

Summary
Hemoglobin breakdown produces an iron-dependent neuronal injury after experimental CNS hemorrhage that may be attenuated by heme oxygenase (HO) inhibitors. The HO enzymes are phosphoproteins that are activated by phosphorylation in vitro. While testing the effect of kinase inhibitors in cortical cell cultures, we observed that HO activity was consistently decreased by the MEK inhibitor U0126. The present study tested the hypothesis that MEK/ERK pathway inhibitors reduce HO activity and neuronal vulnerability to hemoglobin. The MEK inhibitors U0126 and SL327 and the ERK inhibitor FR180204 reduced baseline culture HO activity by 35–50%, without altering the activity of recombinant HO-1 or HO-2; negative control compounds U0124 and FR180289 had no effect. Hemoglobin exposure for 16 hours produced widespread neuronal injury, manifested by release of 59.2±7.8% of neuronal lactate dehydrogenase and a twelve-fold increase in malondialdehyde; kinase inhibitors were highly protective. HO-1 induction after hemoglobin treatment was also decreased by U0126, SL327, and FR180204. These results suggest that reduction in HO activity may contribute to the protective effect of MEK and ERK inhibitors against heme-mediated neuronal injury.
Keywords: cell culture, free radical, hemoglobin toxicity, intracerebral hemorrhage, mouse, oxidative stress
Introduction
A considerable body of experimental and clinical evidence suggests that toxins released from an intracerebral hematoma may contribute to cell injury in adjacent tissue (Xi, et al., 2006). One putative neurotoxin is hemoglobin, the most abundant protein in blood, which is released from lysed erythrocytes in the days after hemorrhage and contributes to peri-hematomal edema and oxidative stress (Huang, et al., 2002). Investigation of hemoglobin neurotoxicity in cell culture models and in vivo suggests that the hemoglobin molecule per se is not the primary toxin (Sadrzadeh, et al., 1987, Regan, et al., 1993). However, at least under some experimental conditions, the quantity of iron released as a consequence of the breakdown of its heme moieties apparently exceeds the sequestration or export capacity of CNS cells. The result is an injury that is largely selective for neurons, which are highly sensitive to low molecular weight iron (Kress, et al., 2002).
Heme degradation to equimolar quantities of iron, biliverdin, and carbon monoxide is catalyzed by the heme oxygenase (HO) enzymes (Abraham, et al., 2008). Two isoforms have been identified to date in the mammalian CNS (Schipper, 2004). Heme oxygenase-1 is expressed primarily by glial cells and is induced by heat shock, heme, and a variety of oxidants. Heme oxygenase-2 is constitutively expressed by neurons and endothelial cells. The effect of heme oxygenase activity on acute CNS injury has been extensively investigated in studies using either HO inhibitors or genetically modified mice. A protective effect has been consistently observed in models that are relevant to ischemia or trauma (Takizawa, et al., 1998, Panahian, et al., 1999, Chang, et al., 2003), which has been attributed to the antioxidant and anti-inflammatory effects of biliverdin/bilirubin and carbon monoxide (Abraham, et al., 2008, Parfenova, et al., 2008). In contrast, HO activity increased or accelerated injury in most (Wagner, et al., 2000, Koeppen, et al., 2002, Koeppen, et al., 2004, Gong, et al., 2006, Wang, et al., 2006a, Qu, et al., 2007) but not all (Wang, et al., 2006b) experimental models of intracerebral hemorrhage (ICH), presumably due to iron toxicity that negated any benefit of the other breakdown products.
Clinical ICH is a complex injury that may include varying degrees of compressive ischemia, mechanical injury from hematoma expansion or retraction, inflammation, and the toxicity of blood components (Xi, et al., 2006). The disparate effect of HO on heme-mediated and other CNS injuries suggests that it may be a challenging therapeutic target, since any benefit of direct HO inhibitors against hemoglobin neurotoxicity may be negated by their deleterious effects on other injury cascades. An alternative approach to direct enzyme inhibition is to prevent the increase in HO activity produced by hemorrhage, which may be due to HO activation and/or HO-1 induction. Both HO-1 and HO-2 are phosphoproteins, and in vitro are activated by the phosphatidylinositol-3-kinase and protein kinase C/CK2 pathways, respectively (Boehning, et al., 2003, Salinas, et al., 2004). However, we have recently observed that selective inhibitors of these pathways had no effect on HO activity in murine cortical cell cultures (Chen-Roetling, et al., 2008). In the course of these kinase inhibitor experiments, we noted that the MEK 1/2 inhibitor U0126 surprisingly reduced baseline culture HO activity. In the present study, we tested the effect of MEK and ERK inhibitors on HO activity and hemoglobin neurotoxicity in this culture system.
