

Abstract
Recent results in animal models suggest that thrombin may modulate brain injury in Parkinson's disease (PD). High doses of thrombin (∼20 U) can damage dopaminergic neurons, while we have found that low dose thrombin (1 U), given several days before a brain insult (thrombin preconditioning), is protective in models of PD and stroke. However, the effects of such low levels of thrombin at the time of, or after, exposure to the dopamine neurotoxin 6-hydroxydopamine (6-OHDA) have not been examined and are the focus of this study. In the first set of experiments, rats received co-administration of thrombin (1 U) or saline and 6-OHDA (5 μg) into the medial forebrain bundle. 6-OHDA + thrombin resulted in striking increases in behavioral deficits, compared to 6-OHDA + saline. Similarly, co-administration of an agonist to protease-activated receptor (PAR)-1, a thrombin receptor, also resulted in significantly greater behavioral deficits. In a second set of experiments, thrombin (1 U) or saline was administered 1 or 7 days after 6-OHDA to determine the effects of thrombin after 6-OHDA. Surprisingly, the rats that received saline had strikingly increased behavioral and neurochemical deficits resulting from the 6-OHDA lesion, while delayed thrombin administration prevented this effect. The results indicate that thrombin has differential effects in the 6-OHDA model, dependent on the time of administration. The ability of a second cannula insertion with saline infusion to increase dramatically deficits raises questions as to what role physical injury to already susceptible cells might play in the pathogenesis of some cases of PD.
Keywords: Parkinson's disease, brain injury, sensorimotor, protease-activated receptor-1, PAR-1
Introduction
Thrombin derived from blood or brain prothrombin has been implicated in modulating brain injury in hemorrhagic and ischemic stroke [46]. Thrombin is a key mediator of edema formation and cell death in experimental stroke [26,37,41]. Recent evidence also suggests that thrombin may modulate brain injury in Parkinson's disease (PD). High doses of thrombin (∼20 U) can lead to degeneration of dopaminergic neurons in the substantia nigra, although cell-type specificity is still unclear [5,9]. This effect is likely mediated by cell death pathways involving microglial activation [5,8,10,25]. It is also interesting that intracerebral hemorrhage (with resultant thrombin production) in or near the nigrostriatal tract, has been found to produce L-DOPA responsive behavioral deficits similar to those observed in PD [19,30]. Many thrombin effects are receptor mediated, involving protease-activated receptor (PAR) activation [21]. Mice deficient in PAR-1 are protected against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced neurotoxicity [16], which causes PD-like effects in humans and animals [23,24].
In contrast to the detrimental effects of high doses of thrombin, administration of a low dose thrombin several days prior to experimental stroke is highly protective, an effect termed thrombin preconditioning [29,45]. It is well established that a minor insult to the brain can activate cell survival mechanisms that can reduce or prevent tissue destruction normally expected from a severe insult several days later [15]. In ischemia models, resistance to an otherwise major cereberovascular challenge has been called “ischemic tolerance”. Ischemic or thrombin preconditioning does not produce detectable tissue loss, although sensitive behavioral assessment suggests that they may cause transient functional deficits reflecting sub-fatal trauma, which may be sufficient to prepare the brain for more severe insults [3,18].
Recently, a low dose of thrombin (1 U) was also found to be protective in the 6-OHDA model of experimental PD [3]. Such cross-tolerance among other injury models is known to occur [15]. Although the mechanisms of protection remain to be determined, the preconditioning effects of thrombin depend on activation of the protease-activated receptor-1 (PAR-1) [4], a thrombin receptor linked to injury tolerance [21].
These findings suggest a dose and a temporal relationship in the effects of thrombin in neurological disease models. In considering the potential of thrombin to contribute to the pathogenesis of PD it is, therefore, important to determine the effects of both co- and post-administration of thrombin in experimental PD models. The current study, therefore, examined the effects of co-administering a low dose of thrombin (1 U) or a PAR-1 agonist with the neurotoxin 6-hydroxydopamine (6-OHDA). 6-OHDA-induced neurotoxicity in the rat is a commonly used experimental model of PD. The study also examined the effects of thrombin administration at different time points after 6-OHDA. There were differential effects on behavioral and neurochemical endpoints based on administration time. Co-administration of either thrombin or a PAR-1 agonist at the time of the neurotoxin delivery increased behavioral and neurochemical deficits. Interestingly, a second cannula insertion and infusion of saline after 6-OHDA potentiated injury, while thrombin prevented that effect.
Materials and Methods
Materials
Unless otherwise noted, all chemicals were purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA).
Animals
All animal protocols were approved by the University of Michigan Committee on the Use and Care of Animals. For all experiments, adult male Sprague-Dawley rats (250-325 g; Charles River Laboratories, Portage, MI, USA) were used. Rats were allowed free access to food and water. All animals were housed in a temperature controlled room (25°C) with a 12:12 hour light:dark cycle. Behavioral tests were typically conducted in the morning near the beginning of the light cycle.
Surgery
Prior to surgery, animals were fully anesthetized with pentobarbital (50 mg/kg, Abbott Laboratories, North Chicago, IL, USA). Rats were then placed in a stereotaxic frame (Model 5000, Kopf Instruments, Tujunga, CA, USA). A longitudinal incision was made in the scalp, the skull was exposed, and then a cranial burr hole (1 mm) was drilled into the skull at the infusion site. A 26-gauge Hamilton syringe needle (Hamilton Company, Reno, NV, USA) was then lowered into the right medial forebrain bundle (anterior/posterior: -2.2 mm, right/left: 1.9 mm, ventral: 8.0 mm to bregma) and the animals were administered the drug treatments. For those animals that received a second surgery, the procedure was conducted as described above, except a 30-gauge syringe needle was used and the infusion occurred 1 mm dorsal to the site of the first infusion, in order to account for the larger volume (50 μL). Bone wax was used to fill the burr hole and the surgery site was stitched after the infusion. After completion of each infusion, the cannula was allowed to remain in place for 5 minutes to allow for proper absorption and avoid cortical exposure during cannula withdrawal. Core body temperature was maintained at 37°C during the surgery and until the animals were ambulatory, using a rectal probe feedback-controlled heating pad.
For 6-OHDA infusions, 30 minutes prior to lesioning animals received the noradrenergic uptake blocker, desipramine (15 mg/kg, ip.) and the monoamine oxidase inhibitor pargyline (25 mg/kg, ip.) to enhance dopaminergic neurotoxicity, and limit the loss of noradrenergic neurons [36].
Experimental design
This study was divided into three parts. In part I, the effects of different doses of 6-OHDA on behavioral deficits were examined. We have previously used a dose of 10 μg to examine the protective effects of thrombin preconditioning [3]. However, that 6-OHDA dose produces near maximal deficits in the behavioral tests used. Since we expected that thrombin might enhance deficits in some experiments, we examined the behavioral deficits with a lower dose (5 μg; n = 5) in comparison to that found with 10 μg (n = 9), to ensure that the lower dose would lead to detectable, but not maximal, deficits. The total infusion volume was 4 μL and occurred over 8 minutes.
