

Abstract

The hairpin ribozyme is a small, non-coding (nc)RNA that catalyzes a site-specific phosphodiester bond cleavage reaction. Prior biochemical and structural analyses pinpointed the amidine moiety of base Ade38 as a key functional group in catalysis, however base changes designed to probe function resulted in localized misfolding of the active site. To define the requirements for chemical activity using a conservative modification, we synthesized and incorporated N1-deaza-Adenosine into the full-length ribozyme construct. This single-atom variant severely impairs activity, although the active site fold remains intact in the accompanying crystal structures. The results demonstrate the essentiality of the imino moiety, as well as the importance of its interaction with substrate in pre-catalytic and transition-state conformations. This work demonstrates the efficacy of single-atom approaches in the analysis of ncRNA structure-function relationships.
Non-protein-coding (nc)RNAs are an emergent class of nucleic acid molecules that play essential roles in peptide-bond formation, tRNA processing, pre-mRNA splicing and gene regulation.1-8 Ribozymes are a class of ncRNAs that can accelerate biological reactions by shifting the pKas of key nucleobases toward neutrality, by providing functional groups for acid/base catalysis, and by electrostatic stabilization of high-energy intermediates.9-13 A major challenge in the field is to identify key RNA functional groups and to discern how they contribute to rate enhancement.
The hairpin ribozyme is a small RNA enzyme whose family members catalyze 2′-nucleophilic attack on an adjacent phosphorus leading to bond scission.14 The 2′-OH group donates its proton to a base (B:) and the SN2 reaction proceeds via a phosphorane transition state (Figure 1A). A nearby acid (AH) alleviates charge build-up at O5′ via protonation. The hairpin ribozyme performs this reaction without need of a metal hydroxide,15-17 implicating the nucleobases themselves in chemistry. As such, the hairpin serves as a model system to investigate the principles of nucleobase-mediated RNA catalysis.
Figure 1.

Transition-state scheme and activity profiles for adenosine and N1-deazaadenosine. (A) Transition-state stabilization of small ribozymes. (B) Single turnover cleavage of adenosine and N1dA at position 38 of the hairpin ribozyme crystallization construct (Figure S1). The kobs for wild type was 0.7 ± 0.2 min-1. The reaction was monitored for 50 hr and showed no evidence of cleavage. (inset) Bases at position 38 of this
Several lines of evidence suggest important roles for bases G8 and A38 in the hairpin ribozyme mechanism. A series of crystal structures revealed that each base flanks the scissile bond, and resides at a distance consistent with hydrogen-bond stabilization of the transition state.18,19 Computational analyses favor G8 and A38 as key players20,21 and biochemical investigations have delineated various attributes of each base that promote catalysis. Substitution of G8 with nucleobase variants or an imidazole moiety altered pH-rate profiles22,23 suggesting general base catalysis.24 However, replacement with an abasic residue produced only a 350-fold loss in cleavage with little effect on the pH-rate profile.25 Cationic nucleobase variants partially restored activity when added to G8 deficient abasic constructs, supporting electrostatic stabilization and functional group alignment roles for this position.25,26 In contrast, an abasic variant in place of A38 produced a 14,000-fold loss in cleavage, and shifted the titration point of the pH-rate profile by three log units into the basic.27 Like G8 deficient variants, exogenously added ligands with positive character rescued A38 abasic constructs. However, such ligands functioned optimally when protonated in both cleavage and ligation, which does not support the microscopic reversibility of a general acid/base mechanism.27 This led to the suggestion that the A38 amidine moiety (i.e. an exocyclic amine alpha to an imino group) contributes electrostatic stabilization and substrate orientation via protonation of the N1 imino28, but did not rule out a role for A38 as a general acid in cleavage, or the possibility that A38 positions a water molecule that serves as a specific acid.27
To provide insight into the role of A38, a recent investigation from our lab analyzed the structural effects produced by several base substitutions at position 38. Each variant was examined in the context of two conformations. A ‘pre-catalytic’ (PC) state was used in which the 2′-OH group of A-1 was rendered inert by replacement of this nucleophile with an -O-methyl moiety. A ‘transition-state’ (TS) analog was used in which the scissile bond was substituted with an inert 2′,5′ linkage. The results revealed that the most catalytically impaired variants, C38 and G38, exhibited local misfolding when restrained to adopt a TS-like conformation.29 Moreover, each variant at position 38 adopted an anti conformation in contrast to the preferred syn state of the wild type. The anti conformer directs the imino group of the base amidine moiety away from the scissile bond, but leaves the exocyclic amine position relatively unaltered. These observations imply a critical role for the N1 imino group in catalysis29 in support of biochemical data.27 However, no further analysis has been conducted to assess the contribution of the A38 imino group in hairpin ribozyme folding and catalysis, or to probe its putative positive charge in the context of PC or TS conformations.
