

Abstract
Purpose
Malignant rhabdoid tumors (MRT), although rare, are one of the most aggressive pediatric malignancies. Loss of INI1, a tumor suppressor gene and member of the SWI/SNF chromatin remodeling complex, is a recurrent genetic characteristic of these tumors and an important diagnostic marker. We have previously demonstrated a novel interaction between the serine/threonine kinase Akt and INI1, as well as other SWI/SNF subunits. This, coupled with experiments in the literature suggesting that the PI3K/Akt pathway is dysregulated in MRT cells, caused us to investigate the activation and importance of this pathway in this tumor type.
Methods
In this study, we used MTT assays to evaluate the sensitivity of MRT cell lines to PI3K inhibition. Western blot analysis and Raf pulldown assays were used to examine potential mechanisms of PI3K/Akt dysregulation.
Results
Inhibition of the PI3K/Akt pathway caused a significant reduction in the survival of the four MRT cell lines tested, and three cell lines demonstrated constitutively active Akt. Two of these constitutively active Akt cell lines abundantly expressed IGF-1R and an inhibitor of IGF-1R, NVP-AEW541, reduced Akt phosphorylation in one of them. The third constitutively active Akt cell line appeared to express a mutant IGF-1R.
Conclusions
Our data suggests that the PI3K/Akt pathway is a crucial means of maintaining the survival and growth of MRT cells. The cells therefore employ various mechanisms to stimulate this pathway, and growth factor receptor dysregulation appears to be a common method. Drugs that inhibit the PI3K pathway or interfere with IGF autocrine loops may be of great value in treating MRT, which is largely resistant to conventional chemotherapeutic approaches.
Keywords: SWI/SNF, Malignant rhabdoid tumor, PI3K, Akt, Oncogene addiction, NVP-AEW541
Introduction
Malignant rhaboid tumor (MRT) is a rare and aggressive pediatric cancer, often occurring in children less than 2 years of age. It is most often reported in the kidney, but can also occur in the brain, referred to as atypical teratoid/rhabdoid tumor (AT/RT), or in soft tissues. MRT has a high incidence of early metastases, as well as a high resistance to chemotherapy, resulting in a survival rate of only approximately 20%.
Although originally thought to be related to Wilm’s tumor, rhabdoid tumors were later shown to be characterized by monosomy 22 or a partial deletion of chromosome band 22q11.2 [1–3]. This region was further narrowed to the INI1 gene locus, now a known tumor suppressor gene and a member of the SWI/SNF chromatin remodeling complex [4–6]. Heterozygous loss of INI1 in mouse knockout experiments has been shown to predispose mice to tumors consistent with MRT [7–9]. Further, reintroduction of INI1 into MRT cells has been shown to cause cell cycle arrest at the G0–G1 stage, flat cell formation, and expression of senescence-associated markers [10–13].
As a member of the SWI/SNF complex, INI1 may play an important role in cell cycle regulation through interaction with the retinoblastoma protein (pRb). SWI/SNF subunits, Brg and hBrm, have been shown to interact with pRb, and pRb is unable to suppress growth in their absence [14, 15]. Betz et al. [10], however, argue that INI1 is not required for Rb-mediated cell cycle arrest because the growth of MRT cell lines can be suppressed by transient transfection with p16INK4a or constitutively active Rb. They, and others, suggest that INI1 functions upstream of Rb by activating p16INK4a. Chromatin immunoprecipitation (ChIP) assays support this hypothesis by localizing INI1 to the p16INK4a promoter, where its presence correlates with transcriptional activation [16]. ChIPs have also demonstrated that the presence of INI1 is correlated with increased transcription of p21WAF/CIP and decreased transcription of cyclin D [12, 17–19]. Although the number of genes regulated by the SWI/SNF complex in mammals is unclear, the complex appears to be responsible for the regulation of approximately 5% of yeast genes [20, 21] and therefore loss of INI1 has the potential to affect many other targets.
We have previously demonstrated an interaction between SWI/SNF subunits, including INI1, and the serine/threonine kinase Akt [22]. Akt is known to be dysregulated in many types of cancer including breast, ovarian, pancreatic, prostate, and non-small cell lung cancer [23–28] and we sought to characterize Akt signaling in MRT cells. Akt is a downstream effector of phosphoinositol-3-kinase (PI3K). PI3K is activated by various growth factor receptor signaling cascades and generates lipid products, which recruit Akt to the plasma membrane, where it is phosphorylated and activated. Akt then targets many proteins involved in cell survival, cell growth, and the cell cycle. For example, activated Akt protects cells from apoptosis by phosphorylating substrates such as pro-apoptotic enzymes, caspase-9 and BAD, resulting in their inactivation, as well as phosphorylating forkhead-related transcription factors, preventing the transcription of pro-apoptotic genes [29–31]. Akt is also able to phosphorylate cyclin-dependent kinase inhibitors, p27 and p21, causing cytoplasmic retention and inhibiting their role in G1 arrest [32–34].
Here we first investigated the effect of PI3K/Akt inhibition on MRT cells, and upon finding them sensitive, next addressed Akt activation. In the majority of cell lines tested, Akt appeared to be dysregulated and possible causes for dysregulation were investigated. Our results suggest that MRT cells depend on Akt for survival and that one mechanism for its activation may be through aberrant activation of the IGF-1R pathway. Signaling through IGF-1R has been shown to play a role in promoting the malignant phenotype of many human neoplasias (see [35] and references therein). Furthermore, studies have shown that IGF-1 is capable of stimulating the PI3K/Akt pathway in cancer cell lines and that this effect is blocked by the IGF-1R inhibitor, NVP-AEW541 [36]. Studies in MRT cell lines demonstrated that, in at least one cell line, treatment with NVP-AEW541 could reduce constitutive Akt phosphorylation. Taken together, these results indicate that drugs targeting the PI3K/Akt pathway or that block IGF-1R, may be of therapeutic value in cases of MRT, and help to improve the prognosis for this aggressive tumor type.