In part II, the effects of thrombin or a PAR-agonist, when co-administered with 6-OHDA were tested. 6-OHDA (5 μg) and either saline (n = 11), thrombin (1 U; n = 5), or a PAR-1 agonist [n = 6; 5 nanomols; Ala-para-fluorPhe-Arg-Cha-homoArg-Tyr-NH2; NeoMPS, Strasbourg, France; [43]] were infused into the right medial forebrain bundle. The total infusion volume for these studies was 6 μL and occurred over 12 minutes.
In part III, the effects of thrombin when administered after 6-OHDA were examined. 6-OHDA (5 μg) was administered as described above. The total infusion volume was 4 μL and occurred over 8 minutes. A second infusion of either saline (50 μL; n = 5) or thrombin (1 U in 50 μL saline; n = 6) was administered 7 days after 6-OHDA. This concentration of thrombin was chosen because it has previously been found to be protective when administered prior to 6-OHDA [3]; it is, therefore, ideal to further elucidate the temporal effects of thrombin in the 6-OHDA model. Additionally, this dosage of thrombin (1 U/50 μL saline) does not produce brain edema [45]. To evaluate the effects of thrombin at an earlier time-point after 6-OHDA, additional animals were administered either saline (n = 4) or thrombin (n = 5) one day after 6-OHDA.
Neurobehavioral assessment
All animals in these studies underwent behavioral analysis prior to and through 14 to 21 days after 6-OHDA administration. The observer was blinded to the treatment group until completion of all behavioral studies.
Vibrissae-elicited forelimb placing (placing test)
This test has been well described and utilized to measure 6-OHDA-elicited deficits [35,39,44]. Additionally, thrombin preconditioning has been found to provide protection from 6-OHDA-elicited deficits in this test [3].
The ability of each forelimb to reach for a table surface, guided by the corresponding vibrissae, to obtain weight support was evaluated. Vibrissae-elicited forelimb placing was measured by brushing the animals' left or right vibrissae against a tabletop while all limbs hung freely. The forelimb not being tested was discouraged from placing by putting the experimenter's finger in front of that forelimb (details and movies can be found at Schallertlab.org). After training, non-lesioned animals will immediately and consistently place the corresponding forelimb onto the ledge. The percent of unsuccessful placing events for the affected limb was calculated over at least 10 trials. Failure to respond to vibrissae stimulation on the unaffected side did not occur.
Forelimb-use asymmetry during rearing (asymmetry test)
This test has been utilized to assess 6-OHDA-elicited deficits [35,39] and also protection elicited by preconditioning in this model [3,39]. Briefly, the animal was placed in a clear plastic cylinder (height: 30 cm, width: 20 cm) and forelimb asymmetry was determined during rearing behavior. Forelimb contact with the cylinder wall was scored as left, right, or both during explorative rearing behavior. The percent forelimb asymmetry was determined using the following formula:
[(unaffected-side places — affected-side places)/(total places)] × 100.
Catecholamine determination
After completion of behavioral studies, some of the animals were sacrificed for striatal catecholamine determination. Dopamine, 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) were measured as previously described [3]. Briefly, animals were deeply anesthetized and decapitated and the brains were removed and placed in ice-cold saline for five minutes. Using an ice-cold brain slicer, a 2.0 mm coronal section (anterior/posterior: 0.0–2.0 mm to bregma) was then removed. The left and right striata were dissected on a plastic dish over ice, weighed, flash frozen using liquid nitrogen, and then stored at -80 °C until use. The frozen striata were then sonicated on ice in cold 0.1M perchloric acid. After centrifuging for 30 min at 16,000 × g, the supernatant was collected from the sonicate solution. The supernatant was stored at -80 °C until HPLC analysis. A portion of the supernatant (20 μL) was injected into a Waters 1525 HPLC pump (Waters Corp., Milford, MA, USA). The mobile phase consisted of the following: 0.06 M sodium phosphate, 0.03 M citric acid, 1.3 mM 1-octanesulfonic acid, 0.1 mM EDTA, 8% (v/v) methanol, in HPLC grade water, pH 3.5. A Waters Symmetry C18 4.6 × 150 mm column (3.5 μm particle size) was used to separate catecholamines. Detection was achieved using a Waters 2465 electrochemical detector with a glassy working electrode, set at 750mV to a salt-bridge Ag/AgCl reference electrode. The flow rate of the mobile phase was 1.0 mL/min. Catecholamines were quantified by generation of a calibration curve, using peak area of external standards of the highest available purity.
Statistics
Three or more groups of behavioral data were compared using the Kruskal-Wallis nonparametric ANOVA to assess significant differences among groups. Significant results were then subjected to post hoc analysis by the Mann-Whitney U-test with Bonferroni correction for multiple comparisons (p<0.025 was deemed significant for comparison of two groups to control). Two groups of behavioral data were compared using the Mann-Whitney U-test. Neurochemistry data for two groups was analyzed using the Student's t-test (one-tailed). Neurochemistry for three groups was first analyzed by ANOVA, and where significant, post hoc comparison by Dunnett's test was conducted. For all statistical tests p<0.05 was deemed significant unless otherwise noted.
Results
Behavioral deficits with 5 and 10 μg 6-OHDA
Administration of 10 μg of 6-OHDA resulted in marked placing and asymmetry deficits (Fig. 1), including animals that displayed maximal (100%) deficits. At that dose, the deficits reached a plateau after about 7 days. Rats given 5 μg of 6-OHDA had almost no detectable placing deficit, but had a forelimb use asymmetry of about 40% (Fig. 1). Again, that latter deficit reached a plateau after about 7 days. This lower dose of 6-OHDA, which should allow examination of factors that enhance brain injury, was used in subsequent studies.
Figure 1.
