

Abstract
Resistin, an adipocyte-derived hormone, is thought to represent a link between obesity and insulin-resistant diabetes. The potential role of resistin as a cardioprotective agent has not been explored. Our hypothesis is that resistin has a cardioprotective effect that is mediated by the resistin receptor-coupled activation of PI3K/Akt/PKC/KATP dependent pathways. Our studies demonstrated that pretreatment of mouse hearts with 10 nM resistin for 5 min protected the heart against I/R injury in a mouse heart perfusion model. When mouse hearts were subjected to 60 min of LAD ligation followed by 4 h of reperfusion, resistin pretreatment (33 μg/kg) for 30 min or 24 h before ligation was able to significantly reduce the infarct size/risk area. The protective effect of resistin was abolished by wortmannin, as well as by an Akt inhibitor, triciribine. Resistin’s protective effect was absent in Akt kinase-deficient mutant mice. The protective effect was also blocked by chelerythrine, a PKC inhibitor, and εV1-2, a PKCε inhibitor. Finally, the protective effect was blocked by 5-hydroxydecanoate, which blocks the opening of mitoKATP channels. Resistin-induced Akt phosphorylation in HL-1 cells was inhibited by wortmannin and triciribine. Resistin also induced PKCε phosphorylation, which was blocked by triciribine. These studies demonstrate that resistin’s cardioprotective effect is mediated by PI3K/Akt/PKC dependent pathways. In addition to cardiomyocytes, resistin also induced Akt phosphorylation in endothelial cells and smooth muscle cells, suggesting that resistin receptors are present in these cells. The effect of resistin on apoptosis was assessed in hearts subjected to 30 min of ischemia and 3 h of reperfusion. There were significantly fewer in situ oligo ligation-positive myocyte nuclei in mice treated with resistin. Our results show that resistin can dramatically reduce apoptosis and infarct size, thus protecting the heart against I/R injury.
Keywords: Resistin, myocardial protection, apoptosis
Introduction
Adipose tissue is an endocrine organ that secretes a large number of proteins under normal and pathophysiological conditions. Among these proteins is resistin, a new adipocyte hormone that was discovered independently by three different groups [1–3]. Mouse resistin mRNA encodes a cysteine-rich 114 amino acid protein containing a 20 amino acid signal sequence [1–3]. Resistin is expressed in white adipose tissue, with the highest levels in abdominal and female gonadal adipose tissue [4]. It is also expressed in brown fat tissue, placenta, pancreatic islet, pituitary, and a number of other rat tissues. The tissue level of resistin is decreased by insulin, fasting, somatotropin, thyroid hormone, tumor necrosis factor-α, endothelin-1, epinephrine, isoproterenol, PPARγ, estrogen, and insulin-like growth factor I (IGF-I). It is upregulated by growth hormone, dexamethasone, neuropeptide Y, hyperglycemia, and aging [see review 5].
Serum resistin levels are significantly elevated in genetically obese mice and diabetic mice as well as diet-induced models of diabetic obesity [3]. Earlier studies in rodents showed that resistin impairs glucose tolerance and insulin action, in addition to inhibiting adipogenesis in murine 3T3-L1 cells [1, 2, 6]. Overexpression of resistin in mice leads to insulin resistance and causes dyslipidemia [7–9]. Because of these findings, many researchers believe that resistin is a potential link between obesity and insulin resistance. However, several human studies have not shown differences in resistin expression between normal, insulin-resistant or type 2 diabetic samples [10,11]. Thus, the role of resistin in human diabetes remains controversial.
Obesity-related disorders are generally believed to be associated with the development of ischemic heart disease. Recently, it has been shown that resistin activates human saphenous vein endothelial cells by promoting endothelin-1 release and upregulating VCAM-1 and MCP-1 [12]. In human aortic endothelial cells, resistin induces the expression of VCAM-1, ICAM-1 and pentraxin 3, a marker of inflammation [13]. A number of other studies show that resistin may be involved in promoting atherosclerosis [14–18].
Despite all of this previous research on resistin, the effect of resistin on acute myocardial infarction has not been explored. Since insulin exerts a cardioprotective effect by activating Akt [19,20], resistin might be expected to inhibit this pathway by blocking insulin signaling. The present experiments demonstrate that resistin protects against myocardial infarction by activating PI3K/Akt/PKCε/KATP-dependent pathways.
Materials and Methods
Materials and animals
Wortmannin (PI3K inhibitor) and triciribine (Akt inhibitor) were purchased from Calbiochem (San Diego, CA). Chelerythrine (PKC inhibitor) and 5-hydroxydecanoate (blocks mitoKATP channels) were products of Sigma (St. Louis, MO). PKCε inhibitor-εV1-2 and TAT (carrier peptide) were obtained from KAI Pharmaceuticals, Inc. (South San Francisco, CA). Recombinant mouse resistin was purchased from Cell Sciences, Inc. (Canton, MA). Resistin was dissolved in water to get a stock solution of 250 μg/ml. The stock solution of resistin was further dissolved in 100 μl of saline and injected i.p. to mice 15 or 30 min before LAD (left anterior descending coronary artery ligation). Control mice received the same volume of saline or vehicle. An ApopTag in situ oligo ligation kit was obtained from Chemicon International (Temecula, CA). All other reagents were of the highest grades commercially available.
Male ICR mice (25–30 g) were purchased from Harlan (Indianapolis, IN). Akt kinase-deficient mutant mice were obtained from Dr. S. Izumo’s laboratory, Harvard University [21]. The animal protocols were approved by the East Tennessee State University Animal Care and Use Committee.