Materials and methods
Cell culture and treatments
Four malignant rhabdoid tumor cell lines; TM87-16, TTC549, STM91-01, and RT4E, were used in this work. TM87-16, STM91-01, TTC549 (kind gifts from Dr. Timothy Triche, Los Angeles Children’s Hospital), and RT4E (established by the MUSC tumor bank) were established and maintained as previously described [37, 38]. Cells were grown in RPMI 1640 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco). Control cell lines, HeLa and MCF7, were grown in minimum essential medium (MEM) (Gibco, Carlsbad, CA) supplemented with 10% FBS. For various assays, cells were treated with LY294002 (Calbiochem, San Diego, CA, USA), rapamycin (Sigma, St Louis, MO, USA), and NVP-AEW541 (kind gift from Dr. Steve Rosenzweig, MUSC).
MTT assay
Mitochondrial function was determined by the reduction of 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide. MRT cells were plated in rows (6 replicates per cell line) of a 96-well plate at a density of 1 × 104 cells per well and cultured for 24 h in routine culture media. The cells were then incubated in media with serum containing vehicle (DMSO) or LY294002 at concentrations of 5, 10, 15, or 20 μM for 48 h. The media and drug were removed from the cells, replaced with 200 μl drug-free media and 50 μl of a MTT solution (100 ng MTT, 20 ml phosphate-buffered saline (PBS) [140 mM NaCl, 2.7 mM KCl, 10 mM NA2HPO4, 1.8 mM KH2PO4, pH 7.4]), and incubated for 4 h. This solution was then removed and cells were incubated overnight with 100 μl lysing buffer (125 ml DMF, 125 ml water, 50 g SDS, 5 ml 80% acetic acid, 5 ml 1 M HCl). The plate was read at a wavelength of 570 nm by a μQuant Microplate Spectrophotometer (BioTek Instruments, Winooski, VT, USA). The entire experiment was performed independently twice for a total of 12 readings per cell line.
Western blot analysis
Cells were rinsed in cold PBS and scraped into PBS containng 1 mM sodium orthovanadate and incubated for 20 min on ice. Cells were then pelleted and whole cell extracts were prepared by incubating the cell pellet in cell lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton-X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate), plus 0.5 mM phenyl-methylsulphonylfluoride (PMSF) and 1 mM sodium orthovanadate, on ice for 30 min. The resulting lysate was clarified by centrifugation at 11,000 rpm for 10 min using a JA-18.1 rotor and a Beckman J2-MI centrifuge at 4°C. Extracts were fractionated on SDS-polyacrylamide gels and electroblotted onto Immobilon PVDF membranes (Millipore, Billerica, MA, USA). Western blots were performed using the following antibodies: anti-Akt 1,2 (H-136, Santa Cruz, Santa Cruz, CA), anti-phospho Akt (S473, Cell Signaling #9271, Beverly, MA, USA), anti-Akt (Cell Signaling #9272), anti-β-actin (Figs. 2, 3, 4, A5441, Sigma, St Louis, MO, USA), goat anti-actin (Fig. 5, Santa Cruz #sc-1615), anti-EGF Receptor (Cell Signaling, #2232), anti-phospho-Erk (Cell Signaling, #3371), anti-p44/42 MAP Kinase (total Erk, Cell Signaling #9102), anti-IGF-I Receptor β (Cell Signaling, #3027), anti-PTEN (Cell Signaling #9552), anti-pan-Ras (Ab-3, Oncogene Research Products, Calbiochem #OP40), and anti-phospho-S6 Ribosomal Protein (ser235/236, Cell Signaling, #4856). Secondary antibodies were horseradish peroxidase-conjugated anti-rabbit (#172–1019, Bio-Rad, Hercules, CA, USA) or anti-mouse (#NA9310 V, Amersham Biosciences, Piscataway, NJ, USA) and were detected using chemiluminescence (ECL: Amersham Biosciences) and exposure of blots to X-ray film. For stripping, blots were incubated with RestoreTM Western Blot Stripping Buffer (#21059, Pierce Biotechnology, Inc., Rockford, IL, USA).
Fig. 2.

Three out of four MRT cell lines have constitutively active Akt that can be modulated by LY294002 or rapamycin. Cells were grown to sub-confluency and then incubated in media in the presence of serum (RC for randomly cycling), in serum-free media (SS), or in serum-free media with either 20 μM LY294002 (LY) or 150 ng/ml rapamycin (RA) for 24 h. Duplicate aliquots from cell lysates harvested from each sample containing 30 μg of protein were separated by 10% sodium dodecyl sulfate (SDS)-PAGE. Western blotting was performed and the resulting membranes cut into strips. For one membrane, the top portion was probed with antibodies to Akt1,2 and the bottom was probed with antibodies to phosphorylated S6 Ribosomal Protein (P-S6). The top portion of the other membrane was probed with anti-phospho-Akt (Akt-P) antibodies and the bottom with anti-actin antibodies. Cell lines are designated above the panels. The experiment was performed independently multiple times for each cell line
Fig. 3.

PTEN expression in MRT cell lines. 30 μg of protein from TM87-16 (TM), STM91-01 (STM), TTC549 (549), RT4E (4E) and HeLa (HL) cell lines were separated by 10% sodium dodecyl sulfate (SDS)-PAGE. Western blotting was performed using an anti-PTEN antibody and reprobed with an anti-actin antibody to confirm equal loading. The positions of molecular mass markers run in parallel are indicated to the left of the panels
Fig. 4.