Methods
Cortical cell cultures
All procedures on animals were conducted in accordance with a protocol approved by the Thomas Jefferson University Institutional Animal Care and Use Committee (IACUC). Mixed neuron–glia cortical cell cultures were prepared from fetal B6129 mice (gestational age 13- to 15-days), using a previously described protocol (Rogers, et al., 2003). After cell dissociation by trituration, cultures were plated on confluent glial feeder cultures in 24-well plates (Falcon, Becton Dickinson, Franklin Lakes, NJ), at a density of three hemispheres/plate. Plating medium contained Minimal Essential Medium (MEM, Invitrogen, Carlsbad, CA), 5% equine serum (Hyclone, Logan, UT), 5% fetal bovine serum (Hyclone), 23 mM glucose, and 2 mM glutamine. On day 5 in vitro, two-thirds of the culture medium was aspirated and replaced with feeding medium, which was similar to plating medium except that it contained 10% equine serum and no fetal bovine serum. This procedure was repeated on day 9 or 10 and then daily beginning on day 11. Glial feeder cultures were prepared from postnatal day 1–3 mice, using plating medium similar to that described above, except that it was supplemented with 10 ng/ml epidermal growth factor (Sigma, St. Louis, MO), 10% equine serum and 10% fetal bovine serum. Glial culture medium was partially changed twice weekly.
MEK/ERK pathway inhibitors and negative controls
U0126 (Promega, Madison, WI) is a potent inhibitor of both MEK1 and MEK2 (Favata, et al., 1998). It differs from the commonly-used MEK inhibitor PD98059 by directly inhibiting MEK enzyme activity, rather than by preventing its activation by Raf, and also by having much greater activity against MEK2 (Alessi, et al., 1995). U0124 (Tocris Bioscience, Ellisville, Missouri) is a similar compound that does not inhibit MEK, and is marketed as a negative control. SL327 (Tocris) also selectively inhibits both MEK1 and MEK 2 (Scherle, et al., 2000). FR180204 (EMD/Calbiochem, San Diego, CA) is inactive against MEK, but directly inhibits ERK1 (MAPK3) and ERK2 (MAPK3(Ohori, et al., 2005). FR180289 (EMD) differs from FR180204 only by substitution of a hydroxyl group for the 3′ amine (Ohori, et al., 2005), but does not inhibit ERK1 or ERK2 and is marketed as its negative control.
HO activity assay
HO activity was quantified using a modification of the method of Vreman and Stevenson (Vreman, et al., 1988). Concentrated inhibitor stock solutions were freshly prepared in dimethylsulfoxide (DMSO). Dilutions were adjusted so that all conditions including sham-wash controls and hemoglobin without inhibitors had the same DMSO concentration (0.25%). Cultures were treated for either 30 minutes or 4 hours with enzyme inhibitors, hemoglobin, or both in MEM containing 10 mM glucose (MEM10) at 37°C in a 5% CO2 incubator; control cultures received MEM10 with DMSO vehicle only. At the end of the exposure interval, cultures were washed and harvested in ice-cold Dulbecco’s Phosphate Buffered Saline (DPBS) with 3X concentration of inhibitors or DMSO vehicle. Samples of the cell suspension (40 μl) were placed in amber septum-sealed glass vials on ice, and were diluted with equal volumes of freshly prepared 75 μM hemin and 4.5 mM NADPH (final reactant concentrations 25 μM hemin, 1.5 mM NADPH, total volume 120 μl); control vials lacked NADPH. Vials were purged for 4 sec with CO-free air at a flow rate of 250 ml/min. Reactions were then run for 15 minutes at 37°C in a water bath under reduced light, and were terminated by quick-freezing vials on dry ice. CO was quantified in the vial head space by gas chromatography (Peak Laboratories, Mountain View, CA). HO activity was expressed as nanomoles CO produced per hour per milligram protein. Protein concentrations were determined by the BCA method (Pierce, Rockford, IL).
Immunoblotting
Cells were lysed in ice-cold lysis buffer (210 mM mannitol, 70 mM sucrose, 5 mM HEPES, 1 mM EDTA, 0.1 % sodium dodecyl sulfate, 0.1 % Triton X-100). After sonication, debris was removed by centrifugation, and the protein concentration of the supernatant was quantified (BCA method, Pierce, Rockford, IL). Samples (20 μg protein each) were then boiled in sample buffer (Tris-Cl 60 mM, β-mercaptoethanol 5%, sodium dodecyl sulfate 2%, glycerol 10%, and bromophenol blue 0.05%) for 3 min. Proteins were separated on 12 % SDS polyacrylamide gels at 80–100 V. The gels were then soaked in transfer buffer (glycine 39mM, Tris-CL 48mM, SDS 0.037% and methanol 20%), and were transferred to polyvinylidene difluoride membranes with a semidry transfer apparatus for 40 minutes at 20V. Completion of transfer was assessed by observing the transfer of the pre-stained protein marker (Bio-Rad Laboratories, Hercules, CA, Cat. No. 161–0375). After blocking with 5% nonfat dry milk, membranes were exposed overnight at 4 °C to rabbit anti-HO-1 or anti-HO-2 antibodies (Assay Designs, Ann Arbor, MI, 1:5000 and 1:2000 dilutions, respectively) combined with rabbit anti-actin antibody (1/1500, Sigma, St. Louis, MO ) as a gel loading control. After washing, they were then exposed to HRP-conjugated goat anti-rabbit IgG antibody (1:3500) for 1h at room temperature. Immunoreactive proteins were visualized using Super Signal West Femto Reagent (Pierce) and Kodak Gel Logic 2200.
