential orientation, while waters near a larger surface adopt a broad distribution. (C) A schematic showing how (r,ω,μ) are defined. We further classify the water molecules in S1 according to their orientation to the solute. Water molecules that point the water dipole toward the solute are denoted as “1a”, and water molecules that point the water dipole away are denoted as “1b”. It is important to note that the tilt angle is unaffected by rotation about the μ and r vectors.
All water molecules within the radius of S1 and with ω < 90° are labeled “1a”, and all water molecules with ω > 90° are labeled “1b” (Figure 3C).
Is the orientation of S1 water molecules influenced by the local geometry of C60? Overall, the average number of 1a and 1b water molecules occupying S1 in a MD frame is 102.3 ± 4.9 and 106.7 ± 4.9, respectively. A slight preference (1a/1b = 0.96) for 1b (with the dipole pointing away) is not surprising considering the hydrophobic nature of the solute. The ratio of pentagonal to hexagonal faces in C60 is 12/20 or 1.67. We measure the distance of all S1 waters to the nearest hexagon and pentagon geometric face center and determine whether the water is closest to a hexagonal face center or a pentagonal face center. We found on average 54 ± 5.4 1a and 56.5 ± 5.4 1b water molecules closest to hexagonal face centers and 48.3 ± 5.3 1a and 50.3 ± 5.3 1b water molecules closest to pentagonal face centers. This shows a bias for water molecules to be near pentagonal faces. There is a 1.12 ratio of waters closer to hexagonal face centers over waters closest to pentagonal face centers. This is smaller than the expected 1.67 ratio, especially if we remember the smaller surface area of pentagons; with unit edges, the surface area is 21 for the pentagons and 52 for the hexagons. There is no preference vis-à-vis the orientation of the water to the solute (1a or 1b) to be closer to a hexagonal or pentagonal face center. A pentagon face has 5 vdW centers with a surface area of 21, giving a ratio of 0.238 vdW centers per unit area, whereas a hexagon face has 6 vdW centers with a surface area of 52, giving a ratio of 0.115 vdW centers per unit area. Therefore, there are more attractive interactions near a pentagon face, explaining the bias of water molecules.
3.2. Water–Water Structure
Instead of defining strict geometric criteria for hydrogen bonding, for each water pair considered, we look at the distribution of water O···O distance, and the distribution of the smallest of the four possible OOH angles between two water molecules. This is effectively the “best” hydrogen-bonding geometry of the pair. We do this for all water pairs within an 8 Å distance, and then classify the contacts by shell.30
Figure 4 shows the two-dimensional distribution of contact distances and angles, g(r,θ), for S1···S1, S1···S2, S2···S2, S2···B, and B···B water···water contacts surrounding C60 and CH4. There is a characteristic shape to the contact distance/angle distribution g(r,θ)24 with a sharp peak corresponding to hydrogen-bonding water molecules, centered at (r = 2.7 Å, θ = 10°), and a second broad peak at (r = 4.8 Å, θ = 45°). The first peak corresponds to waters that are hydrogen bonded. The second peak corresponds to close O···O pairs that are in contact but are not hydrogen bonded.
Figure 4.

Water···water pairwise interactions are characterized by the O···O distance in Å and the smallest O···OH angle in degrees. The radial distribution function g(r,θ) shows the water···water contact length/angle distribution, and these pairwise interactions are classified by shell. The distributions g(r,θ) of interactions between S1···S1, S1···S2, S2···S2, S2···B, and B···B are shown in parts A–E for C60 and in parts F–J for CH4. The first sharp peak at (r = 2.7 Å, θ = 10°) corresponds to a “good” linear hydrogen-bonding geometry, and the second broad peak at (r = 4.8 Å, θ = 45°) corresponds to close O···O pairs that are in contact but are not hydrogen bonded. There is an increase in hydrogen-bonded waters within S1, particularly for C60, and a disruption of hydrogen bonding between S1···S2 contacts.