Global ischemia in vitro
Male ICR mice were injected with sodium heparin (500 units/kg body weight, i.p.) 30 min prior to anesthetization with sodium pentobarbital (120 mg/kg). Hearts were rapidly excised and perfused retrogradely at 60 mm Hg by the Langendorff technique with Krebs-Henseleit bicarbonate buffer. After 30 min of preliminary perfusion, a range of end-diastolic pressures were tested to construct a functional curve. After 45 min of ischemia, hearts were reperfused for 45 min. At the end of perfusion, another functional index was measured.
Functional analysis
Ventricular function of isolated mouse hearts was measured by inserting a tiny plastic-wrapped balloon connected to a pressure transducer into the left ventricle via the mitral valves. Cardiac functional parameters, such as left ventricular developed pressures (LVDP), ±dP/dt, heart rates, and coronary flow rates, recorded before ischemia and 45 min after reperfusion, were used for comparison. Percentages of functional recovery were calculated by dividing each of the functional recovery values at the end of reperfusion by their corresponding pre-ischemic values. End diastolic pressures prior to ischemia and after 45 min of reperfusion were calculated from the functional curves. The perfusate flowing out of the heart was collected and measured. Coronary flow rate was determined by the amount of perfusate measured in a specific time period.
Regional ischemia in vivo
Mice weighing 25–30 g were anesthetized with tribromoethanol (275 mg/kg, i.p.). An endotracheal tube (PE 90) was inserted 5–8 mm from the larynx, and the mice were ventilated with room air (a tidal volume of 0.5 ml) using a rodent respirator (Columbus Instruments International, Columbus, OH) set at 110–120 beats/min. LAD ligation was performed as described previously [22]. After 60 min of LAD ligation, the heart was reperfused for 4 h. Mice were anesthetized with tribromoethanol (275 mg/kg, i.p.), and hearts were perfused as Langendorff preparations for 5 min. The left coronary artery was re-occluded, and 1% Evans blue was infused into the aorta and coronary arteries to determine the area at risk. Hearts were transversely cut into 5 sections, with one section made at the site of the ligature. Macroscopic staining with triphenyltetrazolium chloride (TTC) was used to quantitate the infarct sizes as described previously [22]. The area at risk was expressed as the percentage of the left ventricle, and the area of infarct was expressed as the percentage of the area at risk.
Assessment of apoptosis- ISOL Analysis
In this series of experiments, we reduced the ischemia time from 60 min to 30 min to study apoptosis in the absence of myocardial infarction. Staining of DNA strand breaks was performed in 5 μm sections of hearts by the ApopTag in situ oligo ligation (ISOL) kit using oligo A according to manufacturer’s instructions with modifications. Endogenous biotin was blocked with an Avidin/Biotin blocking kit (BioGenex, San Ramon, CA). The peroxidase substrate TACS blue (KPL, Gaithersburg, MD) was used to label positive nuclei, and nuclear fast red was used to counterstain normal nuclei. The percentage of ISOL-positive cardiac cells was determined by counting 10 random fields/section.
Cell culture
Mouse aortic-derived endothelial cells and rat heart smooth muscle cells were kindly provided by Dr. Clement Diglio (Wayne State University). These cells were cultured in DMEM and were supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA) at 37°C in a humidified atmosphere of 5% CO2.
Murine cardiomyocyte HL-1 cells were generously provided by Dr. William Claycomb (Louisiana State University). These cells were cultivated as monolayer in Claycomb Media™ (JRH Biosciences, Lenexa, KS) and were supplemented with 10% fetal bovine serum (JRH Biosciences), 0.1 mM norepinephrine, and 2 mM glutamine. All culture dishes were pre-coated with 0.0005% fibronectin (Sigma) in 0.02% gelatin (23).
Western blot analysis
At 70% confluency, HL-1 cells were incubated for 16 h in Claycomb medium containing 0.5% BSA. Mouse aortic endothelial cells and rat heart smooth muscle cells were incubated for 16 h with DMEM supplemented with 0.5% fetal bovine serum. Resistin was added directly to the conditioned medium at a final concentration of 18 nM for 10 min. In some experiments, cells were pretreated with inhibitors for designated periods before resistin treatment. Cells were harvested in a buffer containing 20 mM Tris-HCl (pH 7.8), 137 mM NaCl, 15% glycerol, 1% Triton X-100, 2 μg/ml each of leupeptin, aprotinin, and pepstatin, 2 mM benzamidine, 20 mM NaF, 10 mM sodium pyrophosphate, 1 mM sodium vanadate, 25 mM beta-glycerophosphate, and 1 mM phenylmethylsulfonylfluoride. Protein concentration was determined by Bio-Rad protein assay (BioRad, Hercules, CA). Aliquots of 30 μg of lysates were electrophoresed on 12% SDS-PAGE and transferred to nitrocellulose membranes. Western blot analysis was carried out with antibodies against phospho-Akt (Ser473), total Akt (Cell Signaling Technology, Beverly, MA), phospho-PKCε (Ser729) (Upstate Biotechnology Inc, Lake Placid, NY) or PKCε (BD Biosciences) at 4°C overnight. Appropriate secondary antibodies conjugated to horseradish peroxidase were then added for 1.5 h. Antigen-antibody complex was detected using enhanced chemiluminescence reagent (GE Healthcare Bio-Sciences, Piscataway, NJ). Band density was measured by AlphaEase Image Analysis Software (Alpha Innotech Corp., San Leandro, CA).