To dissect the contribution of the A38 N1-imino from other nucleobase substituents, we synthesized30 and incorporated N1-deazaadenosine (N1dA) - an isostere of adenosine (Figure 1B, inset) - at position 38 of the hairpin ribozyme. This change resulted in undetectable activity (Figure 1B) representing ∼107-fold reduction in rate relative to the uncatalyzed reaction.31
We then asked whether loss of activity resulted from improper local folding or the absence of an essential functional group in the active site. To distinguish between these possibilities, we determined crystal structures of N1dA38 variants in the context of a minimal construct (Figure S1). The structures revealed that the global and local folds were intact relative to wild type. Simulated-annealing omit electron-density maps for N1dA38 and flanking residues support a syn conformation for the N1-deaza base in which the Watson-Crick face points toward the scissile bond (Figure 2). The syn conformation illustrates the compatibility of the N1-deaza substitution with retention of local folding, unlike the C38 and G38 variants.29 Our results further confirm the importance of the A38 amidine moiety identified in biochemical studies,27 but pinpoint the imino group as essential for chemistry. Specific spatial differences between the N1dA38 and wild type structures were then considered in light of these findings.
Figure 2.
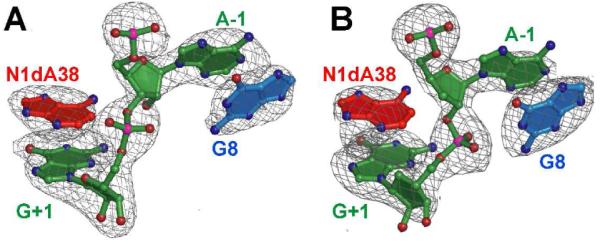
The mFo-DFc omit electron density maps of N1dA38 hairpin ribozyme structures from this investigation. (A) The pre-catalytic variant. (B) The transition-state (TS) analog. Maps are contoured at the 3σ level.
The N1dA38 base position and the 2′-nucleophile at A-1 were regions of notable change compared to the wild type PC conformation (Figure 3A). The 2′-nucleophile was unprotected in crystallization, thus revealing the ribose pucker in an unbiased state. Prior structures harbored a 2′-O-methyl moiety at A-1 to prevent cleavage and were dubbed ‘pre-catalytic’ due to in-line attack angles, τ, of ∼160°.18,32 Without the -O-methyl group, the 2′-nucleophile moves 4 Å deeper into the active site core coincident with a change in the A-1 pucker from C2′-endo to C3′-endo (Figure 3A). MD simulations predicted the latter pucker and support hydrogen bonding between N6 of A9 and the 2′-OH of A-1.33 Recent MD analysis suggested the C3′-endo pucker of A-1 is ideal for 2′-nucleophile activation by N1 of A38 and subsequent proton transfer to the O5′-leaving group.34 We find this new hypothesis intriguing, but see structural parallels between the proposed ‘activating’ C3′-endo pucker interaction and a cleavage--inactivating conformation observed for variants G8A and G8U.32 In the latter structures, the proximity of the unprotected 2′-OH nucleophile to the A38 N1 imino (3.4 Å) was hypothesized to stabilize non-in-line attack geometry where τ is ∼97°. This interaction was also perceived to block a mode of TS stabilization involving the pro-Rp oxygen of the scissile-bond.32
Figure 3.