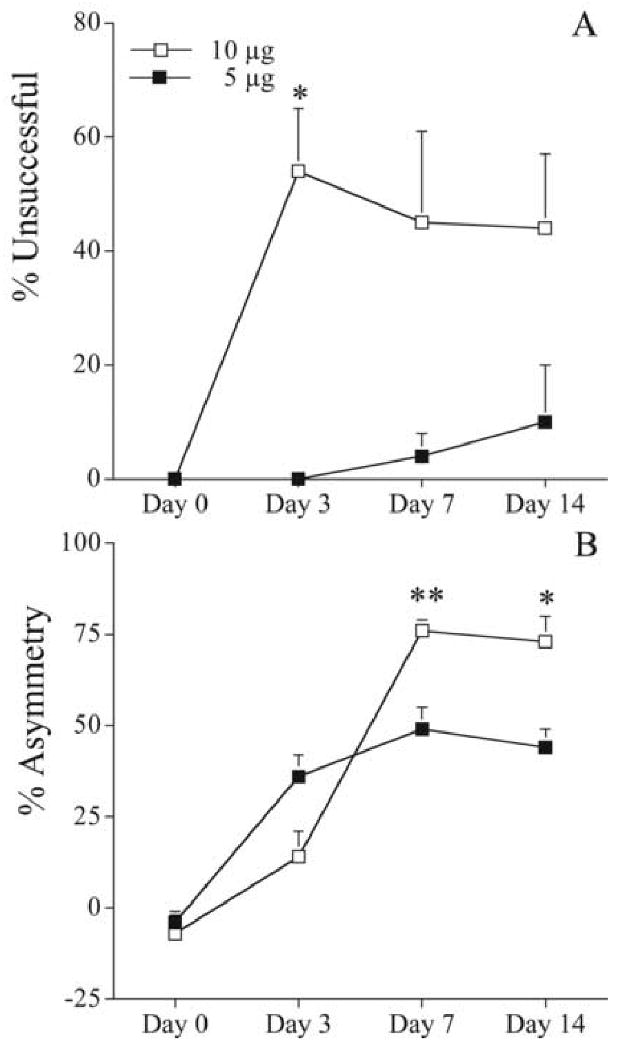
Behavioral deficits in rats receiving either five (filled squares) or ten μg (open squares) 6-OHDA (n= 5 and 9, respectively). The total infusion volume was 4 μL. A. Vibrissae-elicited forelimb placing test. The percent unsuccessful forelimb reaches onto a table surface is reported. B. Forelimb-use asymmetry test. Asymmetrical use of the forelimb for weight support during vertical/lateral exploration in the cylinder. The percent asymmetry is reported. Error bars indicate SEM. * = p<0.05, **=p<0.01, Mann-Whitney U-test.
Co-administration of thrombin or a PAR-1 agonist with 6-OHDA
Behavioral Analysis
The co-administration of thrombin or a PAR-1 agonist (5 nanomols) significantly increased 6-OHDA-elicited (5 μg) deficits compared to saline control (Fig. 2). Placing deficits were significantly increased in thrombin and the PAR-1 agonist groups, compared to saline (Fig. 2A) on days 3 [34 ± 19 and 28 ± 15 vs. 0 ± 0; % unsuccessful (mean) ± SEM; thrombin and PAR-1 agonist vs. saline; p<0.025 Mann-Whitney U-test with Bonferroni correction, after p<0.05 Kruskal-Wallis nonparametric ANOVA] and 7 (54 ± 23 and 55 ± 21 vs. 3 ± 3%). Similarly, forelimb-use asymmetry deficits were greater in thrombin and the PAR-1 agonist groups (Fig. 2B), with the thrombin group reaching statistical significance on day 7 [74 ± 8 vs. 29 ± 10; % asymmetry (mean) ± SEM; thrombin vs. saline; p<0.025 Mann-Whitney U-test with Bonferroni correction, after p<0.05 Kruskal-Wallis nonparametric ANOVA).
Figure 2.
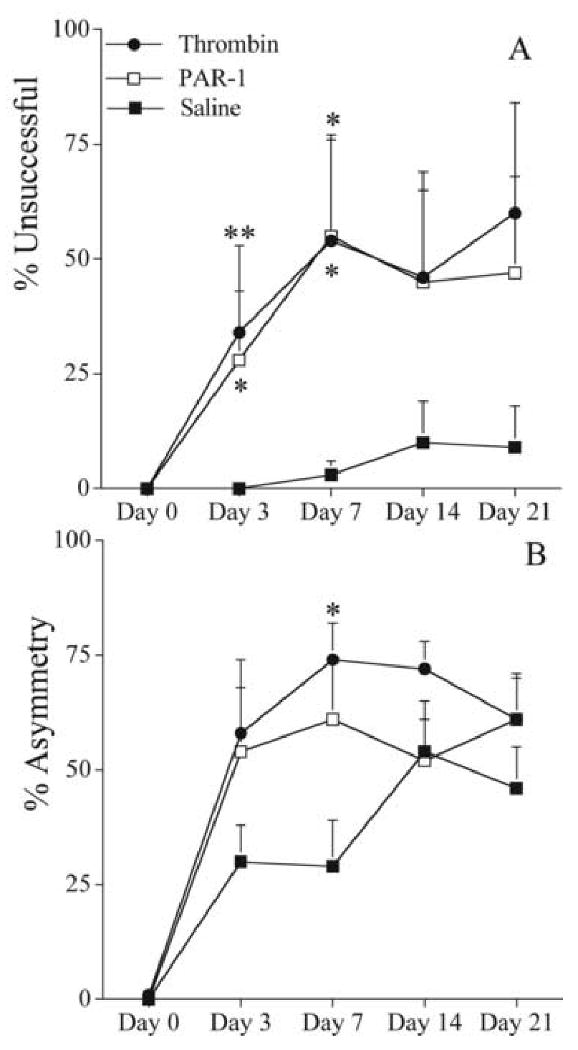
Effects co-administration of thrombin or a PAR-1 agonist with 6-OHDA (5 μg) on behavioral deficits. Either thrombin (1 U; n = 5), a PAR-1 agonist peptide (5 nanomols; n = 6), or saline (n = 11) was co-administered with 6-OHDA (total injection volume = 6 μL). A. Vibrissae-elicited forelimb placing test. The percent unsuccessful forelimb reaches onto a table surface is reported. B. Forelimb-use asymmetry test. Asymmetrical use of the forelimb for weight support during vertical/lateral exploration in the cylinder. The percent asymmetry is reported. Error bars indicate SEM. * = p<0.025, ** = p<0.01 Mann-Whitney U-test with Bonferroni correction after Kruskal-Wallis nonparametric ANOVA is significant (p<0.05).
Neurochemistry
The co-administration of thrombin or the PAR-1 agonist (5 nanomols) with 6-OHDA (5 μg) did not result in statistically significant decreases in dopamine or dopamine metabolites, as compared to the co-administration of saline with 6-OHDA (Table 1). Additionally, the absolute values of ipsilateral (Table 1) and contralateral (data not shown) dopamine, DOPAC, and HVA were not significantly different between groups.
Table 1.
Catecholamine depletion after 6-hydroxydopamine (6-OHDA; 5μg): The effects of co-administration of thrombin or a PAR-1 agonist.
| Dopamine | DOPAC | HVA | |
|---|---|---|---|
| 6-OHDA+saline | 78±8% (4.4±1.7) | 63±13% (0.57±0.21) | 63±9% (0.29±0.07) |
| 6-OHDA+thrombin | 75±17% (3.7±2.7) | 59±22% (0.87±0.53) | 57±15% (0.31±0.13) |
| 6-OHDA+PAR-1 agonist | 89±7% (2.2±0.15) | 80±15% (0.23±017) | 81±8% (0.15±0.06) |
Results expressed as % depletion compared to contralateral. The absolute ipsilateral catecholamine levels (ng/mg wet tissue weight) are given in parentheses. Values are shown as mean ± SEM. Saline (n = 11), thrombin (1 U; n = 5), or a PAR-1 agonist (5 nanomols; n = 6) was co-administered with 6-OHDA (total volume 6 μL). There were no significant differences among the groups.