Statistical Analysis
Statistical analysis for all experiments was assessed by Neuman-Keuls multiple-range test followed by analysis of variance. Data were presented as means ± SEM of 5–6 hearts. Differences were considered significant at P<0.05.
Results
Resistin can protect against I/R injury
Our first series of experiments were designed to explore the direct effect of resistin on I/R injury using a mouse heart perfusion model. Our studies demonstrated that pretreatment of mouse hearts with 10 nM resistin for as short as 5 min protected the heart against I/R injury. The functional recovery was significantly improved in resistin-treated hearts. The percentages of recovery for control vs. resistin-treated hearts were 29 ± 5% vs. 67 ± 6% for dP/dt, 32 ± 5% vs. 66 ± 6% for -dP/dt, 38 ± 7% vs. 72 ± 7% for LVDP, and 29 ± 2% vs. 77 ± 4% for LVDP x HR (Figure 1A). There was no difference between the pre-ischemic left ventricular end-diastolic pressures (LVEDP) of the two groups. The LVEDP of the resistin-treated hearts after 45 min of reperfusion was significantly lower than that of the control hearts (42 ± 4 vs. 66 ± 6 mm Hg, P< 0.05; Figure 1B). Neither the heart rates nor the coronary flow rates of the two groups were significantly different. Our results demonstrate that a protective effect can be observed upon administration of 10 nM resistin.
Figure 1.

Panel A. Effect of resistin administered in vitro on the post-ischemic recovery of cardiac function, expressed as the percentage of pre-ischemic value. Hearts were perfused with 10 nM resistin for 5 min and subjected to 45 min of ischemia followed by 45 min of reperfusion. Values are means ± SEM of six hearts. a=P<0.05 vs. control. LVDP, left ventricular developed pressure; ±dP/dt, first derivatives of left ventricular pressure; HR, heart rate. Panel B. Improvement of left ventricular end-diastolic pressure (LVEDP) after 45 min of global ischemia and 45 min of reperfusion in hearts pretreated for 5 min with 10 nM resistin. Values are means ± SEM of 6 hearts. a=P<0.05 vs. pre-ischemic LVEDP; b=P<0.05 vs. Control, after 45 min of reperfusion.
Resistin minimizes infarct size
The next series of experiments were carried out to examine the dose and time-effect of resistin treatment on myocardial infarction in mice subjected to LAD ligation. This in vivo study was important because certain blood components, such as leukocytes, are also potential contributors to ischemic damage during reperfusion. When mouse hearts were subjected to 60 min of LAD ligation followed by 4 h of reperfusion, resistin pretreatment for 30 min was able to minimize infarct size at 330 ng/kg. Maximal protection was achieved at 3.3 μg/kg; there was no additional protection at 33 μg/kg (Figure 2A). The areas at risk, expressed as a percentage of LV, were not different between the resistin-treated group (3.3 μg/kg) vs. untreated group (40 ± 1.7% vs. 48 ± 2.4%, p<0.05).
Figure 2.

Protective effect of resistin on myocardial infarction in mice. Panel A. Mice were pretreated with varying doses of resistin for 30 min. Panel B. Mice were pretreated with resistin (33 μg/kg) 30 min or 24 h prior to ligation. Mice were then subjected to 60 min LAD ligation followed by 4 h of reperfusion. Values are means ± SEM of 6 hearts, a=P<0.05 vs. Control.
Resistin pretreatment (33 μg/kg) 30 min or 24 h before ligation was able to reduce the infarct size/risk area from 66 ± 4% in the control to 26 ± 5% and 32 ± 1%, respectively (P<0.05; Figure 2B). This intriguing data suggest that resistin has a late preconditioning effect, which could have great clinical significance.
Involvement of the PI3K pathway in resistin’s cardioprotective effect
To determine whether resistin exerts its protective effects through PI3K pathways, wortmannin (0.4 mg/kg, i.p.) was administered 15 min before the administration of resistin. Our results demonstrate that the protective effect of resistin can be blocked by wortmannin (Figure 3). We also found that DMSO, a solvent used commonly to dissolve hydrophobic inhibitors, is a strong cardioprotective agent. Administration of only 1 μl per mouse is enough to confer significant reduction of infarct size. Therefore, wortmannin as well as triciribine were resuspended in a buffer containing 0.2% carboxymethylcellulose and 0.25% Tween-80. Changing to this buffer had no effect on infarct size (data not shown).
Figure 3.

Blocking of the protective effect of resistin by an inhibitor of PI3K, wortmannin. Mice were injected with wortmannin (W) (0.4 mg/kg, i.p.) 15 min before the administration of resistin (3.3 μg/kg, i.p.). After 15 min, mice were subjected to LAD ligation followed by 4 h of reperfusion. n=6, a=P<0.05 Resistin vs. others.
Involvement of Akt in resistin’s cardioprotective effect
Since Akt is the downstream target of PI3K, the involvement of Akt in the protective effect of resistin was explored by using triciribine, a highly selective Akt inhibitor. Mice were preteated with triciribine (6.6 mg/kg, i.p.) for 15 min followed by resistin (3.3 μg/kg, i.p.). Our studies showed that resistin’s protective effect was abolished by triciribine (Figure 4A). To verify these results, the protective effect of resistin was evaluated in kinase-deficient Akt mice (kdAkt mice). The protective effect of resistin was absent in kdAkt mice (Figure 4B).
Figure 4.