Immunocytochemistry
Cultures were washed with MEM10 and then fixed with ice-cold 4% paraformaldehyde for one hour. After washing with tris-buffered saline (TBS), cultures were serially treated with: 0.25% Triton X-100 for 10 min, 10% normal goat serum for 15 min, 1:100 dilutions of primary antibodies (anti-NeuN, clone A60, Alexa Fluor®488 conjugated, Millipore, Billerica, MA or rabbit anti-glial fibrillary acidic protein (GFAP), Invitrogen, Carlsbad, CA) overnight. Cultures treated with anti-NeuN were then washed and imaged. Cultures treated with anti-GFAP were treated for 30 min with biotinylated anti-rabbit IgG (1:200, Vector Laboratories, Burlingame, CA) followed by NeutrAvidin Rhodamine Red-X conjugate (Invitrogen) 5 μg/ml for 30 min.
ERK activity assay
The effect of inhibitors and negative controls was confirmed using the p44/42 MAP Kinase assay kit (Cell Signaling Technology, Danvers, MA), following the manufacturer’s instructions. Cell lysates were incubated overnight at 4°C with immobilized phospho-p44/42 ERK monoclonal antibody. Immunoprecipitation pellets were then collected by centrifugation and were incubated in kinase buffer containing substrate (Elk-1 fusion protein) and 200 μM ATP at 30°C for 30 minutes. Phosphorylated Elk-1 was detected by immunoblotting as described above, using a 1:1000 dilution of rabbit anti-phospho-Elk (Ser 383) primary antibody.
Cytotoxicity experiments
Cultures were used for experiments on 12–16 days in vitro. Neurons are easily distinguished from glial cells during this interval by their phase-bright cell bodies and extensive network of processes. All exposures were conducted in MEM10 at 37°C in a 5% CO2 atmosphere. As described above, the dilution of all inhibitors was adjusted so that all conditions contained 0.25% DMSO vehicle. Control cultures in each experiment were subjected to medium exchange only, following which they were treated with an equal concentration of DMSO vehicle. The hemoglobin exposure concentration was determined from prior studies using this model, which demonstrated that 10 μM hemoglobin (tetramer concentration) produced widespread neuronal injury with overnight treatment (Rogers, et al., 2003), without injuring astrocytes (Chen-Roetling, et al., 2006).
Assessment of injury
Neuronal death was quantified by assaying lactate dehydrogenase (LDH) activity in the culture medium, a method that correlates well with cell counts after trypan blue staining in mixed cortical cultures (Koh, et al., 1988), as previously described (Regan, et al., 1998). In order to compare results from experiments using cultures prepared in different platings, all LDH values were normalized to those in sister cultures from the same plating treated with 300 μM NMDA, which releases virtually all neuronal LDH without injuring glial cells. The mean baseline LDH activity of sister cultures subjected to medium exchange and vehicle treatment only was subtracted from all values to quantify the signal that was specific to the cytotoxic exposure, following the protocol of Koh and Choi (Koh, et al., 1988).
Malondialdehyde is a sensitive marker of oxidative injury in this model (Regan, et al., 1998). After sampling for LDH assay, cultures were harvested, proteins were precipitated with 4.5% trichloroacetic acid, and malondialdehyde was quantified as previously detailed (Regan, et al., 1998). Protein content of each sample was estimated by the BCA method (Pierce, Rockford, IL); all malondialdehyde values were expressed as nanomoles/milligram protein.
Statistical Analysis
Data were analyzed with one-way analysis of variance. Differences between groups were then assessed with the Bonferroni Multiple Comparisons test.
Results
MEK and ERK inhibitors attenuate culture HO activity
In initial experiments, the effect of 30 min incubation with MEK or ERK inhibitors on culture HO activity was assessed. HO activity in control cultures treated with DMSO vehicle only for this interval was 1.01±0.07 nmol CO/h/mg protein (Fig. 1A). In cultures treated with the MEK inhibitors U0126 or SL327 or the ERK inhibitor FR180204, a significant reduction in HO activity was observed. U0124 and FR180289, structural analogs of U0126 and FR180204 that are marketed as negative controls for these compounds, had no effect on HO activity at the same concentrations. Kinase activity assays confirmed that, at the concentrations used, U0126, SL327, and FR180204 inhibited phosphorylation of Elk-1 by ERK, while U0124 and FR180289 had no such effect (Fig. 1B, C).
Figure 1.

MEK and ERK inhibitors reduce culture heme oxygenase (HO) activity. A) Mean HO activity (nmol CO/h/mg protein ±SEM, n = 5–12/condition) in cultures washed and then treated with DMSO vehicle only (Veh) or with U0126 (30 μM), SL327 (30 μM), FR180204 (FR, 50 μM), U0124 (negative control for U0126, 30 μM), or FR180289 (FRN, negative control for FR180204, 50 μM) for 30 minutes. ***P<0.001 and **P<0.01 v. activity in vehicle-treated cultures, Bonferroni multiple comparisons test. B) Immunoblot demonstrating effect of MEK inhibitors and negative control U0124 on kinase activity at these concentrations, as assessed by phosphorylation of ERK substrate Elk-1. Cultures were treated with hemoglobin (Hb) 10 μM alone or with inhibitors or control for 4 hours before lysis and immunoprecipation of active ERK for kinase assay. C) Immunoblot demonstrating inhibition of active ERK by FR180204 (FR, 50 μM) but not by FR180289 (FRN, 50 μM).