Superpositions of hairpin ribozyme structures. (A) The pre-catalytic N1dA38 variant and wild-type structure. (B) The N1dA38 variant in the context of the TS analog and the A38 TS analog structure.
Prior crystal structures of the hairpin ribozyme suggest that the transition state utilizes a C2′-endo pucker at A-1. This conformation was reported to facilitate proton abstraction from the 2′-nucleophile and to permit simultaneous stabilization of the pro-Rp and pro-Sp oxygens by A38, A9 and G8,18,19 consistent with the electrostatic stabilization role ascribed to these bases.21,22,25-27 The C2′-endo pucker at A-1 also supports bifurcated hydrogen bonding between its 2′-OH to the N1 and N2 groups of G8, which was proposed to lower the pKa of the 2′-nucleophile.32 Small molecule structures support the plausibility of this interaction as well as proton transfer from the 2′-nucleophile to a nearby water molecule (Figure S2). Collectively, these observations suggest that the C2′-endo pucker at A-1 remains a plausible conformation for catalysis, even though our N1dA38 structure with an unprotected 2′-nucleophile adopts the C3′-endo state. Further work is needed to differentiate between these catalytic proposals, but either mechanism must recognize the essentiality of the A38 imino moiety in cleavage chemistry.
To provide functional insight into the A38 imino moiety, we superimposed the N1dA38 PC structure upon that of wild type (Figure 3A). This comparison revealed that the N1dA38 base recoils from the active site, widening a gap between the Watson-Crick face of A38 and the scissile bond (Figure S3). This recoil does not occur in the G8A or G8U variants that possess a free 2′-OH at A-1 suggesting the effect is not due to uncapping of the nucleophile. The N1dA38 base pivots such that its N6 exocyclic amine shifts 1.7 Å relative to the wild type (Figure 3A, inset). The magnitude and direction of this change suggest that the recoil arises from a steric clash between the N1dA38 C1 atom and the scissile bond. As such, we determined a structure harboring N1dA38, but no scissile phosphate (i.e. a product mimic).19 In this context, no appreciable change of the N1dA38 base was observed relative to the N1dA38 PC state (not shown). Moreover, the N6 distance between the N1dA38 product-mimic and the matched wild-type structure was 1.9 Å (Figure S4). In both the PC and product-mimic structures containing N1dA38, the N6 amine of the base moves within ∼3.6 Å of A24 N1 (Figure S3). This result demonstrates how the ribozyme accommodates the non-polar deaza base as it withdraws from the active site. At a minimum, this result suggests that the N1 imino group draws the A38 base toward the scissile bond and plays a vital role in substrate positioning. In light of N1dA38’s relatively modest local rearrangements in the PC state, the catastrophic loss of cleavage activity supports an essential role for the N1 moiety in the reaction that cannot be fulfilled by any other chemical group.
To investigate the compatibility of the N1dA38 variant with TS geometry, we determined its structure in the context of an inert 2′,5′ linkage in place of the scissile bond (Figure 2B). This linkage restrained the pro-Rp and pro-Sp non-bridging oxygens into a TS-like conformation in the context of wild type Ade38.19 Electron density maps of the N1dA38 variant revealed a similar orientation in which the non-bridging oxygens form 3.1 Å and 2.7 Å hydrogen bonds to the respective exocyclic amines of G8 and A9 (Figures 2B & 3B). However, absence of the N1 imino moiety again resulted in recoiling of the deaza base from the active site, yielding a stable Hoogsteen base pair with A24 (Figure S5). The displacement of the N1dA38 N6 amine was 2.6 Å relative to wild-type (Figure 3B, inset), which is 0.9 Å longer than the movement of the N6 atom of N1dA38 in the PC state (Figure 3A). This difference may originate from a strong requirement to protonate the imino group of A38 in the TS to ameliorate charge repulsion between N1 and the non-bridging oxygens of the scissile bond.18,19,24,27,35 Such repulsive contacts are far less prominent in the PC conformation making comparable steric and electrostatic accommodations unnecessary.