The first peak is significantly higher for S1···S1 contacts. This indicates an increase in hydrogen-bonded waters within S1. This effect is very pronounced in C60, where the g(r,θ) first peak reaches a height of 0.220. In CH4, the first peak reaches a height of 0.19, whereas, in bulk water, the first peak has a maximum height of 0.137. There is also a significant decrease in the height of the second peak in S1···S1 contacts for C60, indicating less non-hydrogen-bonding contacts. The first peak of C60 g(r,θ) of S1···S2 contacts is lower than bulk, indicating a smaller number of hydrogen-bonded waters between the first and second hydration shell. This is reflected in the one-dimensional RDF gsol–O(r), where C60 shows a great deal of structure in S1, and a greater depletion of water between S1 and S2. The distribution g(r,θ) is similar in all other classes, with the least hydrogen-bonding contacts seen in S2···B, and some enhanced hydrogen bonding in S2···S2.
The one-dimensional distributions shown in Figure 5 confirm the ordering of waters in S1. There is an increase in the S1··· S1 peak at 2.7 Å in the water···water contact distance distribution, g(r), for both solutes. We see a notable difference in the distribution of water···water contact angles, g(θ), with a large increase in small angle contacts for C60 in S1···S1, a decrease in the large angle contacts for C60 in S1···S1, and an increase in large angle contacts for C60 in S1···S2. We conclude that hydrogen bonding is sensitive to the size of the solute, with a greater enhancement of S1···S1 hydrogen bonding in C60 when compared to CH4, benzene, and cyclohexane,30 and with a larger disruption of the hydrogen bonding between S1···S2.
Figure 5.

The water···water contact length/angle distribution g(r,θ) was summed over all angles θ for each distance r to give g(r) and over all distances r for each angle θ to give g(θ). The contact distribution g(r) of contacts between S1···S1 (in green), S1···S2 (in red), S2···S2 (in cyan), S2···B (in magenta), and B···B (in blue) is shown for (A) CH4 and (B) C60. The smallest O···OH angle distribution g(θ) of water···water contacts is shown for (C) CH4 and (D) C60 with the same colors for classes. There is a dramatic increase in C60 S1··· S1 near-linear contacts, and in C60 S1···S2 wide angle contacts.
In this and previous studies, we have investigated pairwise water···water contacts. We are also interested in investigating three-body water interactions, particularly between the hydrogen-bonded waters in the first peak of g(r,θ) and the contacting but not hydrogen-bonded waters in the second peak of g(r,θ). That is, we would like to explore the network of water interactions that are mediated by hydrogen bonds that lead to an emergent grid of waters around the solute.
We look more closely at the triangular structures water forms through hydrogen bonding in S1 only. A pair of water molecules in S1 with a “long” contact, defined as a distance of 3.6 and 5.08 Å, and not interacting through hydrogen bonds, are the two vertices of a triangle. We then look for a third mediating water molecule, which must be hydrogen bonded to the other vertices according to geometric hydrogen-bonding criteria defined by an O···O distance less than 3.3 Å and an O···H distance less than 2.2 Å (Figure 6). This forms a triangle with an angle of 80–130° at the apex. Surrounding C60, 42% of the waters with long contacts were hydrogen bonded to a third mediating molecule. Surrounding CH4, 43% of long contacts were mediated through hydrogen bonds. In bulk water, 49% of long contacts were mediated through hydrogen bonds (Table 3).