Panel A. Blocking of the protective effect of resistin by an inhibitor of Akt, triciribine (TRI). Mice were injected TRI (6.6 mg/kg, i.p.), 15 min before the administration of resistin (3.3 μg/kg, i.p.). Control and resistin treated data are the same as that shown in Figure 3.
Panel B. The protective effect of resistin was nullified in Akt kinase-deficient mutant mice. Non-transgenic mice (NTg) and Akt kinase-deficient mutant mice (Tg) were injected with resistin (3.3 μg/kg, i.p.) 15 min before they were subjected to LAD ligation followed by 4 h of reperfusion. n=6, a=P<0.05 Resistin vs. others.
Involvement of PKC in resistin’s cardioprotective effect
To explore the involvement of PKC in the protective effect of resistin, a highly specific PKC inhibitor, chelerythrine, was used. Our results show that chelerythrine (5 mg/kg, i.p.) was able to block the protective effect of resistin (Figure 5A). Since the translocation of PKCε is known to be responsible for the preconditioning effect of a number of protective agents, we administered Tat-εV1-2 (0.2 mg/kg, i.p.), a specific PKCε inhibitor that inhibits the translocation of PKCε from cytosol to membrane [24]. Our results demonstrate that resistin’s protective effect was blocked by Tat-εV1-2, but not the carrier Tat peptide (Figure 5B).
Figure 5.

Blocking of the protective effect of resistin by an inhibitor of PKC, chelerythrine (CHE; Panel A) or PKCε, εV1-2 (Panel B). Mice were injected with CHE (5 mg/kg, i.p.), εV1-2 (0.2 mg/kg, i.p) or Tat carrier (83 μg/kg, i.p.) 15 min before the administration of resistin (3.3 μg/kg). After 15 min, mice were subjected to LAD ligation followed by 4 h of reperfusion. n=6, a=P<0.05 vs. Control. Control and resistin treated data are the same as that shown in Figure 3.
Involvement of mitochondrial KATP channel in resistin’s cardioprotective effect
Since the activation of PKC leads to the opening of mitochondrial KATP (mitoKATP) channels, we administered 5-hydroxydecanoate (5-HD; 5 mg/kg, i. p.), which blocks the opening of mitoKATP channels. Our results showed that 5-HD blocked the protective action of resistin (Figure 6).
Figure 6.

Blocking of the protective effect of resistin by an inhibitor of mitoKATP channels, 5-HD. Mice were injected with 5-HD (5 mg/kg, i.p.) 15 min before the administration of resistin (33 μg/kg). After 15 min, mice were subjected to LAD ligation followed by 4 h of reperfusion n=6, a=P<0.05 vs. Control. Control and resistin treated data are the same as that shown in Figure 3.
Signal transduction studies in cardiac cells
HL-1 cells were used to investigate the effectiveness of various inhibitors in blocking resistin-induced Akt phosphorylation. Addition of resistin to HL-1 cells caused a time-dependent phosphorylation of Akt. The maximal level of Akt phosphorylation occurred at 10 min (results not shown). Wortmannin and triciribine were used to block PI3K and Akt, respectively. HL-1 cells were pretreated with 100 nM wortmannin (10 min) or 1 μM triciribine (30 min) before incubating with resistin for 10 min. Phospho-Akt level was examined by Western blot analysis. Quantification was performed with densitometric analysis of phospho-Akt and total Akt. Figure 7A shows that resistin upregulated phospho-Akt by 1.8-fold. In the presence of wortmannin or triciribine, the increase in phospho-Akt level was completely blocked.
Figure 7.

The effect of inhibitors on phospho-Akt and phospho-PKCε levels in HL-1 cells. HL-1 cells were pretreated with wortmannin (W) for 10 min or with triciribine (TRI) for 30 min before treatment with resistin for 10 min. Western blot analyses were carried out with 30 μg of homogenate with phospho-Akt (Ser473) antibody. Duplicate blot was probed with antibodies against total Akt (A). The cells were treated resistin in the absence or presence of triciribine. Phospho-PKCε (Ser729) and total PKCε levels were determined by Western blot analysis (B). Bar graph shows densitometric analysis of relative intensity of phospho-Akt normalized against total Akt (A) or phospho-PKCε normalized against PKCε (B). Data represent means ± SE from 4 samples. aP<0.05, versus control cells, bP<0.05, versus resistin-treated cells.
Phosphorylation of PKCε at Ser729 has been shown to correlate with increased PKCε kinase activity (25). In our study, resistin-induced phospho-PKCε was determined by Western blot analysis. Treatment with resistin for 10 min upregulated phospho-PKCε level by 1.7-fold. In the presence of triciribine, an Akt inhibitor, the resistin-induced increase in phospho-PKCε level was attenuated (Figure 7B). These results suggest that Akt is upstream of PKCε.
Resistin receptors are present in smooth muscle cells and endothelial cells
Our next series of experiments were designed to determine which cell types in the heart contain resistin receptors, other than cardiocytes. Mouse aortic endothelial cells and rat heart smooth muscle cells were treated with resistin for 10 min. Phosphorylated Akt levels were measured by Western blot analysis. Figure 8 shows that resistin upregulated phospho-Akt levels in both cell types.
Figure 8.