Thrombin administered after 6-OHDA
Behavioral Analysis
An unanticipated finding was that infusion of saline one week after the moderate 6-OHDA infusion significantly increased behavioral deficits (Fig. 3). Saline or thrombin was infused 7 days after 6-OHDA (5μg) administration. Neither group exhibited significant placing deficits prior to the second infusion (Fig. 3A). However, in the group given saline as the second infusion, marked placing deficits emerged, as assessed at days 14 and 21 [72 ± 19 and 72 ± 18; % unsuccessful (mean) ± SEM, respectively; Fig. 3A]. This enhanced placing deficit caused by the second cannula insertion and saline infusion was virtually absent in rats where thrombin (1U) was present in the second infusion (at day 14: 12 ± 12 vs. 72 ± 19; % unsuccessful; thrombin vs. saline; p<0.05, Mann Whitney U-test, and at day 21: 2 ± 2 vs. 72 ± 18%; Fig. 3A). A similar pattern was found in forelimb-use asymmetry (Fig. 3B). A striking increase in the degree of forelimb use asymmetry was observed after a second infusion of saline-only, while inclusion of thrombin prevented this effect (thrombin vs. saline at day 14: 32 ± 7 vs. 73 ± 9; % asymmetry ± SEM; p<0.05, Mann-Whitney U-test; at day 21: 27 ± 6 vs. 85 ± 8).
Figure 3.
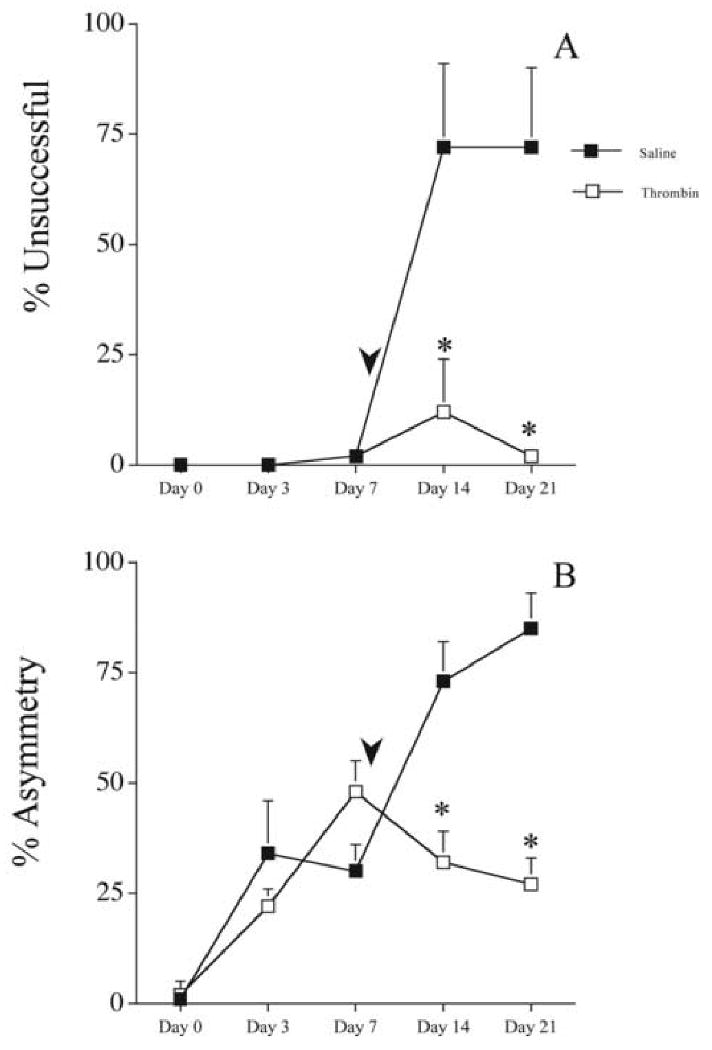
The effects of delayed (7 days) saline or thrombin administration on 6-OHDA (5 μg) induced behavioral deficits. Saline (50 μL; filled squares; n=5) or thrombin (1 U/50 μL; open squares; n=6) was administered at the lesion site 7 days after 6-OHDA. Arrowheads indicate approximate time of second surgery. A. Vibrissae-elicited forelimb placing test. The percent unsuccessful forelimb reach onto a table surface is reported. B. Forelimb-use asymmetry test. Asymmetrical use of the forelimb for weight support during vertical/lateral exploration in the cylinder. The percent asymmetry is reported. Error bars indicate SEM. * = p<0.05 Mann-Whitney U-test.
In a follow-up set of experiments, the effect of giving a second infusion of either saline or thrombin one day after 6-OHDA was examined. Again, the cannula insertion and infusion of saline was associated with increased behavioral deficits (Fig. 4) and these deficits were prevented when the second infusion contained thrombin (1 U). In placing (Fig. 4A), the differences between thrombin and saline groups reached significance on days 7 [0 ± 0 vs. 53 ± 28; % unsuccessful (mean) ± SEM; p<0.05 Mann-Whitney U-test) and 21 (0 ± 0 vs. 60 ± 24%). This effect was also observed in the forelimb-use asymmetry test (Fig. 4B), with the thrombin group exhibiting significantly less asymmetry on days 7 (-4 ± 4 vs. 72 ± 9 % asymmetry in saline controls), 14 (3 ± 8 vs. 75 ± 13%) and 21 (12 ± 12 vs. 79 ± 15%).
Figure 4.