The smallest OOH angle at each vertex determines what we have termed the triangle “arrangement”. For example, in the O···H–O–H···O arrangement, the apical (mediating) water is a double hydrogen-bond donor. In the O–H···O–H···O arrangement, the apical water is a hydrogen-bond donor/acceptor. In the O–H···O···H–O arrangement, the apical water is a double hydrogen-bond acceptor. All triangles in S1 are also classified as 1a···1a, 1a···1b, or 1b···1b according to the orientation of the two water molecules in the long contact (the triangle vertices) relative to the solute. We find that water molecules will preferentially create triangles in S1 that minimize the net dipole of the triangle. Figure 7A shows the log odds of the frequencies of the three hydrogen-bonded triangle arrangements in S1 relative to those in B plotted against the mean total dipole moments of the water triangle. There is a linear correlation between the log-likelihood and the total dipole of the triangle. Triangles with smaller total dipole moment are energetically more favorable and occur more often (positive log-likelihood), whereas triangles with a large total dipole moment are energetically less favorable and occur less often (negative log-likelihood). The triangles that experience the largest increase and decrease in likelihood in S1, with the smallest and largest net dipole, respectively, are shown in Figure 7B.
Figure 7.

The frequency of water triangle structures in bulk water are compared with those found in shell 1 around CH4 and C60, which are further classified by the orientation of the waters in the triangle to the solute (Table 3). The log odds of the ratio of frequencies of the triangle arrangements relative to those in bulk water are plotted against the mean total dipole moments of the water triangle, where each water dipole is taken as a unit vector. Triangles shown are apical double acceptor (O–H···O···H–O, in blue), apical donor/acceptor (O···H–O···H–O, in red), and apical double donor (O···H–O–H···O, in green). Triangles surrounding CH4 are shown as squares, and triangles surrounding C60 are shown as circles. Class 1a···1a triangles are shown as filled, class 1a···1b triangles are shown as striped, and class 1b··· 1b triangles are unfilled. The log-likelihood shows a linear correlation with the total dipole of the triangle. Triangles with smaller total dipole moment are energetically more favorable and occur more often (positive log-likelihood, above the dashed line), whereas triangles with a large total dipole moment are energetically less favorable and occur less often (negative log-likelihood, below the dashed line). (B) The corresponding points marked (1–4) in panel A represent symmetrical water triangles involving the water pairs 1a···1a and 1b···1b with the smallest and largest total dipole moments, respectively. Likewise, the likelihood of these triangles has the largest increase and largest decrease in S1 over B, respectively. Triangles in S1 are most stable in class 1a···1a with the arrangement O–H···O···H–O and in class 1b···1b with the arrangement O···H–O–H···O and are least stable for class 1a···1a with the arrangement O···H–O–H···O and in class 1b···1b with the arrangement O–H···O···H–O. It is interesting to note that triangles surrounding CH4 are the most affected.
Table 3 summarizes the distribution of water triangle arrangements in S1 classified by the orientation of the long contact water molecules and compared to the distribution of triangle arrangements in B. The composition of triangle arrangements in S1 is similar to that in B, although we do see some difference in S1 surrounding CH4 perhaps due to the small volume of the shell. We see that in S1 the preferred triangle arrangement is greatly influenced by the orientation of the water molecules to the solute. In S1, the arrangement O–H···O···H–O is enhanced among class 1a···1a triangles but reduced among class 1b···1b triangles. In class 1a···1a triangles with the O–H···O···H–O arrangement, the two long contact water molecules (with dipole oriented toward the solute) reduce the net dipole of the triangle by canceling the dipole of the mediating apical water. In class 1b···1b triangles with the same O–H···O···H–O arrangement, we find exactly the opposite, where the two long contact water molecules (with dipole oriented away from the solute) strengthen the dipole of the mediating apical water. Class 1b···1b triangles with the arrangement O···H–O–H···O are greatly enhanced in S1 because the net water dipole is reduced in this arrangement. In class 1a···1a triangles with the same O···H–OH···O arrangement, the dipole would be strengthened, and their number therefore decreases. The increase in the relative frequency of triangles with small net dipoles and decrease in the relative frequency of triangles with large net dipoles is more pronounced in S1 surrounding CH4. This may be due to the larger conformational flexibility of waters near the CH4 surface, allowing the water molecules to explore more energetically favorable three-body configurations.