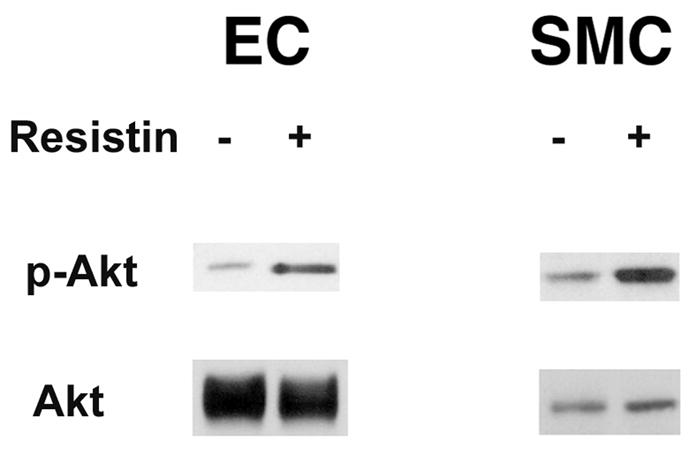
Resistin receptors are present in mouse aortic-derived endothelial cells and rat heart smooth muscle cells. Cells in 100 mm dishes were treated in the presence or absence of resistin for 10 min. Western blot analyses were carried out with 30 μg of homogenate with phospho-Akt antibodies. Total Akt levels were included for protein loading. Representative Western blots are shown.
Resistin inhibits apoptosis
Since programmed myocyte cell death has been linked to I/R injury, the effect of resistin on apoptosis was assessed in hearts subjected to 30 min of ischemia and 3 h of reperfusion. The number of in situ oligo ligation (ISOL)-positive myocyte nuclei was significantly attenuated by resistin (5.4 ± 0.8% vs. 18.3 ± 1.0% control, P<0.05, Figure 9).
Figure 9.

Protective effect of resistin on myocardial apoptosis in mice subjected to 30 min of ischemia and 3 h of reperfusion. Paraffin-embedded myocardial sections were stained by ISOL procedure. Immunolabeled nuclei of myocytes were determined by random counting of 10 fields per section. Values are means ± SEM of 5–6 hearts. a=P<0.05 vs. sham-operated. b=P<0.05 vs. Control.
Discussion
Novel function of resistin
In this study, we demonstrate that resistin has a cardioprotective effect, adding to the research that has studied the role of resistin in glucose homeostatis, adipogenesis, and inflammation. Our studies show for the first time that resistin can dramatically reduce the infarct size and thus protect the heart against I/R injury. We provide evidence that the mechanism of this cardioprotective effect occurs by a PI3K/Akt/PKCε/KATP- channel-dependent pathway.
Mechanism of resistin’s cardioprotective effects
Our in vitro studies demonstrated that resistin rapidly phosphorylates Akt in cardiac cells (Figure 7). The cardioprotective effect of resistin is also abolished by wortmannin (Figure 3), suggesting that resistin activates the PI3K pathway.
There are two lines of evidence to support the involvement of Akt in resistin’s cardioprotective effect. First, we demonstrated that the protective effect of resistin was blocked by triciribine (Figure 4). Triciribine is highly selective for Akt and does not inhibit the activation of PI3K, PDK1, PKC, PKA, STAT-3, ERK-1/2, or c-jun NH2-terminal kinase [26]. Second, the effect of resistin was not present in kdAkt mice (Figure 4B).
Since the PI3K pathway can activate PKC, it is feasible that resistin-induced activation of PKC by PI3K could lead to mitoKATP channel opening and myocardial protection. The activation of PI3K leads to the production of PtdIns(3,4,5)P3, which targets phosphoinositide-dependent kinases (PDK), atypical PKCs such as PKC ε, η, ζ, and λ, phospholipase Cγ, Akt, and other kinases [for Reviews see 27–30]. The relevance of this kinase activation to myocardial protection is two-fold. First, several isoforms of the atypical PKCs have been shown to play an important role in myocardial protection by modulating mitoKATP channels [29, 31–34]. Second, it has been shown recently that overexpression of PKCε protects against myocardial I/R injury [35]. Given these studies, it is possible that the activation of PI3K by resistin could activate various PKCs, which in turn could protect against myocardial I/R injury by modulating mitoKATP channels. In support of this contention, we demonstrated that a specific PKC inhibitor, chelerythrine, blocks the protective effect of resistin (Figure 5A). In addition, since the translocation of PKCε is known to be responsible for the preconditioning effect of a number of protective agents, a specific PKCε inhibitor (Tat-εV1-2) that inhibits the translocation of PKCε from cytosol to membrane was employed [24, 36]. In our studies, Tat-εV1-2 blocked resistin’s cardioprotective effect (Figure 5B).
It is generally believed that PKC-mediated opening of the mitochondrial KATP (mitoKATP) channel plays a pivotal role in the protection conferred during the acute and second window of protection in preconditioned myocardium [37–39]. Diazoxide, a specific and highly selective mitoKATP channel opener, has a cardioprotective effect that can be blocked by 100 μM 5-hydroxydecanoic acid (5-HD) [40]. The opening of the mitoKATP channel by diazoxide is mediated by the PKC signaling pathway [41]. In this study, we showed that resistin’s effect was abolished by 5-HD (Figure 6), suggesting that the mitoKATP channel is one of the important targets of resistin’s act.
The molecular pathways involved in acute cardioprotection have been reviewed extensively (42–44). At present, the evidence indicates that cardioprotection involves at least two molecular signaling pathways, the PI3K and mitogen-activated kinase pathways. The present study aimed to identify potential upstream activators of resistin-induced Akt phosphorylation. We conclude that wortmannin-sensitive PI3K is the upstream activator (Figure 7A).