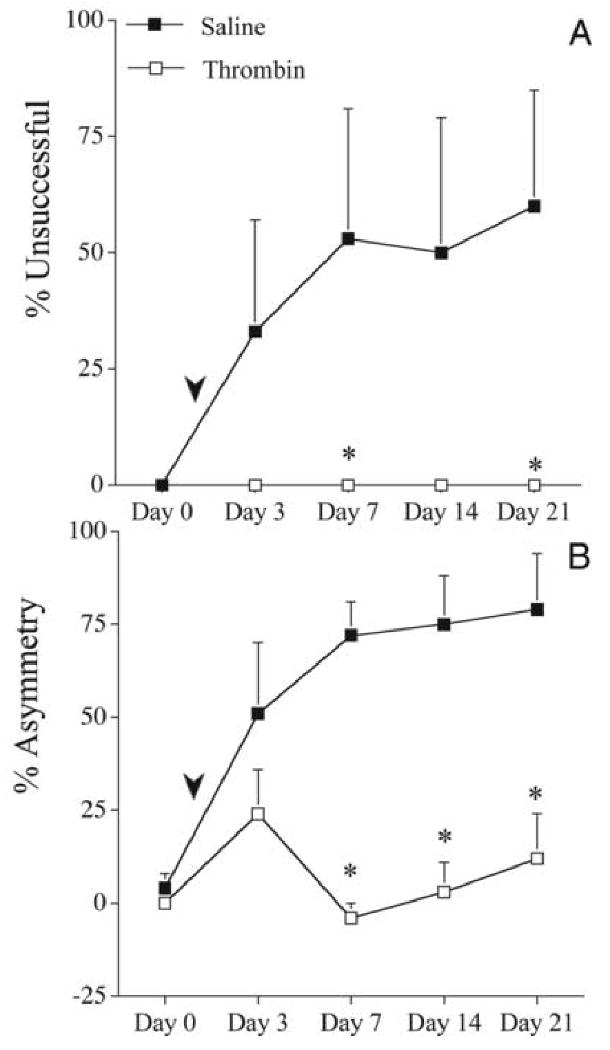
The effects of delayed (1 day) saline or thrombin administration on 6-OHDA (5 μg) induced behavioral deficits. Saline (50 μL; filled squares, n=5) or thrombin (1 U/50 μL; open squares, n=6) was administered at the lesion site one day after 6-OHDA. Arrowheads indicate approximate time of second surgery. A. Vibrissae-elicited forelimb placing test. The percent unsuccessful forelimb reach onto a table surface is reported. B. Forelimb-use asymmetry test. Asymmetrical use of the forelimb for weight support during vertical/lateral exploration in the cylinder. The percent asymmetry is reported. Error bars indicate SEM. * = p<0.05 Mann-Whitney U-test.
We examined whether the adverse effects of the second saline infusion on behavioral deficits were dependent on prior 6-OHDA induced neurotoxicity. Rats were subjected to infusion of saline (4 μL) followed by a second saline infusion (50 μL) 7 days later. This protocol did not result in behavioral deficits (data not shown) in contrast to the effects of infusion of 6-OHDA followed by a second saline infusion shown in Figure 3.
Neurochemistry
Because a secondary cannula insertion/saline infusion 7 days after 6-OHDA (5 μg) resulted in the most striking increases in behavioral deficits, animals were evaluated for dopamine depletion. Compared to the thrombin (1 U) group (n = 5), the saline group (n = 6) exhibited significantly more depletion in dopamine (60 ± 13 vs. 90 ± 8; % depletion; thrombin vs. saline; p<0.05, one-tailed Student's t-test) and HVA (29 ± 14 vs. 77 ± 13%; Fig. 5). Absolute differences in ipsilateral DOPAC (1.76 ± 0.61 vs. 0.41 ± 0.29; ng/mg wet tissue wt.; Fig. 5C) and HVA (1.34 ± 0.33 vs. 0.23 ± 0.12; Fig. 5D) were also evident.
Figure 5.
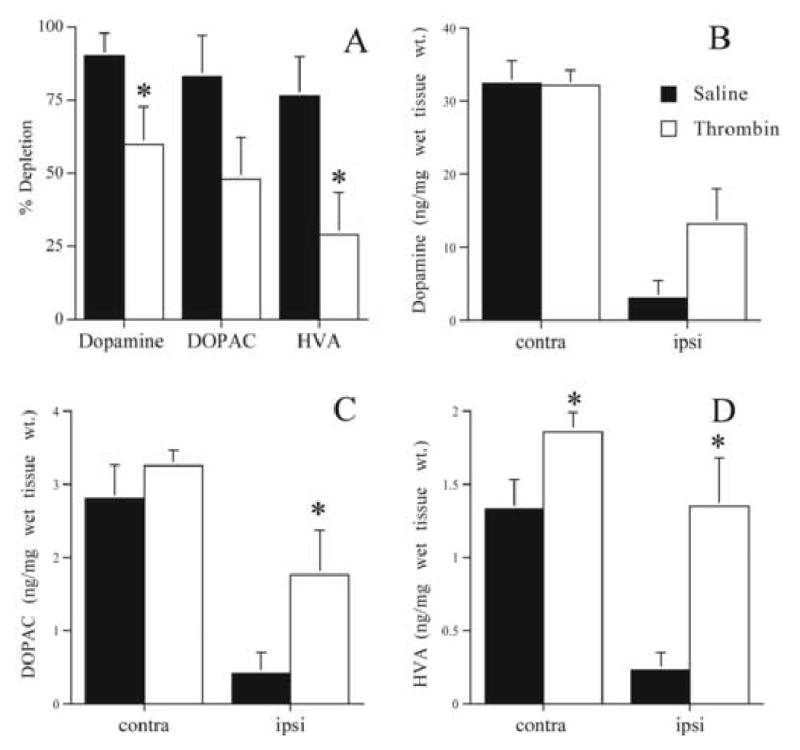
Striatal catecholamines 21 days after 6-OHDA (5 μg): The effects of a second cannula insertion and delayed thrombin administration. Either saline (50 μL; filled bars; n = 5) or thrombin (1 U/50 μL; open bars; n = 6) was administered at the lesion site 7 days after 6-OHDA. A. Percent depletion for dopamine, DOPAC, and HVA. B. Absolute dopamine (ng/mg wet tissue wt.) contralateral (contra) and ipsilateral (ipsi) to 6-OHDA infusion. C. Absolute DOPAC. D. Absolute HVA. Error bars indicate SEM. * = p<0.05, one-tailed student's t-test.
Discussion
There are three main findings in this study. First, co-administration of thrombin or a PAR-1 agonist with 6-OHDA increases behavioral deficits. Second, a cannula insertion after 6-OHDA potentiates the lesion, while third, delayed thrombin infusion protects against this effect. These data provide additional information about the effects of thrombin in experimental PD. Previously, we have found that thrombin given before the 6-OHDA lesion (thrombin preconditioning) is protective [3]. Thus, it is apparent that the effects of thrombin on brain injury in the 6-OHDA model of PD are very dependent upon timing.
While the role of thrombin in the pathogenesis of human PD remains to be determined, several studies have found that thrombin at high doses (∼20 U) can damage dopaminergic neurons in vivo [5,8,9] and in vitro [40 U/mL; [9,25]]. Here, however, we provide data indicating that a much lower dose of thrombin (1 U) can, when given at the time of a 6-OHDA lesion, increase behavioral deficits.
With the interest in multi-factorial causes of cell death in PD, the role of thrombin should be considered as one potential pathogen. There are several potential scenarios where thrombin could contribute to the pathogenesis of PD. For example, blood-brain-barrier (BBB) disruption occurs in human PD [22] and experimental PD [6]. Under the condition of BBB disruption, prothrombin can enter the parenchyma and then be converted to thrombin [46]. Conversely, thrombin itself can cause BBB disruption [27], whereby harmful exogenous agents could enter the parenchyma.