We then asked how loss or gain of a charge at the N1 position of A38 would affect the active site conformation and function. Structural evidence that the wild-type TS analog exists as the A38(H+) species is based on the observation that the N1 atom of A38 is within 2.9 Å of atoms O2′ and O5′ of the scissile bond in the wild-type structure (Figure S5B)19 and 2.7 Å from O5′ in oxo vanadium TS mimic complexes.18,19 Comparable N(H+) to O—P distances derived from small molecule structures were 2.72 ± 0.09 Å, whereas N to O—P distances were 3.61 ± 0.37 Å (Supporting Information). In contrast, the methine group of N1dA38 is isosteric with A38(H+), but electrostatically disparate. This distinction is born out by retreat of the N1dA38 base away from the 2′,5′-bond producing an N1dA38(C1)-to-O2′ distance of 3.8 Å; comparable -C(H)-to-O distances are 3.61 ± 0.18 Å in small molecule structures (Supporting Information). Recoil of the N1dA38 base away from the scissile bond of the TS analog also results in loss of a hydrogen bond between N6 of position 38 and the pro-Rp oxygen of the scissile bond (i.e. a distance change from 2.8 Å to 5.6 Å depicted in Figure S5). This interaction was shown to be important for TS stabilization and cleavage activity18,27,29,36 and has been posited to fine-tune the N1 pKa of A38 by shifting it toward neutrality.19,29 It has been hypothesized that the N1(H+) imino group and N6 amine of A38 work in concert to stabilize a monoanionic phosphorane transition state.37 The importance of the A38 N6 amine is reinforced by the observation that purine substitution at this position reduces cleavage 102- to 103-fold.27,36 The inability to detect activity for the N1dA38 variant however suggests the imino group is more important than N6, which is consistent with the imino group’s absolute requirement to promote phosphorane formation.37
We then asked what the charge state of the A38 N1 moiety might be in the PC conformation, since a pre-existing charge has been described as a prerequisite to phosphorane formation.37,38 The N1 to O-P distances for PC and TS analog structures in the context of A38 were 3.0 Å and 2.9 Å, respectively (Figures S3B & S5B). Comparable C1 to O-P distances for the N1dA38 variants were 3.5 Å and 4.1 Å (Figures S3A & S5A). Whereas the former distances are consistent with protonation of A38 N1 in the PC and TS constructs (2.72 ± 0.09 Å), the latter distances are expected for deprotonation or substitution of the N1 with a methine (3.61 ± 0.37 Å). These results imply that a positive charge on A38 N1 exists prior to TS formation and is dictated by proximity to the scissile bond in PC complexes.
Finally, it has been hypothesized that the A38 N1 group serves to position a catalytic water27, which donates a proton to the O5′ leaving group.27 The results here provide no evidence for water at this position, even though the gap between the scissile bond and the A38 base is widened in the N1dA38 structures. Prior results with more conservative, polar changes on the Watson-Crick face of A38 support this observation as well.29 As such, it seems more plausible that proximity of the A38 N1 group to the scissile bond generates the N1(H+) species upon hairpin ribozyme domain docking, and that this species is operative in cleavage.