The function and conformation of PKCε are regulated by phosphorylation in the activation loop (Thr566), COOH-terminal autophosphorylation (Thr710), and a C-terminal hydrophobic site (Ser729). Several studies in the literature report that PI3K is involved in the autophosphorylation of PKCε. Specifically, the PI3K-dependent kinase-1 (PDK-1) is thought to phosphorylate threonine, leading to autophosphorylation of PKCε (45). Phosphorylation of the COOH-terminal priming site (Ser729) of PKCε increases kinase activity (25). In our study, an inhibitor of Akt (triciribine) blocked resistin-induced phosphorylation of PKCε (Ser729), which suggests that Akt is upstream of PKCε (Figure 7B). Recently, it has been demonstrated that ischemic and pharmacological preconditioning involve the activation of PI3K, Akt, endothelial nitric oxide synthase, guanylyl cyclase, and protein kinase G. Activation of protein kinase G opens mitochondrial KATP channels and provides cardioprotection via a PKCε-dependent pathway (46). In this study, we provide data to demonstrate that resistin’s effect is mediated by a PI3K/Akt/PKCε/KATP channel-dependent signaling cascade. However, a fuller understanding of how resistin actives PI3K awaits cloning of resistin receptors.
Potential role of resistin in obesity-induced heart diseases
Obesity-related disorders are generally believed to be associated with the development of ischemic heart disease. Serum resistin levels are significantly elevated in genetically obese mice and diabetic mice as well as diet-induced models of diabetic obesity [3]. Although recent evidence suggests that resistin is a pro-atherogenic factor [12–18, 47], our results demonstrate that administration of resistin has a beneficial effect on acute myocardial infarction. Interestingly, two other adipocyte-derived hormones, leptin and adiponectin, also have cardioprotective effects [48–51].
In our study, pre-treatment with resistin lasted only 15–30 min, and the in vivo studies lasted less than 5 h. As such, the cardioprotection conferred by resistin can be attributed to the acute effects of resistin, which are dissimilar to the chronic pro-atherogenic effects of resistin. In our study, immunohistochemical staining by CD45 and H & E staining showed little inflammatory infiltration in hearts subjected to I/R injury with or without resistin (data not shown). Furthermore, the area of risk in the resistin-treated heart was the same as the control heart, suggesting that the cardioprotective effect of resistin cannot be explained by neovasculariztion, which likely occurs at least a few days later.
It remains to be established whether resistin confers cardiac protection in humans as well as mice. The bioactive action of resistin between humans and mice appears to be different. For example, human resistin is expressed mainly in circulating mononuclear cells, rather than adipocytes [52]. Since both insulin and resistin activate Akt (19,20), it is quite possible that resistin has a cardioprotective effect in humans.
Potential clinical application of resistin
Resistin has three main properties that would make it an excellent therapeutic agent. First, it is a naturally occurring hormone that should have minimal side effects. Second, resistin has a delayed cardioprotective effect; indeed, we showed that cardioprotection still remained even 24 h after resistin injection. Such a delayed effect would be of great benefit for many patients, especially those undergoing cardiac bypass surgery or other procedures in which cardiac arrest is needed. Finally, we demonstrated previously that resistin promotes angiogenesis [53]. Such an angiogenic effect would improve the recovery of cardiac function after myocardial infarction.
Acknowledgments
We thank Dr. William Claycomb for his generous gift of HL-1 cells and Dr. Clement Diglio for mouse aortic-derived endothelial cells and rat heart smooth muscle cells. This study was supported by a grant-in-aid from the American Heart Association Southeast Affiliate and grants from the Department of Veterans Affairs Merit Review and NIH (HL-087271).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Holcomb IN, Kabakoff RC, Chan B, Baker TW, Gurney A, Henzel W, et al. FIZZ1, a novel cysteine-rich secreted protein associated with pulmonary inflammation, defines a new gene family. EMBO J. 2000;19:4046–55. doi: 10.1093/emboj/19.15.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim KH, Lee K, Moon YS, Sul HS. A cysteine-rich adipose tissue-specific secretory factor inhibits adipocyte differentiation. J Biol Chem. 2001;276:11252–11256. doi: 10.1074/jbc.C100028200. [DOI] [PubMed] [Google Scholar]
- 3.Steppan CM, Bailey ST, Bat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 4.Oliver P, Pico C, Serra F, Palou A. Resistin expression in different adipose tissue depots during rat development. Mol Cell Biochem. 2003;252:397–400. doi: 10.1023/a:1025500605884. [DOI] [PubMed] [Google Scholar]
- 5.Adeghate E. An update on the biology and physiology of resistin. CMLS Cell Mol Life Sci. 2004;61:2485–96. doi: 10.1007/s00018-004-4083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steppan CM, Lazar MA. Resistin and obesity-associated insulin resistance. Trends Endocrinol Metab. 2002;13:18–23. doi: 10.1016/s1043-2760(01)00522-7. [DOI] [PubMed] [Google Scholar]