In clinical case reports of hemorrhage near the nigrostriatal tract, PD-like and L-DOPA responsive symptoms have been documented [19,30]. This effect has also been observed in hemorrhages remote to the nigrostriatal tract [28]. Hemorrhage in or near the nigrostriatal tract would be one potential way that dopaminergic cells could be exposed to thrombin. Indeed, after intracerebral hemorrhage, there is the immediate production of large amounts of thrombin [46]. Thrombin has also been thought to play a pathogenic role in other neurodegenerative diseases. Thrombin results in cognitive impairment and pathological features similar to those in Alzheimer's disease in animals [31] and is also found at high levels in neuritic plaques in human Alzheimer's disease [1].
The high doses of thrombin (∼20 U) used in previous studies indicating that thrombin can damage dopaminergic neurons in vivo [5,9] are similar to the amount that may be produced by a 100 μL cerebral hemorrhage [26]. The current study, however, indicates that a much smaller dose (1 U) can exacerbate brain injury at the time of 6-OHDA infusion. Such doses of thrombin can be generated in the brain without a hemorrhage. Thus, Hua et al. [17] have reported about 0.6 U/g in the brain after focal cerebral ischemia. Experiments using doses in addition to 1 or ∼20 U of thrombin will be required to more fully characterize the dose response relationship of the effects of thrombin on the nigrostriatal dopamine system.
Microglial activation has been found to be an important mechanism by which thrombin could damage dopaminergic neurons [8,10,25]. While the direct contribution of microglia to the pathogenesis of PD has remained a controversial topic, a great deal of research suggests that they do indeed play a role [38]. It is possible here that low dose thrombin at the time of 6-OHDA increases the injury through some level of microglial activation.
Before discussing the results of co-administration of the PAR-1 agonist, the specificity of the drugs used should be addressed. PAR activation occurs by cleavage of the extracellular N-terminus of the receptor, resulting in a new N-terminus that then acts as a tethered peptide ligand, binding to the receptor and resulting in activation [43]. Synthetic peptides mimicking the tethered peptide ligand of the specific PAR are able to activate selectively the receptor without the proteolysis step [12,13]. Indeed, the PAR-1 agonist peptide utilized here has been successfully used to elicit such a specific response [43].
Similarly to thrombin, the PAR-1 agonist, when co-administered with 6-OHDA, increased behavioral deficits. There was a tendency for the PAR-1 group to have greater depletion of dopamine, DOPAC and HVA, but this did not reach statistical significance. Hamill et al. in an abstract (2004) have also reported that a PAR-1 knockout is protective against MPTP-induced neurotoxicity, another experimental PD model. These results suggest that PAR-1 could play a role in PD. However, as with thrombin itself, it is likely that the effects of PAR-1 activation will depend on timing and degree of activation. Thus, the effects of PAR-1 activation in the brain have been found to range from neuroprotective to promoting cell death [42]. In a 6-OHDA rat model of PD, we have found that PAR-1 mediates thrombin preconditioning-elicited protection, while PAR-4 activation increases 6-OHDA-induced behavioral deficits [4]. It is also interesting that PAR-1 has been found to be upregulated in human PD cases [20].
Surprisingly, a cannula insertion and saline infusion delivered 1 or 7 days after 6-OHDA resulted in marked behavioral deficits. This was most noticeable in the rats receiving the saline infusion seven days after 6-OHDA. These rats showed no detectable deficits prior to the infusion, but had marked placing deficits thereafter (Fig. 3). This temporal pattern is very different from rats that received only 6-OHDA, where deficits plateau at about day 7 (Fig. 1; [33]). It should be noted that this effect of cannula insertion plus saline infusion is not simply an additive effect of two injuries but depends on the timing. In our previous experiments, rats underwent cannula insertion/saline infusion prior to the 6-OHDA infusion (5 μg). In those experiments, animals did not exhibit placing deficits [4].
The finding that a cannula insertion after 6-OHDA potentiates the lesion raises questions about the role of physical injury in the pathogenesis of PD. The data here suggest that a traumatic event, such as a cannula insertion, into vulnerable tissue increases the severity of the impairment. Indeed, there has been recent interest in the role of traumatic brain injury (TBI) in PD. For example, in experimental TBI, aggregation of α-synuclein, a pivotal protein in PD, occurs [40]. It is possible that a non-specific form of injury to the nigrostriatal pathway may exaggerate PD symptoms in patients with partial dopamine degeneration, or even precipitate symptoms in people who are otherwise at a sub-clinical stage. Of some relevance to the issue of neurotoxin-induced tissue vulnerability may be the finding that after recovery from unilateral partial dopamine depletion in young adulthood, there is a re-emergence of symptoms during old age (and only on the impaired side of the body) [33,34]. Further research combining neurotoxic, genetic and/or TBI models of disease will be needed to assess the contribution of TBI to the pathogenesis of PD.
Delayed thrombin administration protected against the behavioral deficits induced by the cannula insertion/saline infusion after 6-OHDA. Thrombin also improved striatal catecholamine levels compared to saline treated-rats. These findings were unexpected, as our previous research suggests that thrombin delivered at the time of the 6-OHDA infusion is detrimental and, to be protective, pretreatment several days before toxin delivery is required. There are multiple potential explanations for how delayed thrombin administration could provide protection from a second cannula insertion. It is possible that the clotting cascade is activated by thrombin, generating fibrin that could seal the wound area. Indeed thrombin-soaked sponges are used to maintain hemostasis in clinical neurosurgery [2]. However, it should be noted that in the rat, such sponges have been found to produce edema [11]. Additionally, thrombin is a potent mitogen for astrocytes and induces morphological changes [7,14,32,41]. Whether astrocytic alterations or microglial activation after the injury would be beneficial remains to be determined. Further research will need to be conducted to discern how thrombin is able to protect against the effects of a second cannula insertion in this PD model.
The current study has assessed neurobehavioral and neurochemical deficits after 6-OHDA. Morphological or stereological analysis was not conducted. In future studies, it will be useful to determine whether thrombin at the time of injury or cannula insertion after 6-OHDA results in increased loss of dopaminergic neurons and terminals. Additionally, it would be advantageous to determine if PAR-1 activation is also able to prevent the effects of a second cannula insertion.
In summary, our findings indicate that thrombin at the time of 6-OHDA increases the severity of the behavioral impairment. Additionally, a cannula insertion with saline infusion, occurring after 6-OHDA increased behavioral and neurochemical deficits, and thrombin administration protected against this effect.
Acknowledgments
This work was supported by NS-39866 (G.X.), NS-19608 (T.S.) and NS-34709 (R.F.K.) from the National Institutes of Health (NIH). J.R.C was supported by the training grant T32ES07062 (R.J.R).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Jason R. Cannon, Email: cannonjr@upmc.edu.
Ya Hua, Email: yahua@umich.edu.