3.3. Diffusion Properties
The diffusion coefficient D is predicted using the DSE, eq 1. We set a = 5.0 Å for C60 and a = 1.8 Å for CH4 and take the viscosity of water as η = 0.852 g m−1 s−1, corresponding to a temperature of T = 300 K. We used the Einstein relation in eq 2 to measure D in our simulations, and this gave us values similar to those predicted by the DSE equation. The hard sphere solutes diffuse with the same D value as the solute. The D value of water is calculated by eq 1 to be 2.762 nm2 ns−1 under those conditions (a = 1.4 Å), and has been measured experimentally at 2.3 nm2 ns−1.36 The D value of SPC water measured from our simulations was twice as fast, and this difference has been documented elsewhere.37 However, this did not seem to affect the diffusion rate of the solutes, which diffused at rates similar to those predicted. Diffusion rates are summarized in Table 2.
In order to find the rotational diffusion coefficient, we ran an additional 10 × 1 ns simulations under the conditions described in Methods section 2.1 but output the coordinates every 10 fs. We calculated the orientational correlation C2 from these additional finely sampled simulations using eq 6 and fit the first 10 ps of the correlation function to a single exponential as in eq 4.
For C60, we predicted τ1 = 323 ps and τ2 = 108 ps using the DSE equation for the rotational diffusion coefficient, eq 3. In our simulation, we found τ1 = 625 ps and τ2 = 286 ps for C60. For CH4, we predicted from the DSE equation τ1 = 31.5 ps and τ2 = 10.5 ps, and in our simulation, we found τ1 = 21.6 ps and τ2 = 7.07 ps. The ratio between τ1 and τ2 is preserved for CH4 (τ2 ~ 3 τ1); however, for C60, the ratio is more like τ2 ~ 2τ1. The ratio between the volumes of CH4 and C60 is approximately 12, and we therefore expect the same ratio in the rotational correlation times. In fact, the ratio is 2–3 times more than that, and this is because CH4 rotates at approximately the rate we would expect, but C60 is found to rotate more slowly than predicted by its volume alone (has a 3 times longer τ2 value in simulation). The DSE equation as presented in eqs 1 and 3 predicts the diffusion coefficient under complete stick boundary conditions. In fact, we can rewrite the DSE equation as38
where ζ is a parameter representing the boundary conditions; ζ can take values from 0 to ⅓ corresponding to complete stick and slip boundaries, respectively. We might therefore conclude that C60 diffuses under a mixture of stick–slip boundary conditions, rather than complete stick boundary conditions.
The NMR measurements τ2 of C60 in water are experimentally impossible because of the negligible solubility of C60 (1.3 × 10−11 mg/mL39). Experiments performed on C60 dissolved in tetrachloroethane (viscosity of 1.1 g m−1 s−1 at 293 K) measured a reorientational correlation time of τ2 = 15.5 ps at 283 K. NMR measurements of solid-state C60 measured τ2 = 6.8–14.9 ps in the temperature range 241–331 K.40 We have shown that our results simulated a longer τ2 value by an order of magnitude than those measured experimentally in the tetrachloroethane solution and in the solid state, and 3 times longer than that predicted from the DSE equation, corresponding to a slower rate of rotational diffusion in simulation. Figure 8 illustrates the diffusive behavior of a randomly selected water molecule near the solute surface and the rotational diffusion of the solute. Water diffuses laterally much faster than the time it takes for the solute to rotate completely, and it also becomes apparent that the water molecule diffuses more slowly near the solute surface.7
Figure 8.

Showing how a single, randomly selected water molecule moves as it diffuses over 100 ps (thick line). We also follow the rotation of C60 by tracing the path of C1 in C60 (thin line). In both cases, the time along the path is marked using color going from blue to red and points are drawn every 2 ps. It is clear that the water molecule moves more slowly when it is close to the surface of C60. The translation motion of the center of mass motion is eliminated for greater clarity.