In order to determine if these MEK and ERK inhibitors were direct HO inhibitors, their effect on the activity of recombinant HO-1 and HO-2 (both purchased from Assay Designs, Ann Arbor, MI) was determined. In the presence of DMSO vehicle only, recombinant HO-1 and HO-2 had activities of 297.0±39.0 nmol CO/h/mg protein and 504.2±44.1 nmol CO/h/mg protein, respectively (Fig. 2). These activities were not significantly altered by U0126, SL327, or FR180204. Not surprisingly, the activity of both recombinant isoenzymes was strongly inhibited by the HO inhibitor zinc protoporphyrin IX (50 μM).
Figure 2.

Effect of MEK and ERK inhibitors on activity of recombinant HO-1 and HO-2. HO activity of recombinant rat HO-1 and HO-2 with DMSO vehicle alone (Veh) or with U0126 (30 μM), SL327 (30 μM), FR180204 (FR, 50 μM), or zinc protoporphyrin IX (ZnPPIX, 50 μM). Reaction vial contained 0.25μg cytochrome P450 reductase with 2.5 μg HO-1 or 1 μg HO-2. **P<0.01, **P<0.001 v. corresponding vehicle condition, Bonferroni multiple comparisons test.
MEK and ERK inhibitors protect neurons from hemoglobin
We have previously reported that reducing HO activity by either HO-2 gene knockout or treatment with an HO inhibitor protects neurons in this culture system from hemoglobin (Rogers, et al., 2003). Since MEK and ERK inhibitors rapidly reduced HO activity, their effect on hemoglobin neurotoxicity was assessed. Consistent with prior observations, widespread neuronal injury was observed in cultures treated for 16h with 10 μM hemoglobin plus DMSO vehicle only (Fig. 3); the glial monolayer remained intact and morphologically normal throughout the course of the experiment, also as previously reported (Chen-Roetling, et al., 2006). Cell death, as quantified by LDH release into the culture medium, was 59.2±7.8% of that in sister cultures treated with 300 μM NMDA, which releases essentially all neuronal LDH (Fig. 4A). Concomitant treatment with U0126, SL327, or FR180204 consistently had a robust protective effect (Fig. 3, 4A).
Figure 3.
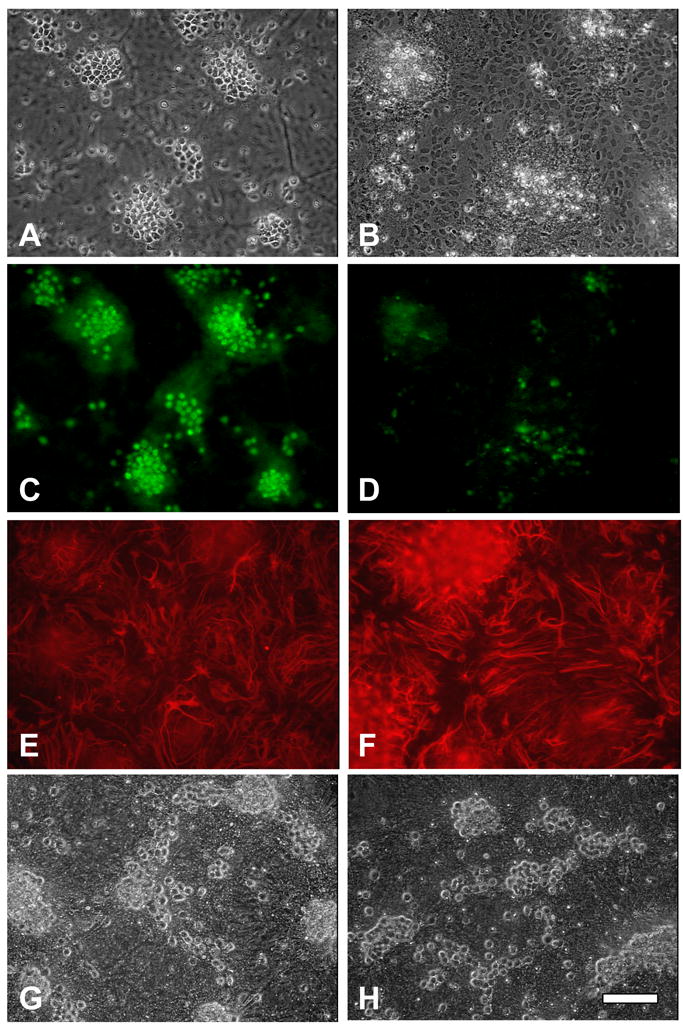
Morphologic appearance of mixed neuron-astrocyte cultures 24h after: A, C, E) Treatment with vehicle only. Neurons with phase-bright cell bodies appear primarily in clusters on the glial feeder layer monolayer, which is in a different plane and therefore slightly out of focus. Phase-bright cells stain with Alexa Fluor®488 conjugated anti-NeuN (C), confirming neuronal identity, while background glial monolayer stains with anti-GFAP (E). B, D, F) Treatment with hemoglobin 10 μM with vehicle. Most neurons have degenerated to debris, astrocyte monolayer is intact (B), NeuN immunoreactivity is diminished and limited to areas of degenerating cells (D), GFAP immunoreactivity persists and is increased, particularly near degenerating neurons (E). G, H) Unfixed cultures treated with hemoglobin 10 μM plus U0126 (30 μM) or FR180204 (50 μM), respectively; neuronal morphology is preserved. Scale bar = 100 μm.