Growth factor receptor expression varies between MRT cell lines. Duplicate aliquots of 30 μg of protein from TM87-16 (TM), STM91-01 (STM), TTC549 (549), RT4E (4E), MCF7, and HeLa (HL) cell lines were separated by 8% sodium dodecyl sulfate (SDS)-PAGE. Western blotting was then performed. One membrane was probed with anti-EGFR antibodies followed by antibodies to actin, the other was probed with anti-IGF-1R antibodies
Fig. 5.

Akt phosphorylation can be modulated by NVP-AEW541 in one MRT cell line. Cells were grown to sub-confluency and then incubated in media in the presence of serum (RC), in serum-free media (SS) or in serum-free media containing 0.5 μM NVP-AEW541 (NV) for 24 h. Aliquots from cell lysates harvested from each sample containing 50 μg of protein were separated by 10% SDS-PAGE. Western blotting was performed by probing with anti-phospho-Akt (Akt-P) antibodies; the blot was then stripped and probed with anti-actin antibodies and, lastly, with antibodies to total Akt. Cell lines are designated above the panels
RAF pull-down assay
Cells were cultured for 24 h in routine culture media or serum-free media. A 50% slurry of glutathione sepharose (GSH) beads (Amersham Biosciences) in PBS was prepared. Whole cell extracts of 200 μg were then diluted with 100 mM IP Buffer (100 mM KCl, 50 mM Tris, pH 7.5, 5 mM MgCl2, 0.1% NP-40), plus 0.5 mM PMSF and 1 mM sodium orthovanadate, and precleared with 10 μl of empty GSH beads in 250 μl total volume by rolling for 30 min at 4°C. After the beads were pelleted, 10 μl of Raf-1 RBD agarose beads (#14–278, Upstate, Millipore, Billerica, MA, USA) were incubated with the supernatants at 4°C rolling for two hours. Beads were washed 3 times for 2 min with 100 mM IP buffer, containing 0.5 mM PMSF and 1 mM sodium orthovanadate. Proteins were boiled off the beads in Laemmli sample buffer, fractionated on 10% SDS-poly-acrylamide gels and Western blotted with a pan-Ras antibody.
Ras sequencing
DNA was extracted from STM91-01, TM87-16, TTC549, and RT4E cells to fragments of the Ras genes were generated in 25 μl PCR reactions containing 300 ng DNA, 1.5 mM MgCl2, 0.1 mM dNTPs, PCR Buffer (Roche, India-napolis, IN, USA), 0.15 μM each of forward and reverse oligos, and 1 U of AmpliTaq Gold (Roche). Primers were used to amplify codons 12 and 13 of H-Ras (forward 5′-GAG GAG CGA TGA CGG AAT-3′ and reverse 5′-CTG CAG CCA GCC CTA TCC T-3′), K-Ras (forward 5′-GAC TGA ATA TAA ACT TGT GG-3′ and reverse 5′-CTG TAT CAA AGA ATG GTC CT-3′), and N-Ras (forward 5′-GAC TGA GTA CAA ACT GGT GG-3′ and reverse 5′-TGC ATA ACT GAA TGT ATA CCC-3′). Primers were also used to amplify codon 61 of H-Ras (forward 5′-CTG TCT CCT GCT TCC TCT AGA-3′ and reverse 5′-GTA CTG GTG GAT GTC CTC AA-3′), K-Ras (forward 5′-GAC TGT GTT TCT CCC TTC T-3′ and reverse 5′-TGG CAA ATA CAC AAA GAA AG-3′), and N-Ras (forward 5′-CAA GTG GTT ATA GAT GGT GAA ACC-3′ and reverse 5′-AAG ATC ATC CTT TCA GAG AAA ATA AT-3′). Amplification was performed using the following cycling parameters: 10 min at 94°C; 40 cycles of 94°, 65°, and 72°C for 30 s each; and a single final extension at 72°C for 7 min. Products were then run on a 1% agarose gel and extracted using a Marligen Matrix Gel Extraction System (Marligen Biosciences, Ijamsville, MD, USA). Sequencing was performed by the MUSC Nucleic Acids Analysis and Molecular Biology Core Facility.
Results
PI3K inhibition
Previous experiments have suggested that the PI3K/Akt pathway may play a significant role and possibly be dysregulated in MRT cells. For example, inhibition of the PI3K pathway has been shown to sensitize MRT cell lines to apoptosis induced by radiation, vinblastine, and TRAIL [39, 40]. Also, although treatment with Gefitinib, an EGFR-tyrosine kinase inhibitor, has been shown to inhibit the phosphorylation of Akt in some cell lines, it is unable to do so in MRT cell lines [41]. We therefore sought to investigate the importance of the PI3K/Akt pathway in MRT cells using the methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay. Cells growing in the presence of serum were assayed alone or treated with increasing concentrations of LY294002, a PI3K inhibitor, for 48 h. All cell lines tested exhibited a significant and dose-dependent reduction in cell survival after the treatment (Fig. 1). These experiments were repeated on serum-starved cells and the results were similar except for the TM87-16 cell line, which was resistant to doses of LY294002 above 10 μM (data not shown).
Fig. 1.