The structural observations and absence of detectable activity for N1dA38 herein suggest that any mechanism of hairpin ribozyme cleavage must consider: (i) an A38N1(H+) species in the pre-catalytic and transition state, (ii) the unlikelihood of a specific-acid water at O5′, and (iii) N6 of A38 is not sufficient to support catalysis, thus favoring the N1 imino in phosphorane stabilization and leaving group protonation.27,37 Overall, our single-atom analysis of the hairpin ribozyme demonstrates a conservative strategy to probe structure-function relationships and the charge character of bases, which has broad implications for elucidating the mechanism of action of more complex ncRNAs.
Supplementary Material
Acknowledgment
We thank the Hauptman-Woodward Institute Inc. (Buffalo, NY) for technical resources and the Cornell High Energy Synchrotron Source (CHESS, Ithaca, NY). CHESS is supported by NSF award DMR-0225180 and by the Public Health Services via NIH/NCRR award RR-01646. Funding was provided in part by grants to JEW from NIH (GM63162) and the donors of the PRF (45534 AC4). RCS was supported in part by an Elon Huntington-Hooker graduate fellowship.
References
- (1).Noller HF, Hoffarth V, Zimniak L. Science. 1992;256:1416–9. doi: 10.1126/science.1604315. [DOI] [PubMed] [Google Scholar]
- (2).Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. Science. 2000;289:920–30. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- (3).Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. Cell. 1983;35:849–57. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- (4).Valadkhan S. Biol Chem. 2007;388:693–7. doi: 10.1515/BC.2007.080. [DOI] [PubMed] [Google Scholar]
- (5).Ferat JL, Michel F. Nature. 1993;364:358–61. doi: 10.1038/364358a0. [DOI] [PubMed] [Google Scholar]
- (6).Cech TR, Zaug AJ, Grabowski PJ. Cell. 1981;27:487–96. doi: 10.1016/0092-8674(81)90390-1. [DOI] [PubMed] [Google Scholar]
- (7).Grundy FJ, Henkin TM. Mol Microbiol. 1998;30:737–49. doi: 10.1046/j.1365-2958.1998.01105.x. [DOI] [PubMed] [Google Scholar]
- (8).Winkler W, Nahvi A, Breaker RR. Nature. 2002;419:952–6. doi: 10.1038/nature01145. [DOI] [PubMed] [Google Scholar]
- (9).Doudna JA, Lorsch JR. Nat. Struct. Mol. Biol. 2005;12:395–402. doi: 10.1038/nsmb932. [DOI] [PubMed] [Google Scholar]
- (10).Bevilacqua PC, Yajima R. Curr Opin Chem Biol. 2006;10:455–64. doi: 10.1016/j.cbpa.2006.08.014. [DOI] [PubMed] [Google Scholar]
- (11).Ferré-D’Amaré AR. Biopolymers. 2004;73:71–8. doi: 10.1002/bip.10516. [DOI] [PubMed] [Google Scholar]
- (12).Bevilacqua PC. Biopolymers. 2004;73:69–70. doi: 10.1002/bip.10522. [DOI] [PubMed] [Google Scholar]
- (13).Walter NG. Mol Cell. 2007;28:923–9. doi: 10.1016/j.molcel.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cochrane JC, Strobel SA. Acc Chem Res. 2008;41:1027–35. doi: 10.1021/ar800050c. [DOI] [PubMed] [Google Scholar]
- (15).Nesbitt S, Hegg LA, Fedor MJ. Chem. Biol. 1997;4:619–30. doi: 10.1016/s1074-5521(97)90247-7. [DOI] [PubMed] [Google Scholar]
- (16).Hampel A, Cowan JA. Chemistry & Biology. 1997;4:513–517. doi: 10.1016/s1074-5521(97)90323-9. [DOI] [PubMed] [Google Scholar]