- 7.Kitagawa Y, Bujo H, Takahashi K, Shibasaki M, Ishikawa K, Yagui K, et al. Impaired glucose tolerance is accompanied by decreased insulin sensitivity in tissues of mice implanted with cells that overexpress resistin. Diabetologia. 2004;47:1847–53. doi: 10.1007/s00125-004-1530-4. [DOI] [PubMed] [Google Scholar]
- 8.Sato N, Kobayashi K, Inoguchi T, Sonoda N, Imamura M, Sekiguchi N, et al. Adenovirus-mediated high expression of resistin causes dyslipidemia in mice. Endocrinology. 2005;146:273–9. doi: 10.1210/en.2004-0985. [DOI] [PubMed] [Google Scholar]
- 9.Satoh H, Nguyen MT, Miles PD, Imamura T, Usui I, Olefsky JM. Adenovirus-mediated chronic “hyper-resistinemia” leads to in vivo insulin resistance in normal rats. J Clin Invest. 2004;114:224–31. doi: 10.1172/JCI20785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iqbal N, Seshadri P, Stern L, Loh J, Kundu S, Jafar T, et al. Serum resistin is not associated with obesity or insulin resistance in humans. Eur Rev Med Pharmacol Sci. 2005;9:161–5. [PubMed] [Google Scholar]
- 11.Kusminski CM, McTernan PG, Kumar S. Role of resistin in obesity, insulin resistance and Type II diabetes. Clin Sci (Lond) 2005;109:243–56. doi: 10.1042/CS20050078. [DOI] [PubMed] [Google Scholar]
- 12.Verma S, Li SH, Wang CH, Fedak PW, Li RK, Weisel RD, et al. Resistin promotes endothelial cell activation: further evidence of adipokine-endothelial interaction. Circulation. 2003;108:736–40. doi: 10.1161/01.CIR.0000084503.91330.49. [DOI] [PubMed] [Google Scholar]
- 13.Kawanami D, Maemura K, Takeda N, Harada T, Nojiri T, Imai Y, et al. Direct reciprocal effects of resistin and adiponectin on vascular endothelial cells: a new insight into adipocytokine-endothelial cell interactions. Biochem Biophys Res Commun. 2004;314:415–9. doi: 10.1016/j.bbrc.2003.12.104. [DOI] [PubMed] [Google Scholar]
- 14.Burnett MS, Lee CW, Kinnaird TD, Stabile E, Durrani S, Dullum MK, et al. The potential role of resistin in atherogenesis. Atherosclerosis. 2005;182:241–8. doi: 10.1016/j.atherosclerosis.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 15.Calabro P, Samudio I, Willerson JT, Yeh ET. Resistin promotes smooth muscle cell proliferation through activation of extracellular signal-regulated kinase 1/2 and phosphatidylinositol 3-kinase pathways. Circulation. 2004;110:3335–40. doi: 10.1161/01.CIR.0000147825.97879.E7. [DOI] [PubMed] [Google Scholar]
- 16.Jung HS, Park KH, Cho YM, Chung SS, Cho HJ, Cho SY, et al. Resistin is secreted from macrophages in atheromas and promotes atherosclerosis. Cardiovasc Res. 2006;69:76–85. doi: 10.1016/j.cardiores.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 17.Kougias P, Chai H, Lin PH, Lumsden AB, Yao Q, Chen C. Adipocyte-derived cytokine resistin causes endothelial dysfunction of porcine coronary arteries. J Vasc Surg. 2005;41:691–8. doi: 10.1016/j.jvs.2004.12.046. [DOI] [PubMed] [Google Scholar]
- 18.Kougias P, Chai H, Lin PH, Yao Q, Lumsden AB, Chen C. Effects of adipocyte-derived cytokines on endothelial functions: implication of vascular disease. J Surg Res. 2005;126:121–9. doi: 10.1016/j.jss.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 19.Kovacic S, Soltys C–L, Barr AJ, Shiojima I, Walsh K, Dyck JRB. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem. 2003;278:39422–27. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- 20.Yang J, Holman GD. Insulin and contraction stimulate exocytosis, but increased AMP-activated protein kinase activity resulting from oxidative metabolism stress slows endocytosis of GLUT4 in cardiomyocytes. J Biol Chem. 2005;280:4070–78. doi: 10.1074/jbc.M410213200. [DOI] [PubMed] [Google Scholar]
- 21.Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, et al. Akt/protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol. 2002;22:2799–809. doi: 10.1128/MCB.22.8.2799-2809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Z, Chua CC, Ho Y–S, Hamdy RC, Chua BHL. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am J Physiol Heart Circ Physiol. 2001;280:H2313–H2320. doi: 10.1152/ajpheart.2001.280.5.H2313. [DOI] [PubMed] [Google Scholar]
- 23.Claycomb WC, Lanson NA, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocytes. Proc Natl Acad Sci USA. 1998:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schechtman D, Craske ML, Kheifets V, Meyer T, Schechtman J, Mochly-Rosen D. A critical intramolecular interaction for protein kinase Cepsilon translocation. J Biol Chem. 2004;279:15831–40. doi: 10.1074/jbc.M310696200. [DOI] [PubMed] [Google Scholar]
- 25.Parekh DB, Ziegler W, Parker PJ. Multiple pathways control protein kinase C phosphorylation. EMBO J. 2000;19:496–503. doi: 10.1093/emboj/19.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang L, Dan HC, Sun M, Liu Q, Sun XM, Feldman RI, et al. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 2004;64:4394–4399. doi: 10.1158/0008-5472.CAN-04-0343. [DOI] [PubMed] [Google Scholar]
- 27.Good LA, Ziegler WH, Parekh DB, Alessi DR, Cohe P, Parker J. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–2045. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- 28.Toker A, Cantley LC. Signaling through the lipid products of phosphoinositide-3- OH kinase. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 29.Tong H, Chen W, Steenbergen C, Murphy E. Ischemic preconditioning activates phosphoinositide-3-kinase upstream of protein kinase C. Circ Res. 2000;87:309–315. doi: 10.1161/01.res.87.4.309. [DOI] [PubMed] [Google Scholar]