Rudy J. Richardson, Email: rjrich@umich.edu.
Guohua Xi, Email: guohuaxi@umich.edu.
Richard F. Keep, Email: rkeep@umich.edu.
Timothy Schallert, Email: tschallert@mail.utexas.edu.
References
- 1.Akiyama H, Ikeda K, Kondo H, McGeer PL. Thrombin accumulation in brains of patients with Alzheimer's disease. Neurosci Lett. 1992;146:152–154. doi: 10.1016/0304-3940(92)90065-f. [DOI] [PubMed] [Google Scholar]
- 2.Arand AG, Sawaya R. Intraoperative chemical hemostasis in neurosurgery. Neurosurgery. 1986;18:223–233. doi: 10.1227/00006123-198602000-00022. [DOI] [PubMed] [Google Scholar]
- 3.Cannon JR, Keep RF, Hua Y, Richardson RJ, Schallert T, Xi G. Thrombin preconditioning provides protection in a 6-hydroxydopamine Parkinson's disease model. Neurosci Lett. 2005;373:189–194. doi: 10.1016/j.neulet.2004.10.089. [DOI] [PubMed] [Google Scholar]
- 4.Cannon JR, Keep RF, Schallert T, Hua Y, Richardson RJ, Xi G. Protease-activated receptor-1 mediates protection elicited by thrombin preconditioning in a rat 6-hydroxydopamine model of Parkinson's disease. Brain Res. 2006;1116:177–186. doi: 10.1016/j.brainres.2006.07.094. [DOI] [PubMed] [Google Scholar]
- 5.Carreno-Muller E, Herrera AJ, de Pablos RM, Tomas-Camardiel M, Venero JL, Cano J, Machado A. Thrombin induces in vivo degeneration of nigral dopaminergic neurones along with the activation of microglia. J Neurochem. 2003;84:1201–1214. doi: 10.1046/j.1471-4159.2003.01634.x. [DOI] [PubMed] [Google Scholar]
- 6.Carvey PM, Zhao CH, Hendey B, Lum H, Trachtenberg J, Desai BS, Snyder J, Zhu YG, Ling ZD. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur J Neurosci. 2005;22:1158–1168. doi: 10.1111/j.1460-9568.2005.04281.x. [DOI] [PubMed] [Google Scholar]
- 7.Cavanaugh KP, Gurwitz D, Cunningham DD, Bradshaw RA. Reciprocal modulation of astrocyte stellation by thrombin and protease nexin-1. J Neurochem. 1990;54:1735–1743. doi: 10.1111/j.1471-4159.1990.tb01228.x. [DOI] [PubMed] [Google Scholar]
- 8.Choi SH, Joe EH, Kim SU, Jin BK. Thrombin-induced microglial activation produces degeneration of nigral dopaminergic neurons in vivo. J Neurosci. 2003;23:5877–5886. doi: 10.1523/JNEUROSCI.23-13-05877.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi SH, Lee DY, Ryu JK, Kim J, Joe EH, Jin BK. Thrombin induces nigral dopaminergic neurodegeneration in vivo by altering expression of death-related proteins. Neurobiol Dis. 2003;14:181–193. doi: 10.1016/s0969-9961(03)00085-8. [DOI] [PubMed] [Google Scholar]
- 10.Choi SH, Lee da Y, Chung ES, Hong YB, Kim SU, Jin BK. Inhibition of thrombin-induced microglial activation and NADPH oxidase by minocycline protects dopaminergic neurons in the substantia nigra in vivo. J Neurochem. 2005;95:1755–1765. doi: 10.1111/j.1471-4159.2005.03503.x. [DOI] [PubMed] [Google Scholar]
- 11.Colon GP, Lee KR, Keep RF, Chenevert TL, Betz AL, Hoff JT. Thrombin-soaked gelatin sponge and brain edema in rats. J Neurosurg. 1996;85:335–339. doi: 10.3171/jns.1996.85.2.0335. [DOI] [PubMed] [Google Scholar]
- 12.Coughlin SR. Protease-activated receptors start a family. Proc Natl Acad Sci U S A. 1994;91:9200–9202. doi: 10.1073/pnas.91.20.9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dery O, Corvera CU, Steinhoff M, Bunnett NW. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am J Physiol. 1998;274:C1429–1452. doi: 10.1152/ajpcell.1998.274.6.C1429. [DOI] [PubMed] [Google Scholar]
- 14.Ehrenreich H, Costa T, Clouse KA, Pluta RM, Ogino Y, Coligan JE, Burd PR. Thrombin is a regulator of astrocytic endothelin-1. Brain Res. 1993;600:201–207. doi: 10.1016/0006-8993(93)91374-2. [DOI] [PubMed] [Google Scholar]
- 15.Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7:437–448. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- 16.Hamill CE, Caudle WM, Richardson JR, Miller GW, Traynelis SF. Abstract Viewer/Itinerary Planner. Washington, DC: Society for Neuroscience; 2004. Role for protease-activated receptor 1 (PAR1) in MPTP-induced dopaminergic neurotoxicity. Program No. 562.6: Online. [Google Scholar]
- 17.Hua Y, Wu J, Keep RF, Hoff JT, Xi G. Thrombin exacerbates brain edema in focal cerebral ischemia. Acta Neurochir Suppl. 2003;86:163–166. doi: 10.1007/978-3-7091-0651-8_34. [DOI] [PubMed] [Google Scholar]
- 18.Hua Y, Wu J, Pecina S, Yang S, Schallert T, Keep RF, Xi G. Ischemic preconditioning procedure induces behavioral deficits in the absence of brain injury? Neurol Res. 2005;27:261–267. doi: 10.1179/016164105X25270. [DOI] [PubMed] [Google Scholar]
- 19.Inoue H, Udaka F, Takahashi M, Nishinaka K, Kameyama M. Secondary parkinsonism following midbrain hemorrhage. Rinsho Shinkeigaku. 1997;37:266–269. [PubMed] [Google Scholar]
- 20.Ishida Y, Nagai A, Kobayashi S, Kim SU. Upregulation of protease-activated receptor-1 in astrocytes in Parkinson disease: astrocyte-mediated neuroprotection through increased levels of glutathione peroxidase. J Neuropathol Exp Neurol. 2006;65:66–77. doi: 10.1097/01.jnen.0000195941.48033.eb. [DOI] [PubMed] [Google Scholar]
- 21.Jiang Y, Wu J, Hua Y, Keep RF, Xiang J, Hoff JT, Xi G. Thrombin-receptor activation and thrombin-induced brain tolerance. Journal of Cerebral Blood Flow & Metabolism. 2002;22:404–410. doi: 10.1097/00004647-200204000-00004. [DOI] [PubMed] [Google Scholar]
- 22.Kortekaas R, Leenders KL, van Oostrom JC, Vaalburg W, Bart J, Willemsen AT, Hendrikse NH. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol. 2005;57:176–179. doi: 10.1002/ana.20369. [DOI] [PubMed] [Google Scholar]