Figure 4.

MEK and ERK inhibitors protect neurons from hemoglobin. A) Cultures were treated for 16h with hemoglobin (Hb) 10 μM plus DMSO vehicle alone or with U0126 (30 μM), SL327 (30 μM), or FR180204 (FR, 50 μM). LDH values are scaled to mean values in sister cultures treated with 300 μM N-methyl-D-aspartate (NMDA), which produces near-100% neuronal death, after subtraction of mean LDH in sister cultures subjected to wash and vehicle treatment only, in order to yield the LDH signal specific for hemoglobin neurotoxicity. B) Culture malondialdehyde (MDA) after treatment as in A. ***P<0.001. v. Hb, Bonferroni multiple comparisons test, n = 8–23/condition.
MEK and ERK inhibitors reduce expression of oxidative injury markers after hemoglobin treatment
Malondialdehyde is a sensitive marker of the oxidative injury produced by hemoglobin in CNS cells (Sadrzadeh, et al., 1987). In cultures treated with hemoglobin plus vehicle only, it was increased by 12.5-fold compared with control cultures subjected to medium exchange and vehicle treatment only (Fig. 4B). Malondialdehyde levels in cultures treated with hemoglobin plus kinase inhibitors were similar to those in vehicle-treated controls.
HO-1 is rapidly induced by oxidants in CNS cells (Schipper, 2004). Since MEK and ERK inhibitors appeared to be acting as antioxidants in this model, their effect on HO-1 expression was determined. In cultures treated with hemoglobin plus DMSO vehicle for 4h, which is a time point prior to the onset of neuronal lysis, HO-1 expression was increased 2.2-fold over control cultures treated with vehicle only (Fig. 5A). Consistent with prior observations (Benvenisti-Zarom, et al., 2006), this increase was reduced by about half by U0126 and SL327, and was completely prevented by the direct ERK inhibitor FR180204. HO-2 expression was not altered by hemoglobin or kinase inhibitor treatment. HO activity was increased approximately 2.5-fold by hemoglobin (Fig. 5B). This increase was completely prevented by concomitant treatment with U0126, SL327, or FR180204.
Figure 5.

MEK and ERK inhibitors prevent HO-1 induction after hemoglobin treatment. A) Expression of HO-1, HO-2 and actin (gel loading control) in cultures treated with vehicle (Veh) only for 4h, with hemoglobin (Hb) 10 μM plus vehicle, or with Hb plus U0126 (30 μM), SL327 (30 μM), or FR180204 (FR, 50 μM). B) HO activity in cultures treated as in A. ###P<0.001. v. vehicle, *P<0.05, **P<0.01, ***P<0.001 v. Hb, Bonferroni multiple comparisons test, n = 5–13/condition.
Discussion
These results suggest the following conclusions. First, baseline HO activity is attenuated by inhibitors of the MEK/ERK pathway in this mixed neuron/glia cell culture system. Second, this decrease is associated with reduced neuronal vulnerability to hemoglobin, in agreement with prior observations that HO activity accelerates hemoglobin neurotoxicity in cell culture and in vivo (Huang, et al., 2002, Rogers, et al., 2003). Third, HO-1 induction in this hemoglobin neurotoxicity model is also reduced by these inhibitors, suggesting that it is mediated at least in part by the MEK/ERK signal transduction pathway.
The inhibition in HO activity after brief incubation with ERK or MEK inhibitors was unexpected, since activation of HO-1 or HO-2 by ERK-catalyzed phosphorylation has not been reported. HO-1 has possible ERK phosphorylation sites at T108, S174, and T252, and an ERK1 binding site at P170, while HO2 has several ERK docking domain sites (Obenauer, et al., 2003). The effect of ERK-catalyzed phosphorylation on HO activity has not been defined and seems worthy of further investigation. It is unlikely that the inhibitors acted directly on HO catalysis, since they had no significant effect on the activity of recombinant HO-1 or HO-2. In assessing these compounds in oxidative injury models, the potentially confounding effect of reduced HO activity should be considered.
The protective effect of MEK and ERK inhibitors provides an additional line of evidence that excessive HO activity may be detrimental to neurons exposed to hemoglobin, and complements prior and more specific observations using HO-2 knockout neurons or HO inhibitors (Huang, et al., 2002, Rogers, et al., 2003). A growing body of experimental evidence suggests that HO activity may have either antioxidant or pro-oxidant effects, with the net effect varying with the cell population and the type of injury. Two factors may account for a pro-oxidant effect in this hemoglobin neurotoxicity model. First, HO substrate was added to cultures at a relatively high concentration (10 μM hemoglobin, containing 40 μM heme). While this concentration is considerably lower than that adjacent to an intracerebral hemorrhage (Letarte, et al., 1993), it is markedly higher than the endogenous heme that is available to cells in models more relevant to CNS ischemia (~ 1 nM, Taketani, 2005), in which HO activity is protective (Takizawa, et al., 1998, Panahian, et al., 1999). Second, neurons are very vulnerable to low molecular weight iron (Kress, et al., 2002), a product of heme breakdown by HO, which likely reflects their limited capacity to sequester it in ferritin (Moos, et al., 2004). Release of supraphysiologic quantities of iron may overwhelm any benefit provided by HO activity (Dennery, et al., 2003), which includes bilirubin and carbon monoxide production (Abraham, et al., 2008, Parfenova, et al., 2008).