- 30.Wymann MP, Pirola L. Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta. 1998;1436:127–150. doi: 10.1016/s0005-2760(98)00139-8. [DOI] [PubMed] [Google Scholar]
- 31.Hu K, Duan D, Li GR, Nattel S. Protein kinase C activates ATP-sensitive K+ current in human and rabbit ventricular myocytes. Circ Res. 1996;78:492–498. doi: 10.1161/01.res.78.3.492. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Gao WD, O’Rourke B, Marban E. Synergistic modulation of ATP-sensitive currents by protein kinase C and adenosine. Implications for ischemic preconditioning. Circ Res. 1996;78:443–454. doi: 10.1161/01.res.78.3.443. [DOI] [PubMed] [Google Scholar]
- 33.Ping P, Zhang J, Qiu Y, Tang XL, Manchikalapudi S, Cao X, et al. Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circ Res. 1997;81:404–14. doi: 10.1161/01.res.81.3.404. [DOI] [PubMed] [Google Scholar]
- 34.Simkhovich BZ, Przyklenk K, Kloner RA. Role of protein kinase C as a cellular mediator of ischemic preconditioning: a critical review. Cardiovasc Res. 1998;40:9–22. doi: 10.1016/s0008-6363(98)00142-4. [DOI] [PubMed] [Google Scholar]
- 35.Ping P, Zhang J, Li RC, Kong D, Tang XL, Qiu Y, et al. PKC-dependent activation of p44/42 MAPKs during myocardial ischemia-reperfusion in conscious rabbits. Am J Physiol Heart Circ Physiol. 1999;276:H1468–81. doi: 10.1152/ajpheart.1999.276.5.H1468. [DOI] [PubMed] [Google Scholar]
- 36.Zhou HZ, Karliner JS, Gray JS. Moderate alcohol consumption induces sustained cardiac protection by activating PKC-epsilon and Akt. Am J Physiol Heart Circ Physiol. 2002;283:H165–H174. doi: 10.1152/ajpheart.00408.2001. [DOI] [PubMed] [Google Scholar]
- 37.Garlid KD. Cation transport in mitochondria - the potassium cycle. Biochim Biophys Acta. 1996;1275:123–126. doi: 10.1016/0005-2728(96)00061-8. [DOI] [PubMed] [Google Scholar]
- 38.Gross GJ, Fryer RM. Sarcolemmal versus mitochondrial ATP-sensitive K+ channels and myocardial preconditioning. Circ Res. 1999;84:973–979. doi: 10.1161/01.res.84.9.973. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y, Sato T, O’Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation. 1998;97:2463–2469. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- 40.Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D’Alonzo AJ, et al. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Circ Res. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y, Ashraf M. Role of protein kinase C in mitochondrial KATP channelmediated protection against Ca2+ overload injury in rat myocardium. Circ Res. 1999;84:1156–1165. doi: 10.1161/01.res.84.10.1156. [DOI] [PubMed] [Google Scholar]
- 42.Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovas Res. 2006;70:240–253. doi: 10.1016/j.cardiores.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 43.Gross ER, Gross GJ. Ligand triggers of classical preconditioning and postconditioning. Cardiovas Res. 2006;70:212–221. doi: 10.1016/j.cardiores.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 44.Armstrong SC. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovas Res. 2004;61:427–436. doi: 10.1016/j.cardiores.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 45.Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–2045. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- 46.Costa AD, Garlid KD, West IC, Lincoln TM, Downey JM, Cohen MV, et al. Protein kinase G transmits the cardioprotective signal from cytosol to mitochondria. Circ Res. 2005;97:329–36. doi: 10.1161/01.RES.0000178451.08719.5b. [DOI] [PubMed] [Google Scholar]
- 47.Reilly MP, Lehrke M, Wolfe ML, Rohatgi A, Lazar MA, Rader DJ. Resistin is an inflammatory marker of atherosclerosis in humans. Circulation. 2005;111:932–9. doi: 10.1161/01.CIR.0000155620.10387.43. [DOI] [PubMed] [Google Scholar]
- 48.Chen Z, Chua CC, Chua SC, Jr, Landy CL, Hamdy R, Chua BHL. A study of leptin receptor isoforms and their role in leptin’s cardioprotective effect. Circulation. 2002;106:II-193. [Google Scholar]
- 49.Liao Y, Takashima S, Maeda N, Ouchi N, Komamura K, Shimomura I, et al. Exacerbation of heart failure in adiponectin-deficient mice due to impaired regulation of AMPK and glucose metabolism. Cardiovasc Res. 2005;67:705–13. doi: 10.1016/j.cardiores.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 50.Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, et al. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med. 2005;11:1096–1103. doi: 10.1038/nm1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takahashi T, Saegusa S, Sumino H, Nakahashi T, Iwai K, Morimoto S, et al. Adiponectin replacement therapy attenuates myocardial damage in leptin-deficient mice with viral myocarditis. J Int Med Res. 2005;33:207–14. doi: 10.1177/147323000503300208. [DOI] [PubMed] [Google Scholar]
- 52.Savage DB, Sewter CP, Klenk ES, Segal DG, Vidal-Puig A, Considine RV, et al. Resistin/Fizz3 expression in relation to obesity and peroxisome proliferator-activated receptor-gamma action in humans. Diabetes. 2001;50:2199–202. doi: 10.2337/diabetes.50.10.2199. [DOI] [PubMed] [Google Scholar]
- 53.Chua CC, Gao J, Diglio C, Hamdy RC, Chua BHL. Resistin is a novel angiogenic factor. FASEB J. 2005;19:A1659. [Google Scholar]