4. Conclusions
In this paper, we examine the structural and dynamic behavior of water surrounding the hydrophobic solute C60 and CH4. C60 is an interesting hydrophobic solute for several reasons. First, it is a real nanoscale molecule rather than a large vdW sphere. Second, it contacts many water molecules, and third, it is spherically symmetric and uncharged. With a diameter of 1 nm, C60 represents a boundary case between large and small hydrophobic solutes, and we find evidence that it behaves as a large hydrophobic solute when treated as a hard sphere. C60 is also of great interest in nanomaterial and nanomedicine applications. CH4 is a real solute as well, and is also spherically symmetric. It is of interest to compare our C60 results to CH4, the smallest hydrophobic solute.
We have shown that the hydration shells surrounding the larger, virtually insoluble C60 are dense and well structured, unlike those surrounding the smaller water-soluble solute CH4. Solute–solvent LJ attractions contribute to both the density and the closeness of the hydration shells. When we rerun 100 ns of MD simulation where the LJ attractive forces have been switched off, we observe drying at the C60 surface, and the surrounding water loses almost all order in both spatial location and orientation. A C60 vdW molecule does not dry; however, its hard sphere counterpart will. Canceling the LJ attractive forces of CH4 to create a hard sphere does not induce drying. There is no noticeable effect when canceling the partial charges on CH4, indicating that the effect of the partial charge of CH4 on water is very small. The dramatic loss of structure for C60 but not for CH4 when turning off the LJ attractive forces is the largest difference we observe between these two solutes of such different sizes. The water···carbon interaction is the same in both cases, but the effective number of carbon atoms contacting a water molecule is very different. More specifically, 43 ± 3 water molecules in the first shell of C60 are within 5 Å of atleast one solute carbon atom and 121 ± 10 water molecules are within 8 Å, whereas 19 ± 0.2 water molecules in the first shell of CH4 are within 5 Å and 69 ± 4 water molecules are within 8 Å of the single solute carbon atom. This explains why turning off the LJ term affects C60 so much more than CH4.
The spherically averaged dipole projection of water molecules surrounding C60 or CH4 alternates from negative throughout the first shell to positive in the second shell, and this oscillation is seen to over 8 Å from the solute surface. This effect is qualitatively the same for both solutes except that the sum of the total averaged dipole moment is twice as large for C60. It has been hypothesized that correlations between water dipoles are the origin of long-range attractions between hydrophobes in water, and that this may be important in protein folding mediated by solvent fluctuations.22,23,41,42 The dipole is positive at the solute surface. However, this involves a very small number of water molecules. It is perhaps surprising that this positive first peak is much more pronounced for CH4, as the four hydrogen atoms carry small positive charges that would be expected to repel the inward pointing water hydrogen atoms. We believe that the smaller effective number of water···carbon interactions and the possibility to create water···water interactions around CH4 makes this solute much softer and hence the water dipole can be oriented toward the center. There is also a negative partial charge on the carbon atom that can interact with extremely close water dipoles.
Within the first hydration shell, water adopts orientations that may range from pointing one OH bond almost directly toward the solute surface to pointing one OH bond directly away. Solute size effects the orientation of the surrounding waters, and we find that water prefers a tangential orientation when near a small surface on the order of one carbon but has equal preference for both the tangential and inward orientations near larger surfaces. We suggest that the size crossover occurs on the order of cyclohexane. Another angle is needed to fully resolve the orientation of water molecules to the solute presented in the two-dimensional gsol–water(r,cos(ω)) because of the rotation about the r and μ vectors, and we plan to explore this in subsequent work.
We looked at the distribution of water–water distances and angles surrounding both solutes as suggested by Gallagher and Sharp,25 instead of defining strict hydrogen-bonding criteria. We found an increase in near-linear, short distance water···water contacts (10°, 2.8 Å) corresponding to an increase in “good” hydrogen-bonding geometries within the first hydration shell when compared to contacts in bulk water. This contributes to the water structure and density surrounding the solutes. The enhancement of near-linear contacts is larger in C60 than in other solutes we have studied, and there is a correspondingly large disruption of hydrogen bonding between S1···S2, where large angle contacts are enhanced.