- 23.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 24.Langston JW, Forno LS, Rebert CS, Irwin I. Selective nigral toxicity after systemic administration of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyrine (MPTP) in the squirrel monkey. Brain Res. 1984;292:390–394. doi: 10.1016/0006-8993(84)90777-7. [DOI] [PubMed] [Google Scholar]
- 25.Lee DY, Oh YJ, Jin BK. Thrombin-activated microglia contribute to death of dopaminergic neurons in rat mesencephalic cultures: dual roles of mitogen-activated protein kinase signaling pathways. Glia. 2005;51:98–110. doi: 10.1002/glia.20190. [DOI] [PubMed] [Google Scholar]
- 26.Lee KR, Colon GP, Betz AL, Keep RF, Kim S, Hoff JT. Edema from intracerebral hemorrhage: the role of thrombin. J Neurosurg. 1996;84:91–96. doi: 10.3171/jns.1996.84.1.0091. [DOI] [PubMed] [Google Scholar]
- 27.Lee KR, Kawai N, Kim S, Sagher O, Hoff JT. Mechanisms of edema formation after intracerebral hemorrhage: effects of thrombin on cerebral blood flow, blood-brain barrier permeability, and cell survival in a rat model. J Neurosurg. 1997;86:272–278. doi: 10.3171/jns.1997.86.2.0272. [DOI] [PubMed] [Google Scholar]
- 28.Ling MJ, Aggarwal A, Morris JG. Dopa-responsive parkinsonism secondary to right temporal lobe haemorrahage. Mov Disord. 2002;17:402–404. doi: 10.1002/mds.10081. [DOI] [PubMed] [Google Scholar]
- 29.Masada T, Xi G, Hua Y, Keep RF. The effects of thrombin preconditioning on focal cerebral ischemia in rats. Brain Research. 2000;867:173–179. doi: 10.1016/s0006-8993(00)02302-7. [DOI] [PubMed] [Google Scholar]
- 30.Matsuda W, Sugimoto K, Sato N, Watanabe T, Yanaka K, Matsumura A, Nose T. A case of primary brain-stem injury recovered from persistent vegetative state after L-dopa administration. No To Shinkei. 1999;51:1071–1074. [PubMed] [Google Scholar]
- 31.Mhatre M, Nguyen A, Kashani S, Pham T, Adesina A, Grammas P. Thrombin, a mediator of neurotoxicity and memory impairment. Neurobiol Aging. 2004;25:783–793. doi: 10.1016/j.neurobiolaging.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 32.Neveu I, Jehan F, Jandrot-Perrus M, Wion D, Brachet P. Enhancement of the synthesis and secretion of nerve growth factor in primary cultures of glial cells by proteases: a possible involvement of thrombin. J Neurochem. 1993;60:858–867. doi: 10.1111/j.1471-4159.1993.tb03230.x. [DOI] [PubMed] [Google Scholar]
- 33.Schallert T. Aging-dependent emergence of sensorimotor dysfunction in rats recovered from dopamine depletion sustained early in life. In: Joseph JA, editor. Central Determinants of Age-related Declines in Motor Function. Vol. 515. New York: Ann N Y Acad Sci; 1988. pp. 108–120. [DOI] [PubMed] [Google Scholar]
- 34.Schallert T. Sensorimotor impairment and recovery of function in brain-damaged rats: reappearance of symptoms during old age. Behav Neurosci. 1983;97:159–164. doi: 10.1037//0735-7044.97.1.159. [DOI] [PubMed] [Google Scholar]
- 35.Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST. CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology. 2000;39:777–787. doi: 10.1016/s0028-3908(00)00005-8. [DOI] [PubMed] [Google Scholar]
- 36.Schallert T, Wilcox RE. Neurotransmitter-selective brain lesions. In: Boulton AA, Baker GB, editors. Neuromethods. Vol. 1. Clifton: Humana Press; 1985. pp. 343–387. [Google Scholar]
- 37.Striggow F, Riek M, Breder J, Henrich-Noack P, Reymann KG, Reiser G. The protease thrombin is an endogenous mediator of hippocampal neuroprotection against ischemia at low concentrations but causes degeneration at high concentrations. Proc Natl Acad Sci U S A. 2000;97:2264–2269. doi: 10.1073/pnas.040552897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teismann P, Schulz JB. Cellular pathology of Parkinson's disease: astrocytes, microglia and inflammation. Cell Tissue Res. 2004;318:149–161. doi: 10.1007/s00441-004-0944-0. [DOI] [PubMed] [Google Scholar]
- 39.Tillerson JL, Cohen AD, Philhower J, Miller GW, Zigmond MJ, Schallert T. Forced limb-use effects on the behavioral and neurochemical effects of 6-hydroxydopamine. Journal of Neuroscience. 2001;21:4427–4435. doi: 10.1523/JNEUROSCI.21-12-04427.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uryu K, Giasson BI, Longhi L, Martinez D, Murray I, Conte V, Nakamura M, Saatman K, Talbot K, Horiguchi T, McIntosh T, Lee VM, Trojanowski JQ. Age-dependent synuclein pathology following traumatic brain injury in mice. Exp Neurol. 2003;184:214–224. doi: 10.1016/s0014-4886(03)00245-0. [DOI] [PubMed] [Google Scholar]
- 41.Vaughan PJ, Pike CJ, Cotman CW, Cunningham DD. Thrombin receptor activation protects neurons and astrocytes from cell death produced by environmental insults. J Neurosci. 1995;15:5389–5401. doi: 10.1523/JNEUROSCI.15-07-05389.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H, Reiser G. Thrombin signaling in the brain: the role of protease-activated receptors. Biol Chem. 2003;384:193–202. doi: 10.1515/BC.2003.021. [DOI] [PubMed] [Google Scholar]
- 43.Wang H, Ubl JJ, Reiser G. Four subtypes of protease-activated receptors, co-expressed in rat astrocytes, evoke different physiological signaling. Glia. 2002;37:53–63. doi: 10.1002/glia.10012. [DOI] [PubMed] [Google Scholar]
- 44.Woodlee MT, Schallert T. The interplay between behavior and neurodegeneration in rat models of Parkinson's disease and stroke. Restor Neurol Neurosci. 2004;22:153–161. [PubMed] [Google Scholar]
- 45.Xi G, Keep RF, Hua Y, Xiang J, Hoff JT. Attenuation of thrombin-induced brain edema by cerebral thrombin preconditioning. Stroke. 1999;30:1247–1255. doi: 10.1161/01.str.30.6.1247. [DOI] [PubMed] [Google Scholar]
- 46.Xi G, Reiser G, Keep RF. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective? Journal of Neurochemistry. 2003;84:3–9. doi: 10.1046/j.1471-4159.2003.01268.x. [DOI] [PubMed] [Google Scholar]