PI3K inhibition results in significant and dose-dependent reduction of cell survival in all MRT cell lines tested. MRT cell lines were incubated in media with vehicle (DMSO) or with LY294002 at 5, 10, 15, or 20 μM for 48 h. The MTT activity was determined by the optical density at 570 nm and results are expressed as the mean of six determinations ± standard deviation of the mean. The experiment was repeated independently twice
Akt pathway in MRT cell lines
Due to the sensitivity of the MRT cell lines to LY294002, we next investigated Akt activation in these cells. In these experiments, we also examined HeLa cells as an unrelated cell line known to have supraphysiologic expression levels of IGF-1R (see below). In agreement with Charboneau et al. [42], we found that all MRT cell lines examined expressed Akt and varying levels of P-Akt (Fig. 2). To address whether Akt may be constitutively active, Akt phosphorylation was determined after serum-starvation overnight. Although Akt phosphorylation did drop in TM87-16 cells upon serum starvation, the remaining three cell lines; TTC549, RT4E, and STM91-01, showed no or only slight reduction in levels of Akt phosphorylation (Fig. 2). This suggested that the PI3K/Akt pathway may be constitutively active in these cells. Treatment with LY294002, however, was able to significantly reduce Akt phosphorylation in all cell lines, suggesting that the mechanisms regulating Akt deactivation remained functional. In the TM87-16 and HeLa cell lines treatment with LY294002 and rapamycin resulted in loss of total Akt through unknown mechanisms. Loss of expression of the PTEN tumor suppressor gene, which dephosphorylates the phosphatidylinositol polyphosphates that recruit Akt to the plasma membrane, is a common cause for dysregulation of the PI3K/Akt pathway. Therefore, western blot analysis was used to examine its presence. PTEN was expressed in all MRT cell lines tested (Fig. 3), which confirms the work of Charboneau et al. [42].
In order to examine other components of the PI3K/Akt pathway, mTOR activity was inhibited by treating the cells with rapamycin. This resulted in lowered phosphorylation of S6 ribosomal protein, a downstream target of mTOR, in all cell lines (Fig. 2). Rapamycin treatment also reduced phosphorylation of Akt. mTOR, complexed with rictor, is thought to be the previously elusive PDK2, responsible for phosphorylating Akt on serine 473 [43, 44]. Long-term rapamycin treatment may inhibit rictor-mTOR activity by sequestering newly synthesized mTOR molecules, thus interfering with rictor-mTOR complex formation [43] and preventing Akt phosphorylation. Taken together, these experiments all suggest that the components of the PI3K/Akt pathway downstream of, and including PI3K, function as expected.
Growth factor receptor expression in MRT cell lines
Overexpression of growth factor receptors was then examined as another possible means for dysregulation of the PI3K/Akt pathway. None of the cell lines tested appeared to express epidermal growth factor receptor (EGFR) at elevated levels (Fig. 4). Interestingly, the TM87-16 cell line had a prominent EGFR band that migrated more rapidly than the signal for full-length EGFR found in most of the cell lines. It is intriguing to speculate that this may represent the previously characterized 110 kDA EGFR isoform; a membrane associated protein produced by alternative splicing and thought to be involved in mediating EGFR function [45]. We next examined insulin-like growth factor 1 receptor (IGF-1R) expression. In accordance with Charboneau et al. [42], we found that TTC549 expressed high levels of mature IGF-1R and also found the same to be true of RT4E, which had not been previously examined. Expression levels in these cell lines are comparable to, if not higher than, in HeLa cells, which have been shown to express IGF-1R at levels that are threefold to fourfold higher than normal ectocervical epithelial cells [46]. Interestingly, the signal for IGF-1R in the STM91-01 lane ran in between the immature and mature IGF-1R bands, perhaps indicating a truncation mutation that is not fully processed to the mature form. Charboneau et al. [42] saw a similar band in the TTC709 cell line.
Inhibition of IGF-1R
Since the TTC549 and RT4E cells potentially overexpressed IGF-1R, and STM91-01 cells had an aberrantly migrating band for IGF-1R by Western bot analysis, it was tempting to speculate that IGF-I/IGF-II:IGF-1R autocrine loops may play a role in promoting growth in these cell lines. Therefore, cells were treated with NVP-AEW541, a compound that has been shown to selectively inhibit the IGF-1R in vivo [47]. Figure 5 shows that Akt phosphorylation in the TTC549 cell line was dramatically reduced by treatment with NVP-AEW541 (lanes 7–9). Visually, the treated cells also detached from the plates and were floating in the media. As seen for Fig. 2, serum starvation slightly reduced Akt phosphorylation in the RT4E cell line, but this was not further reduced by treatment with NVP-AEW541 (lanes 4–6). Akt phosphorylation in STM91-01 cells was constitutive in the absence of serum in the absence or presence of NVP-AEW541 (lanes 1–3).
Ras activation in MRT cell lines
Because Ras signaling can activate the PI3K pathway through direct interaction with the PI3K catalytic subunit, and Ras mutations occur in approximately 20% of all tumors [48], we also investigated whether aberrant Ras activation in the MRT cell lines could contribute to Akt activation. Western blot analysis demonstrated that all cells lines expressed Ras isoforms and that expression levels were unaffected by serum starvation (Fig. 6). Phosphorylation of Erk, a downstream effector of Ras, was examined using a phospho-specific Erk antibody. Serum starvation significantly dropped Erk phosphorylation in the STM91-01 and TTC549 cell lines, slightly in the TM87-16 cell line, and not at all in the RT4E cells. This suggested that the RT4E, and perhaps TM87-16, cells may have constitutively active Ras.
Fig. 6.