We expand our pairwise analysis of water···water structure to look at three-body water interactions. The populations of triangular water structures in S1 with a small net dipole are increased over bulk populations, whereas the frequency of triangular structures with a large net dipole are decreased. This means that if two water dipoles point toward the center of the triangle, the third will be more likely to point away from the center and vice versa. This arrangement is energetically more favorable, minimizing the water dipole near the solute surface. Further work is needed to look at the arrangement of many (greater than three) contacting waters forming a network around the hydrophobic solute. We would like to ask what water pattern emerges in S1 in order to reduce the energy of the system. Future work would also determine the change in free energy, enthalpy, and entropy that accompanies the formation of this network.
The diffusive behavior of these spherically symmetric solutes can be well described with the DSE equation and is faithfully reproduced in our MD simulations, despite the fact that SPC water molecules in simulation are found to diffuse laterally more than twice as fast as is predicted and experimentally measured. We can also use our MD simulations to measure the rotational diffusion of C60 in water, which is experimentally impossible, and this is also in good agreement with that predicted by the DSE equation.
Acknowledgments
This work was supported by a Simbios student fellowship to D.R.W. (National Institutes of Health through the NIH Roadmap for Medical Research Grant U54 GM072970), by a Program in Mathematics and Molecular Biology student fellowship for D.R.W. (Burroughs Wellcome Fund), as well as by an NIH award GM-41455 to M.L.
References and Notes
- 1.Kroto HW, Heath JR, Obrien SC, Curl RF, Smalley RE. Nature. 1985;318:162. [Google Scholar]
- 2.Ruoff RS, Malhotra R, Huestis DL, Tse DS, Lorents DC. Nature. 1993;362:140. [Google Scholar]
- 3.Reed CA, Bolskar RD. Chem Rev. 2000;100:1075. doi: 10.1021/cr980017o. [DOI] [PubMed] [Google Scholar]
- 4.Howard CA, Thompson H, Wasse JC, Skipper NT. J Am Chem Soc. 2004;126:13228. doi: 10.1021/ja046322a. [DOI] [PubMed] [Google Scholar]
- 5.Li LW, Bedrov D, Smith GD. J Chem Phys. 2005;123 doi: 10.1063/1.2121647. [DOI] [PubMed] [Google Scholar]
- 6.Choudhury N. J Chem Phys. 2006;125:034502. doi: 10.1063/1.2217442. [DOI] [PubMed] [Google Scholar]
- 7.Choudhury N. J Phys Chem. 2007;111:2565. doi: 10.1021/jp073571n. [DOI] [PubMed] [Google Scholar]
- 8.Friedman SH, Decamp DL, Sijbesma RP, Srdanov G, Wudl F, Kenyon GL. J Am Chem Soc. 1993;115:6506. [Google Scholar]
- 9.Tokuyama H, Yamago S, Nakamura E, Shiraki T, Sugiura Y. J Am Chem Soc. 1993;115:7918. [Google Scholar]
- 10.Raschke TM, Levitt M. J Phys Chem B. 2004;108:13492. [Google Scholar]
- 11.Lum K, Chandler D, Weeks JD. J Phys Chem B. 1999;103:4570. [Google Scholar]
- 12.Ashbaugh HS, Paulaitis ME. J Am Chem Soc. 2001;123:10721. doi: 10.1021/ja016324k. [DOI] [PubMed] [Google Scholar]
- 13.Wallqvist A, Gallicchio E, Levy RM. J Phys Chem B. 2001;105:6745. [Google Scholar]