In addition to reducing HO activity, several other mechanisms may contribute to the robust neuroprotection provided by U0126, SL327, and FR180204 in this model. Activation of the MEK/ERK pathway may mediate neuronal injury in an oxidative environment by directly activating calpain (Glading, et al., 2000), phospholipase A2 (Geijsen, et al., 2000), and NADPH oxidase (Dewas, et al., 2000), and also by increasing production of inflammatory cytokines (Wang, et al., 2004). In addition, ERK signaling mediates apoptosis in CNS cells by upregulating Bax and p53 while downregulating Akt (Zhuang, et al., 2006). Although the association of reduced HO activity and neuronal resistance to hemoglobin is consistent with prior observations using more specific approaches (Huang, et al., 2002, Rogers, et al., 2003), the downstream mechanistic information provided by inhibiting the MEK/ERK pathway is limited by the myriad effects of ERK signaling.
We have previously reported that HO-1 is expressed at a low level in primary cultured neurons and glia, and that it is rapidly induced in glia by hemoglobin or hemin treatment (Regan, et al., 2000, Benvenisti-Zarom, et al., 2006). In contrast to the effect of HO on neurons, HO-1 expression protects glial cells from hemoglobin (Regan, et al., 2000, Chen-Roetling, et al., 2006). Prolonged hemoglobin treatment produces no injury in glial cultures prepared from wild-type mice, but widespread cell death in HO-1 knockout cultures (Chen-Roetling, et al., 2006), and also in wild-type cultures treated concomitantly with HO inhibitors (Regan, et al., 2000). In the present study, the glial feeder monolayer remained morphologically normal after treatment with hemoglobin plus MEK or ERK inhibitors despite a reduction in culture HO-1 expression and HO activity. Immunostaining demonstrated that most of these glial cells were astrocytes; however, a low level of microglial contamination cannot be excluded (Saura, 2007), and may have contributed to neuronal injury in this model. The disparate effect of direct HO inhibitors and MEK/ERK inhibitors on astrocyte vulnerability to hemoglobin suggests that astrocytes treated with the latter have sufficient residual HO activity to prevent hemoglobin-mediated injury. Alternatively, MEK and ERK inhibitors may antagonize other ERK-activated injury cascades in astrocytes, and thereby compensate for the deleterious effect of reduced HO activity in this cell population.
MEK/ERK pathway inhibitors have been extensively tested in small animal models of CNS ischemia and trauma, and have been found to reduce lesion volume and neurological deficits (Namura, et al., 2001, Wang, et al., 2003, Clausen, et al., 2004, Wang, et al., 2004, Otani, et al., 2007). However, comparable studies have not yet been reported in intracerebral hemorrhage models. The results of the present study suggest that U0126, SL 327, and FR180204 may ameliorate the component of injury produced by the breakdown of extracellular hemoglobin in tissue surrounding an intracerebral hematoma. Investigation of these compounds in vivo therefore seems warranted.
Acknowledgments
Funding for this study was provided by a grant from the National Institutes of Health (NS050662).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev. 2008;60:79–127. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Benvenisti-Zarom L, Chen-Roetling J, Regan RF. Inhibition of the ERK/MAP kinase pathway attenuates heme oxygenase-1 expression and heme-mediated neuronal injury. Neurosci Lett. 2006;398:230–234. doi: 10.1016/j.neulet.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Boehning D, Moon C, Sharma S, Hurt KJ, Hester LD, Ronnett GV, Shugar D, Snyder SH. Carbon monoxide neurotransmission activated by CK2 phosphorylation of heme oxygenase-2. Neuron. 2003;40:129–137. doi: 10.1016/s0896-6273(03)00596-8. [DOI] [PubMed] [Google Scholar]
- Chang EF, Wong RJ, Vreman HJ, Igarashi T, Galo E, Sharp FR, Stevenson DK, Noble-Haeusslein LJ. Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury. J Neurosci. 2003;23:3689–3696. doi: 10.1523/JNEUROSCI.23-09-03689.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Roetling J, Li Z, Regan RF. Hemoglobin neurotoxicity is attenuated by inhibitors of the protein kinase CK2 independent of heme oxygenase activity. Curr Neurovasc Res. 2008;5:193–198. doi: 10.2174/156720208785425684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Roetling J, Regan RF. Effect of heme oxygenase-1 on the vulnerability of astrocytes and neurons to hemoglobin. Biochem Biophys Res Commun. 2006;350:233–237. doi: 10.1016/j.bbrc.2006.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen F, Lundqvist H, Ekmark S, Lewen A, Ebendal T, Hillered L. Oxygen free radical-dependent activation of extracellular signal-regulated kinase mediates apoptosis-like cell death after traumatic brain injury. J Neurotrauma. 2004;21:1168–1182. doi: 10.1089/neu.2004.21.1168. [DOI] [PubMed] [Google Scholar]