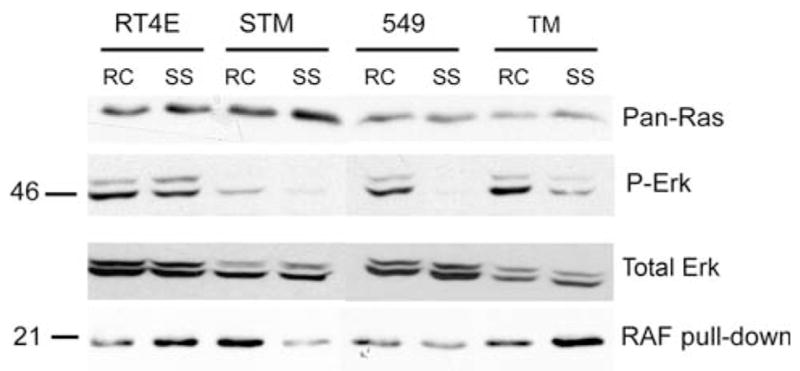
Ras is constitutively activated in 2 MRT cell lines. 30 μg of cell lysates from randomly cycling (RC) and serum-starved (SS) RT4E, STM91-01 (STM), TTC549 (549), and TM87-16 (TM) cell lines were separated by 10% SDS-PAGE. Western blotting was performed by cutting the membrane in half and probing the bottom half with a pan-Ras antibody (top panel) and the top half with an antibody to phosphorylated Erk (P-Erk) (second panel) followed by antibody to total Erk (third panel). Cell lysates of 200 μg were also immunoprecipitated using Raf-1 RBD agarose beads. SDS-PAGE of 10% was used to separate immunoprecipitation reactions and Western blotting was performed with a pan-Ras antibody (bottom panel)
A Raf pulldown assay was employed to more directly address Ras activation. Because Raf binds to active GTP-bound Ras, but not inactive GDP-bound Ras, agarose beads containing the Ras binding domain of Raf were used to specifically immunoprecipitate active Ras. In this assay, serum starvation decreased the amount of active Ras in the STM91-01 and TTC549 cell lines, which correlated with the decreased phosphorylation of Erk. Levels of active Ras actually appeared to increase in both the RT4E and TM87-16 cell lines. In RT4E cells, constitutively activated Ras may therefore be responsible for the observed constitutive activation of Akt. However, in the TM87-16 cell line, Akt does not appear to be constitutively active, despite apparent Ras activation.
Point mutations that occur in codons 12, 13 and 61 are frequently the cause of mutant constitutively active Ras. We therefore sequenced these areas of the H-Ras, K-Ras, and N-Ras genes in the four MRT cell lines examined here, but no mutations were found.
Discussion
Loss of INI1 is a recurrent genetic characteristic of malignant rhabdoid tumor, but because cancer is a multistep process, one would expect to find additional contributing mutations. Here we have demonstrated the importance of the PI3K/Akt pathway in the survival of MRT cells and present it as an example of oncogene addiction. Inhibition of this pathway using LY294002, caused a significant and dose-dependent reduction in survival of all MRT cell lines tested. We further demonstrated that in three of four cell lines tested, Akt was constitutively active. Charboneau et al. [42] suggest that high P-Akt expression in a subset of MRT cell lines and their resultant decreased response to p21, differentiates this group from other MRTs and may be useful in determining a treatment strategy. Indeed, addiction to oncogenes was proposed to be a cancer’s Achilles’ heal [49] and, in at least a subset of MRT cases, dependence on the PI3K/Akt pathway may lead to more effective therapies.
A variety of potential mechanisms were examined to explain the apparent constitutive Akt activation in three of the four MRT cell lines. PTEN loss was first ruled out as a causative factor by Western blot analysis. Overexpression of growth factor receptors was then examined as a means of aberrant activation of the PI3K/Akt pathway. EGFR is able to activate PI3K through a series of reactions: active EGFR binds adaptor protein Grb2, which recruits the docking protein Gab1, which contains pYXXM motifs to which SH2 domains on the regulatory subunit of PI3K (p85) bind, thereby bringing the catalytic subunit of PI3K (p110) in proximity to its membrane-bound lipid substrates [50]. IGF-1R phosphorylates insulin receptor substrate 1 (IRS-1), which also contains pYXXM motifs that bind the SH2 domains of p85 [51].
None of the MRT cell lines examined appeared to over-express EGFR, however, an IGF-1R variant may be contributing to Akt activation in the STM91-01 cell line. Interestingly, these cells were not responsive to treatment with NVP-AEW541; one reason may be that this putative mutated receptor is not recognized by the drug. TTC549 cells expressed high levels of IGF-1R, which may be directly causing constitutive PI3K/Akt activation as evidenced by the reduction in Akt phosphorylation upon treatment with NVP-AEW541. Both the RT4E and TM87-16 cell lines contained constitutively activated Ras, but as in all of the MRT cell lines, did not contain the common activating mutations in any of the Ras isoforms. The mechanism of Ras activation in these cell lines is therefore unknown, but in the RT4E cells Ras may be responsible for activation of the PI3K/Akt pathway through direct activation of PI3K [52].
The functions of INI1 in the cell and the mechanisms by which INI1 loss promotes tumorigenesis are a bit perplexing. Although mice heterozygous for INI1 are predisposed to tumor development, homozygous deletions of INI1 result in peri-implantation lethality. INI1 is therefore known to be essential for embryonic development. It also appears to play a vital role in the survival of normal adult cells. Inactivation of INI1 using a conditional allele in mice has been shown to cause bone marrow aplasia and death, while use of a reversible inverting conditional allele of INI1 demonstrated its necessity for the survival of non-malignant cells [53]. Roberts et al. [53] hypothesized that loss of INI1 may lead to cell death, except in cells that contain additional mutations, which contribute to cancer development. Klochendler-Yeivin et al. [8] have also reported that inactivation of INI1 in mouse embryonic fibroblasts in culture leads to increased sensitivity to DNA damage, growth inhibition, and an increased incidence of apoptosis. Activation of the PI3K/Akt pathway, such as we have reported, may therefore have been necessary for the establishment of these malignant rhabdoid tumors.
Previous work in our lab has demonstrated that SWI/SNF subunits can be immunoprecipitated in a complex with Akt and that at least one subunit, BAF155, can be phosphorylated by Akt [22]. According to Doan et al. [54], loss of INI1 does not affect the expression or assembly of other members of the SWI/SNF complex, which may therefore retain functionality in MRT cells. However, we were unable to identify an interaction between Akt and SWI/SNF components in MRT cells, suggesting an important role for INI1 in this interaction [22]. It is possible that this interaction may still occur, albeit with potentially reduced stability, making it harder to visualize by immunoprecipitation. Therefore, Akt activation in this cell type, and especially in cancers that retain SWI/SNF composition, may cause further dysregulation of SWI/SNF.