- 14.Chandler D. Nature. 2005;437:640. doi: 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- 15.Lee B. Biopolymers. 1991;31:993. doi: 10.1002/bip.360310809. [DOI] [PubMed] [Google Scholar]
- 16.Pratt LR. Annu Rev Phys Chem. 2002;53:409. doi: 10.1146/annurev.physchem.53.090401.093500. [DOI] [PubMed] [Google Scholar]
- 17.Huang X, Margulis CJ, Berne BJ. Proc Natl Acad Sci USA. 2003;100:11953. doi: 10.1073/pnas.1934837100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang X, Margulis CJ, Berne BJ. J Phys Chem B. 2003;107:11742. [Google Scholar]
- 19.Huang DM, Chandler D. J Phys Chem B. 2002;106:2047. [Google Scholar]
- 20.Walther JH, Jaffe RL, Kotsalis EM, Werder T, Halicioglu T, Koumoutsakos P. Carbon. 2004;42:1185. [Google Scholar]
- 21.Hua L, Huang XH, Zhou RH, Berne BJ. J Phys Chem B. 2006;110:3704. doi: 10.1021/jp055399y. [DOI] [PubMed] [Google Scholar]
- 22.Despa F, Berry RS. Biophys J. 2007;92:373. doi: 10.1529/biophysj.106.087023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Despa F, Fernandez A, Berry RS. Phys Rev Lett. 2004;93:228104. doi: 10.1103/PhysRevLett.93.228104. [DOI] [PubMed] [Google Scholar]
- 24.Sharp KA, Madan B, Manas E, Vanderkooi JM. J Chem Phys. 2001;114:1791. [Google Scholar]
- 25.Gallagher KR, Sharp KA. J Am Chem Soc. 2003;125:9853. doi: 10.1021/ja029796n. [DOI] [PubMed] [Google Scholar]
- 26.Hornak J. The Basics of NMR. 1997–1999 http://www.cis.rit.edu/htbooks/nmr/nmr-main.htm.
- 27.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. J Chem Phys. 1995;103:8577. [Google Scholar]
- 28.Chandler D, Weeks JD, Andersen HC. Science. 1983;220:787. doi: 10.1126/science.220.4599.787. [DOI] [PubMed] [Google Scholar]
- 29.Allen M, Tildesley D. Computer Simulation of Liquids. Oxford Science Publications; Oxford, U.K: 1987. [Google Scholar]
- 30.Raschke TM, Levitt M. Proc Natl Acad Sci USA. 2005;102:6777. doi: 10.1073/pnas.0500225102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Astley T, Birch GG, Drew MGB, Rodger PM, Wilden GRH. J Comput Chem. 1998;19:363. [Google Scholar]
- 32.Einstein A. Investigations on the Theory of Brownian Movement. Dover; New York: 1956. [Google Scholar]
- 33.Claridge T. High-Resolution NMR Techniques in Organic Chemistry. Elsevier; 1999. [Google Scholar]
- 34.Mark P, Nilsson L. J Phys Chem A. 2001;105:9954. [Google Scholar]
- 35.Finney JL, Soper AK. Chem Soc Rev. 1994;23:1. [Google Scholar]
- 36.Krynicki K, Green CD, Sawyer DW. Faraday Discuss. 1978:199. [Google Scholar]
- 37.David van der S, Paul JvM, Herman JCB. J Chem Phys. 1998;108:10220. [Google Scholar]
- 38.Chirico G, Placidi M, Cannistraro S. J Phys Chem B. 1999;103:1746. [Google Scholar]
- 39.Bezmel’nitsyn VN, Eletskii AV, Okun’ MV. Usp Fiz Nauk. 1998;168:1195. [Google Scholar]
- 40.Johnson RD, Yannoni C, Dorn HC, Salem J, Bethune DS. Science. 1992;255:1235. doi: 10.1126/science.255.5049.1235. [DOI] [PubMed] [Google Scholar]
- 41.Jensen TR, Jensen MO, Reitzel N, Balashev K, Peters GH, Kjaer K, Bjornholm T. Phys Rev Lett. 2003;90:086101. doi: 10.1103/PhysRevLett.90.086101. [DOI] [PubMed] [Google Scholar]
- 42.Frauenfelder H, Fenimore PW, Chen G, McMahon BH. Proc Natl Acad Sci USA. 2006;103:15469. doi: 10.1073/pnas.0607168103. [DOI] [PMC free article] [PubMed] [Google Scholar]