- Dennery PA, Visner G, Weng YH, Nguyen X, Lu F, Zander D, Yang G. Resistance to hyperoxia with heme oxygenase-1 disruption: role of iron. Free Radic Biol Med. 2003;34:124–133. doi: 10.1016/s0891-5849(02)01295-9. [DOI] [PubMed] [Google Scholar]
- Dewas C, Fay M, Gougerot-Pocidalo MA, El-Benna J. The mitogen-activated protein kinase extracellular signal-regulated kinase 1/2 pathway is involved in formyl-methionyl-leucyl-phenylalanine-induced p47phox phosphorylation in human neutrophils. J Immunol. 2000;165:5238–5244. doi: 10.4049/jimmunol.165.9.5238. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Geijsen N, Dijkers PF, Lammers JJ, Koenderman L, Coffer PJ. Cytokine-mediated cPLA(2) phosphorylation is regulated by multiple MAPK family members. FEBS Letters. 2000;471:83–88. doi: 10.1016/s0014-5793(00)01373-9. [DOI] [PubMed] [Google Scholar]
- Glading A, Chang P, Lauffenburger DA, Wells A. Epidermal growth factor activation of calpain is required for fibroblast motility and occurs via an ERK/MAP kinase signaling pathway. J Biol Chem. 2000;275:2390–2398. doi: 10.1074/jbc.275.4.2390. [DOI] [PubMed] [Google Scholar]
- Gong Y, Tian H, Xi G, Keep RF, Hoff JT, Hua Y. Systemic zinc protoporphyrin administration reduces intracerebral hemorrhage-induced brain injury. Acta Neurochir Suppl. 2006;96:232–236. doi: 10.1007/3-211-30714-1_50. [DOI] [PubMed] [Google Scholar]
- Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96:287–293. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- Koeppen AH, Dickson AC. Tin-protoporphyrin prevents experimental superficial siderosis in rabbits. J Neuropathol Exp Neurol. 2002;61:689–701. doi: 10.1093/jnen/61.8.689. [DOI] [PubMed] [Google Scholar]
- Koeppen AH, Dickson AC, Smith J. Heme oxygenase in experimental intracerebral hemorrhage: the benefit of tin-mesoporphyrin. J Neuropathol Exp Neurol. 2004;63:587–597. doi: 10.1093/jnen/63.6.587. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Vulnerability of cultured cortical neurons to damage by excitotoxins: Differential susceptibility of neurons containing NADPH-diaphorase. J Neurosci. 1988;8:2153–2163. doi: 10.1523/JNEUROSCI.08-06-02153.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress GJ, Dineley KE, Reynolds IJ. The relationship between intracellular free iron and cell injury in cultured neurons, astrocytes, and oligodendrocytes. J Neurosci. 2002;22:5848–5855. doi: 10.1523/JNEUROSCI.22-14-05848.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letarte PB, Lieberman K, Nagatani K, Haworth RA, Odell GB, Duff TA. Hemin: levels in experimental subarachnoid hematoma and effects on dissociated vascular smooth muscle cells. J Neurosurg. 1993;79:252–255. doi: 10.3171/jns.1993.79.2.0252. [DOI] [PubMed] [Google Scholar]
- Moos T, Morgan EH. The metabolism of neuronal iron and its pathogenic role in neurological disease: review. Ann N Y Acad Sci. 2004;1012:14–26. doi: 10.1196/annals.1306.002. [DOI] [PubMed] [Google Scholar]
- Namura S, Iihara K, Takami S, Nagata I, Kikuchi H, Matsushita K, Moskowitz MA, Bonventre JV, Alessandrini A. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci U S A. 2001;98:11569–11574. doi: 10.1073/pnas.181213498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohori M, Kinoshita T, Okubo M, Sato K, Yamazaki A, Arakawa H, Nishimura S, Inamura N, Nakajima H, Neya M, Miyake H, Fujii T. Identification of a selective ERK inhibitor and structural determination of the inhibitor-ERK2 complex. Biochem Biophys Res Commun. 2005;336:357–363. doi: 10.1016/j.bbrc.2005.08.082. [DOI] [PubMed] [Google Scholar]
- Otani N, Nawashiro H, Fukui S, Ooigawa H, Ohsumi A, Toyooka T, Shima K. Role of the activated extracellular signal-regulated kinase pathway on histological and behavioral outcome after traumatic brain injury in rats. J Clin Neurosci. 2007;14:42–48. doi: 10.1016/j.jocn.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Panahian N, Yoshiura M, Maines MD. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J Neurochem. 1999;72:1187–1203. doi: 10.1111/j.1471-4159.1999.721187.x. [DOI] [PubMed] [Google Scholar]