The SWI/SNF complex affects the transcription of many genes that impact tumorigenesis. These include cell cycle regulators such as cyclin D [12], p16INK4a, and p21WAF/CIP [16–19]. SWI/SNF is also involved in the regulation of CD44 and E-cadherin [55, 56] and dysregulation of the transcription of these genes is associated with tumor progression and metastasis formation. SWI/SNF subunits have also been found on steroid receptor targets [57–61]. Akt dysregulation could affect SWI/SNF activity at these promoters and may help explain why malignant rhabdoid tumors are so aggressive and prone to metastasis. Further, given the frequency with which Akt dysregulation occurs in other cancer types, its effects on SWI/SNF activity likely contribute to tumorigenesis in those cases.
Chemotherapy is used in the treatment of MRT after removal of the primary tumor, but there is no accepted standard therapy for the disease. Patients with metastatic MRT are commonly treated with a regimen of ifosfamide-carbo-platin-etoposide (ICE) alternating with vincristine-doxorubicin-cyclophosphamide (VDC), but the prognosis remains poor. Our results suggest that inhibition of the PI3K/Akt pathway may be of therapeutic value in this cancer type. Advances are being made in this direction as SF1126 (Semafore Pharmaceuticals) has recently become the first PI3K inhibitor to enter clinical trials, and perifosine (Keryx Bio-pharmaceuticals), the most clinically advanced AKT inhibitor, is currently in phase II trials. In addition, a subset of MRT cases may be responsive to therapies that block signaling through IGF-1R. NVP-AEW541 has been shown to be readily bioavailable and capable of dose-dependent inhibition of tumor growth in mice [47]. MRT may respond favorably to treatment with these new drugs.
Acknowledgments
We thank Dr. Steve Rosenzweig (MUSC) for helpful discussions and the kind gift of NVP-AEW541. Funding for this work was provided by the MUSC Department of Pathology and Laboratory Medicine, a Hollings Cancer Center Abney Foundation Scholarship (KSJF), a grant from GEO-Centers through the Department of Defense/Hollings Cancer Center Research Program under Subcontract number 42153MK-GC-3532 (CFW, DZ), and by NIH grants CA086860 and CA122023 (DZ). We additionally thank the MUSC Nucleic Acids Analysis and Molecular Biology Core Facility.
References
- 1.Biegel JA, Rorke LB, Packer RJ, Emanuel BS. Monosomey 22 in rhabdoid or atypical tumors of the brain. J Neurosurg. 1990;73:710–714. doi: 10.3171/jns.1990.73.5.0710. [DOI] [PubMed] [Google Scholar]
- 2.Biegel JA, Allen CS, Kawasaki K, Shimizi N, Budarf ML, Bell CJ. Narrowing the critical region for a rhabdoid tumor locus in 22q11. Genes Chromosomes Cancer. 1996;16:94–105. doi: 10.1002/(SICI)1098-2264(199606)16:2<94::AID-GCC3>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 3.Burger PC, Yu IT, Tihan T, Friedman HS, Strother DR, Kepner JL, Duffner PK, Kun LE, Perlman EJ. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a pediatric oncology group study. Am J Surg Pathol. 1998;22:1083–1092. doi: 10.1097/00000478-199809000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Versteege I, Sévenet N, Lange J, Rousseau-Merck M, Ambros P, Handgretinger R, Aurias A, Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 5.Biegel JA, Zhou J, Rorke LB, Stenstrom C, Wainwright LM, Fogelgren B. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999;59:74–79. [PubMed] [Google Scholar]
- 6.DeCristofaro MF, Betz BL, Wang W, Weissman BE. Alteration of hSNF5/INI1/BAF47 detected in rhabdoid cell lines and primary rhabdomyosarcomas but not Wilms’ tumor. Oncogene. 1999;18:7559–7565. doi: 10.1038/sj.onc.1203168. [DOI] [PubMed] [Google Scholar]
- 7.Guidi CJ, Sands AT, Zambrowica BP, Turner TK, Demers DA, Webster W, Smith TW, Imbalzano AN, Jones SN. Disruption of INI1 leads to peri-implantation lethality and tumorigenesis in mice. Mol Cell Biol. 2001;21:3598–3603. doi: 10.1128/MCB.21.10.3598-3603.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klochendler-Yeivin A, Fiette L, Barra J, Muchardt C, Babinet C, Yaniv M. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep. 2000;1:500–506. doi: 10.1093/embo-reports/kvd129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts CWM, Galusha SA, McMenamin ME, Fletcher CDM, Orkin SH. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc Natl Acad Sci USA. 2000;97:13796–13800. doi: 10.1073/pnas.250492697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Betz BL, Strobeck MW, Reisman DN, Knudsen ES, Weissman BE. Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene. 2002;21:5193–5203. doi: 10.1038/sj.onc.1205706. [DOI] [PubMed] [Google Scholar]
- 11.Versteege I, Medjkane S, Rouillard D, Delattre O. A key role of the hSNF5/INI1 tumour suppressor in the control of the G1-S transition of the cell cycle. Oncogene. 2002;21:6403–6412. doi: 10.1038/sj.onc.1205841. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z-K, Davies KP, Allen J, Ahu L, Pestell RG, Zagzag D, Kalpana G. Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol Cell Biol. 2002;22:5975–5988. doi: 10.1128/MCB.22.16.5975-5988.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reincke BS, Rosson GB, Oswald BW, Wright CF. INI1 expression induces cell cycle arrest and markers of senescence in malignant rhabdoid tumor cells. J Cell Physiol. 2003;194:303–313. doi: 10.1002/jcp.10201. [DOI] [PubMed] [Google Scholar]