- (17).Young KJ, Gill F, Grasby JA. Nucleic Acids Res. 1997;25:3760–6. doi: 10.1093/nar/25.19.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Rupert PB, Massey AP, Sigurdsson ST, Ferré-D’Amaré AR. Science. 2002;298:1421–4. doi: 10.1126/science.1076093. [DOI] [PubMed] [Google Scholar]
- (19).Torelli AT, Krucinska J, Wedekind JE. RNA. 2007;13:1052–70. doi: 10.1261/rna.510807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Nam K, Gao J, York DM. J Am Chem Soc. 2008;130:4680–91. doi: 10.1021/ja0759141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Nam K, Gao J, York DM. RNA. 2008;14:1501–7. doi: 10.1261/rna.863108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pinard R, Hampel KJ, Heckman JE, Lambert D, Chan PA, Major F, Burke JM. EMBO J. 2001;20:6434–42. doi: 10.1093/emboj/20.22.6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wilson TJ, Ouellet J, Zhao ZY, Harusawa S, Araki L, Kurihara T, Lilley DM. RNA. 2006;12:980–7. doi: 10.1261/rna.11706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bevilacqua PC. Biochemistry. 2003;42:2259–65. doi: 10.1021/bi027273m. [DOI] [PubMed] [Google Scholar]
- (25).Lebruska LL, Kuzmine II, Fedor MJ. Chem. Biol. 2002;9:465–73. doi: 10.1016/s1074-5521(02)00130-8. [DOI] [PubMed] [Google Scholar]
- (26).Kuzmin YI, Da Costa CP, Fedor MJ. J. Mol. Biol. 2004;340:233–51. doi: 10.1016/j.jmb.2004.04.067. [DOI] [PubMed] [Google Scholar]
- (27).Kuzmin YI, Da Costa CP, Cottrell JW, Fedor MJ. J. Mol. Biol. 2005;349:989–1010. doi: 10.1016/j.jmb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- (28).Cottrell JW, Kuzmin YI, Fedor MJ. J Biol Chem. 2007;282:13498–507. doi: 10.1074/jbc.M700451200. [DOI] [PubMed] [Google Scholar]
- (29).MacElrevey C, Salter JD, Krucinska J, Wedekind JE. RNA. 2008;14:1600–16. doi: 10.1261/rna.1055308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Cristalli G, Franchetti P, Grifantini M, Vittori S, Bordoni T, Geroni C. J Med Chem. 1987;30:1686–8. doi: 10.1021/jm00392a029. [DOI] [PubMed] [Google Scholar]
- (31).Breaker RR, Emilsson GM, Lazarev D, Nakamura S, Puskarz IJ, Roth A, Sudarsan N. RNA. 2003;9:949–57. doi: 10.1261/rna.5670703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Salter J, Krucinska J, Alam S, Grum-Tokars V, Wedekind JE. Biochemistry. 2006;45:686–700. doi: 10.1021/bi051887k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Rhodes MM, Reblova K, Sponer J, Walter NG. Proc Natl Acad Sci U S A. 2006;103:13380–5. doi: 10.1073/pnas.0605090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ditzler MA, Sponer J, Walter NG. RNA. 2009;15:560–75. doi: 10.1261/rna.1416709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ferre-D’Amare AR, Rupert PB. Biochem Soc. Trans. 2002;30:1105–9. doi: 10.1042/bst0301105. [DOI] [PubMed] [Google Scholar]
- (36).Grasby JA, Mersmann K, Singh M, Gait MJ. Biochemistry. 1995;34:4068–76. doi: 10.1021/bi00012a025. [DOI] [PubMed] [Google Scholar]
- (37).Bevilacqua PC, Brown TS, Nakano S, Yajima R. Biopolymers. 2004;73:90–109. doi: 10.1002/bip.10519. [DOI] [PubMed] [Google Scholar]
- (38).Gong B, Chen JH, Chase E, Chadalavada DM, Yajima R, Golden BL, Bevilacqua PC, Carey PR. J. Am. Chem. Soc. 2007;129:13335–42. doi: 10.1021/ja0743893. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.