- Parfenova H, Leffler CW. Cerebroprotective functions of HO-2. Curr Pharm Des. 2008;14:443–453. doi: 10.2174/138161208783597380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Chen-Roetling J, Benvenisti-Zarom L, Regan RF. Attenuation of oxidative injury after induction of experimental intracerebral hemorrhage in heme oxygenase-2 knockout mice. J Neurosurg. 2007;106:428–435. doi: 10.3171/jns.2007.106.3.428. [DOI] [PubMed] [Google Scholar]
- Regan RF, Guo YP, Kumar N. Heme oxygenase-1 induction protects murine cortical astrocytes from hemoglobin toxicity. Neurosci Lett. 2000;282:1–4. doi: 10.1016/s0304-3940(00)00817-x. [DOI] [PubMed] [Google Scholar]
- Regan RF, Jasper E, Guo YP, Panter SS. The effect of magnesium on oxidative neuronal injury in vitro. J Neurochem. 1998;70:77–85. doi: 10.1046/j.1471-4159.1998.70010077.x. [DOI] [PubMed] [Google Scholar]
- Regan RF, Panter SS. Neurotoxicity of hemoglobin in cortical cell culture. Neurosci Lett. 1993;153:219–222. doi: 10.1016/0304-3940(93)90326-g. [DOI] [PubMed] [Google Scholar]
- Rogers B, Yakopson V, Teng ZP, Guo Y, Regan RF. Heme oxygenase-2 knockout neurons are less vulnerable to hemoglobin toxicity. Free Rad Biol Med. 2003;35:872–881. doi: 10.1016/s0891-5849(03)00431-3. [DOI] [PubMed] [Google Scholar]
- Sadrzadeh SMH, Anderson DK, Panter SS, Hallaway PE, Eaton JW. Hemoglobin potentiates central nervous system damage. J Clin Invest. 1987;79:662–664. doi: 10.1172/JCI112865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas M, Wang J, Rosa de Sagarra M, Martin D, Rojo AI, Martin-Perez J, Ortiz de Montellano PR, Cuadrado A. Protein kinase Akt/PKB phosphorylates heme oxygenase-1 in vitro and in vivo. FEBS Lett. 2004;578:90–94. doi: 10.1016/j.febslet.2004.10.077. [DOI] [PubMed] [Google Scholar]
- Saura J. Microglial cells in astroglial cultures: a cautionary note. J Neuroinflammation. 2007;4:26. doi: 10.1186/1742-2094-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherle PA, Ma W, Lim H, Dey SK, Trzaskos JM. Regulation of cyclooxygenase-2 induction in the mouse uterus during decidualization. An event of early pregnancy. The Journal of Biological Chemistry. 2000;275:37086–37092. doi: 10.1074/jbc.M006168200. [DOI] [PubMed] [Google Scholar]
- Schipper HM. Heme oxygenase expression in human central nervous system disorders. Free Radic Biol Med. 2004;37:1995–2011. doi: 10.1016/j.freeradbiomed.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Taketani S. Aquisition, mobilization and utilization of cellular iron and heme: endless findings and growing evidence of tight regulation. Tohoku J Exp Med. 2005;205:297–318. doi: 10.1620/tjem.205.297. [DOI] [PubMed] [Google Scholar]
- Takizawa S, Hirabayashi H, Matsushima K, Tokuoka K, Shinohara Y. Induction of heme oxygenase protein protects neurons in cortex and striatum, but not hippocampus, against transient forebrain ischemia. J Cereb Blood Flow Metab. 1998;18:559–569. doi: 10.1097/00004647-199805000-00011. [DOI] [PubMed] [Google Scholar]
- Vreman HJ, Stevenson DK. Heme oxygenase activity as measured by carbon monoxide production. Anal Biochem. 1988;168:31–38. doi: 10.1016/0003-2697(88)90006-1. [DOI] [PubMed] [Google Scholar]
- Wagner KR, Hua Y, de Courten-Myers GM, Broderick JP, Nishimura RN, Lu SY, Dwyer BE. Tin-mesoporphyrin, a potent heme oxygenase inhibitor, for treatment of intracerebral hemorrhage: in vivo and in vitro studies. Cell Mol Biol. 2000;46:597–608. [PubMed] [Google Scholar]
- Wang J, Doré S. 2006 Neuroscience Meeting Planner. Atlanta, GA: Society for Neuroscience; 2006a. Heme oxygenase 1 exacerbates brain injury after intracerebral hemorrhage. online 583.17. [Google Scholar]
- Wang J, Zhuang H, Doré S. Heme oxygenase 2 is neuroprotective against intracerebral hemorrhage. Neurobiol Dis. 2006b;22:473–476. doi: 10.1016/j.nbd.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang H, Xu L, Rozanski DJ, Sugawara T, Chan PH, Trzaskos JM, Feuerstein GZ. Significant neuroprotection against ischemic brain injury by inhibition of the MEK1 protein kinase in mice: exploration of potential mechanism associated with apoptosis. J Pharmacol Exp Ther. 2003;304:172–178. doi: 10.1124/jpet.102.040246. [DOI] [PubMed] [Google Scholar]
- Wang ZQ, Wu DC, Huang FP, Yang GY. Inhibition of MEK/ERK 1/2 pathway reduces pro-inflammatory cytokine interleukin-1 expression in focal cerebral ischemia. Brain Res. 2004;996:55–66. doi: 10.1016/j.brainres.2003.09.074. [DOI] [PubMed] [Google Scholar]
- Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- Zhuang S, Schnellmann RG. A death-promoting role for extracellular signal-regulated kinase. J Pharmacol Exp Ther. 2006;319:991–997. doi: 10.1124/jpet.106.107367. [DOI] [PubMed] [Google Scholar]