- 14.Dunaief JL, Strober BE, Guha S, Khavari PA, Alin K, Luban J, Begemann M, Crabtree GR, Goff SP. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell. 1994;79:119–130. doi: 10.1016/0092-8674(94)90405-7. [DOI] [PubMed] [Google Scholar]
- 15.Trouche D, LeChalony C, Muchardt C, Yaniv M, Kouzarides T. RB and hbrm cooperate to repress the activation functions of E2F1. Proc Natl Acad Sci USA. 1997;94:11268–11273. doi: 10.1073/pnas.94.21.11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oruetxebarria I, Venturini F, Kekarainen T, Houweling A, Zuijderduijn LM, Mohd-Sarip A, Vries RG, Hoeben RC, Verrijzer CP. P16INK4a is required for hSNG5 chromatin remodeler-induced cellular senescence in malignant rhabdoid tumor cells. J Biol Chem. 2004;279:3807–3816. doi: 10.1074/jbc.M309333200. [DOI] [PubMed] [Google Scholar]
- 17.Kadam S, Emerson BM. Transcriptional specificity of human SWI/SNF BRG1 and BRM chromatin remodeling complexes. Mol Cell. 2003;11:377–389. doi: 10.1016/s1097-2765(03)00034-0. [DOI] [PubMed] [Google Scholar]
- 18.Kang H, Cui K, Zhao K. BRG1 controls the activity of the retinoblastoma protein via regulation of p21CIP1/WAF1/SDI. Mol Cell Biol. 2004;24:1188–1199. doi: 10.1128/MCB.24.3.1188-1199.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee D, Kim JW, Seo T, Hwang SG, Choi EJ, Choe J. SWI/SNF complex interacts with tumor suppressor p53 and is necessary for the activation of p53-mediated transcription. J Biol Chem. 2002;277:22330–22337. doi: 10.1074/jbc.M111987200. [DOI] [PubMed] [Google Scholar]
- 20.Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- 21.Sudarsanam P, Cao Y, Wu L, Laurent BC, Winston F. The nucleosome remodeling complex, Snf/Swi, is required for the maintenance of transcription in vivo and is partially redundant with the histone acetylase, Gcn5. EMBO J. 1999;18:3101–3106. doi: 10.1093/emboj/18.11.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foster KSJ, McCrary WJ, Ross JS, Wright CF. Members of the hSWI/SNF chromatin remodeling complex associate with and are phosphorylated by protein kinase B/Akt. Oncogene. 2006;25:4605–4612. doi: 10.1038/sj.onc.1209496. [DOI] [PubMed] [Google Scholar]
- 23.Bellacosa A, de Feo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, Wan M, Dubeau L, Scambia G, Masciullo V, Ferrandina G, Benedetti Panici P, Mancuso S, Neri G, Testa JR. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64:280–285. doi: 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- 24.Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001;61:3986–3997. [PubMed] [Google Scholar]
- 25.Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, Hamilton T, Tsichlis PN, Testa JR. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proc Natl Acad Sci USA. 1992;89:9267–9271. doi: 10.1073/pnas.89.19.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng JQ, Ruggeri B, Klein WM, Sonoda G, Altomare DA, Watson DK, Testa JR. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci USA. 1996;93:3636–3641. doi: 10.1073/pnas.93.8.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao N, Zhang Z, Jiang BH, Shi X. Role of PI3K/AKT/mTOR signaling in the cell cycle progression of human prostate cancer. Biochem Biophys Res Commun. 2003;310:1124–1132. doi: 10.1016/j.bbrc.2003.09.132. [DOI] [PubMed] [Google Scholar]
- 28.Graff JR, Konicek BW, McNulty AM, Wang ZQ, Houck K, Allen S, Paul JD, Hbaiu A, Goode RG, Sandusky GE, Vessella RL, Neubauer BL. Increased AKT activity contributes to prostate cancer progression by dramatically accelerating prostate tumor growth and diminishing p27Kip1 expression. J Biol Chem. 2000;275:24500–24505. doi: 10.1074/jbc.M003145200. [DOI] [PubMed] [Google Scholar]
- 29.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 30.Franke TF. A difficult Akt to follow. Neural Notes V. 1999;2:3–7. [Google Scholar]
- 31.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–8998. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- 32.Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, Franssen E, Slingerland JM. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;10:1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 33.Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL. PKB/Akt mediates cell-cycle progression by phophorylation of (Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;10:1145–1152. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- 34.Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phophorylation in HER-2/new-overexpressing cells. Nat Cell Biol. 2001;3:245–252. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 35.Baserga R. The contradictions of the insulin-like growth factor 1 receptor. Oncogene. 2000;19:5574–5581. doi: 10.1038/sj.onc.1203854. [DOI] [PubMed] [Google Scholar]
- 36.Slomiany MG, Black LA, Kibbey MM, Tingler MA, Day TA, Rosenzweig SA. Insulin-like growth factor-1 receptor and ligand targeting in head and neck squamous cell carcinoma. Cancer Lett. 2007;248:269–279. doi: 10.1016/j.canlet.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 37.Ota S, Crabbe DC, Tran TN, Triche TJ, Shimada H. Malignant rhabdoid tumor. A study with two established cell lines. Cancer. 1993;71:2862–2872. doi: 10.1002/1097-0142(19930501)71:9<2862::aid-cncr2820710930>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 38.Rosson GB, Vincent TS, Oswald BW, Wright CF. Drug resistance in malignant rhabdoid tumor cell lines. Cancer Chemother Pharmacol. 2002;49:142–148. doi: 10.1007/s00280-001-0398-y. [DOI] [PubMed] [Google Scholar]
- 39.Nocentini S. Apoptotic response of malignant rhabdoid tumor cells. Cancer Cell Int. 2003;3:11. doi: 10.1186/1475-2867-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshida S, Narita T, Koshida S, Ohta S, Takeuchi Y. TRAIL/Apo2L ligands induce apoptosis in malignant rhabdoid tumor cell lines. Pediatr Res. 2003;54:709–717. doi: 10.1203/01.PDR.0000085038.53151.D0. [DOI] [PubMed] [Google Scholar]
- 41.Kuwahara Y, Hosoi H, Osone S, Kita M, Iehara T, Kuroda H, Sugimoto T. Antitumor activity of gefitinib in malignant rhabdoid tumor cells in vitro and in vivo. Clin Cancer Res. 2004;10:5940–5948. doi: 10.1158/1078-0432.CCR-04-0192. [DOI] [PubMed] [Google Scholar]
- 42.Charboneau A, Chai J, Jordan J, Funkhouser W, Judkins A, Biegel J, Weissman B. P-Akt expression distinguishes two types of malignant rhabdoid tumors. J Cell Physiol. 2006;209:422–427. doi: 10.1002/jcp.20737. [DOI] [PubMed] [Google Scholar]
- 43.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 44.Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem. 2005;280:40406–40416. doi: 10.1074/jbc.M508361200. [DOI] [PubMed] [Google Scholar]
- 45.Reiter JL, Maihle NJ. Characterization and expression of novel 60-kDa and 110-kDa EGFT isoforms in human placenta. Ann NY Acad Sci. 2003;995:39–47. doi: 10.1111/j.1749-6632.2003.tb03208.x. [DOI] [PubMed] [Google Scholar]
- 46.Stellar MA, Delgado CH, Bartels CJ, Woodworth CD, Zhiqiang Z. Overexpression of the Insulin-like Growth Factor-1 Receptor and autocrine stimulation in human cervical cancer cells. Cancer Res. 1996;56:1761–1765. [PubMed] [Google Scholar]
- 47.García-Echeverría C, Pearson MA, Marti A, Meyer T, Mestan J, Zimmermann J, Gao J, Brueggen J, Capraro HG, Cozens R, Evans DB, Fabbro D, Furet P, Porta DG, Liebetanz J, Martiny-Baron G, Ruetz S, Hofmann F. In vivo antitumor activity of NVP-AEW541—A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell. 2004;5:231–239. doi: 10.1016/s1535-6108(04)00051-0. [DOI] [PubMed] [Google Scholar]
- 48.Downward J. Targeting RAS signaling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 49.Weinstein IB. Cancer. Addiction to oncogenes—the Achilles heel of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 50.Mattoon DR, Lamothe B, Lax I, Schlessinger J. The docking protein Gab1 is the primary mediator of EGF-stimulated activation of the PI-3K/Akt cell survival pathway. BMC Biol. 2004;2:24. doi: 10.1186/1741-7007-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.LeRoith D, Werner H, Beitner-Johnson D, Roberts CT., Jr Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16:143–163. doi: 10.1210/edrv-16-2-143. [DOI] [PubMed] [Google Scholar]
- 52.Rajalingam K, Schreck R, Rapp UP, Štefan A. Ras oncogenes and their downstream targets. Biochim Biophys Acta. 2007;1773:1177–1195. doi: 10.1016/j.bbamcr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 53.Roberts CWM, Leroux MM, Fleming MD, Orkin SH. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell. 2002;2:415–425. doi: 10.1016/s1535-6108(02)00185-x. [DOI] [PubMed] [Google Scholar]
- 54.Doan DN, Veal TM, Yan Z, Wang W, Jones SN, Imbalzano AN. Loss of the INI1 tumor suppressor does not impair the expression of multiple BRG1-dependent genes or the assembly of SWI/SNF enzymes. Oncogene. 2004;23:3462–3473. doi: 10.1038/sj.onc.1207472. [DOI] [PubMed] [Google Scholar]
- 55.Banine F, Bartlett C, Gunawardena R, Muchardt C, Yaniv M, Knudsen ES, Weissman BE, Sherman LS. SWI/SNF chromatin-remodeling factors induce changes in DNA methylation to promote transcriptional activation. Cancer Res. 2005;65:3542–3547. doi: 10.1158/0008-5472.CAN-04-3554. [DOI] [PubMed] [Google Scholar]
- 56.Gresh L, Bourachot B, Reimann A, Guigas B, Fiette L, Garbay S, Muchardt C, Hue L, Pontoglio M, Yaniv M, Klochendler-Yeivin G. The SWI/SNF chromatin-remodeling complex subunit SNF5 is essential for hepatocyte differentiation. EMBO J. 2005;24:3313–3324. doi: 10.1038/sj.emboj.7600802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Belandia B, Orford RL, Hurst HC, Parker MG. Targeting of SWI/SNF chromatin remodeling complexes to estrogen-responsive genes. EMBO J. 2002;21:4094–4103. doi: 10.1093/emboj/cdf412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DiRenzo J, Shang Y, Phelan M, Sif S, Myers M, Kingston R, Brown M. BRG-1 is recruited to estrogen-responsive promoters and cooperates with factors involved in histone acetylation. Mol Cell Biol. 2000;20:7541–7549. doi: 10.1128/mcb.20.20.7541-7549.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fryer CJ, Archer TK. Chromatin remodeling by the glucocorticoid receptor requires the BRG1 complex. Nature. 1998;393:88–91. doi: 10.1038/30032. [DOI] [PubMed] [Google Scholar]
- 60.Link KA, Burd CJ, Williams E, Marshall T, Rosson G, Henry E, Weissman B, Knudsen KE. BAF57 governs androgen receptor action and androgen-dependent proliferation through SWI/SNF. Mol Cell Biol. 2005;25:2200–2215. doi: 10.1128/MCB.25.6.2200-2215.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Métivier R, Penot B, Hübner MR, Reid G, Brand H, Kos M, Gannon F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]