

Abstract
Arsenic is a human pulmonary carcinogen. Our work indicates that in utero arsenic exposure in mice can induce or initiate lung cancer in female offspring. To define early molecular changes, pregnant C3H mice were given 85 ppm arsenic in drinking water from days 8 to 18 of gestation and expression of selected genes in the fetal lung or in lung tumors developing in adults was examined. Transplacental arsenic exposure increased estrogen receptor-α (ER-α) transcript and protein levels in the female fetal lung. An overexpression of various estrogen-regulated genes also occurred, including trefoil factor-3, anterior gradient-2, and the steroid metabolism genes 17-β-hydroxysteroid dehydrogenase type 5 and aromatase. The insulin growth factor system, which can be influenced by ER and has been implicated in the pulmonary oncogenic process, was activated in fetal lung after gestational arsenic exposure. in utero arsenic exposure also induced overexpression of α-fetoprotein, epidermal growth factor receptor, L-myc, and metallothionein-1 in fetal lung, all of which are associated with lung cancer. Lung adenoma and adenocarcinoma from adult female mice exposed to arsenic in utero showed widespread, intense nuclear ER-α expression. In contrast, normal adult lung and diethylnitrosamine-induced lung adenocarcinoma showed little evidence of ER-α expression. Thus, transplacental arsenic exposure at a carcinogenic dose produced aberrant estrogen-linked pulmonary gene expression. ER-α activation was specifically associated with arsenic-induced lung adenocarcinoma and adenoma but not with nitrosamine-induced lung tumors. These data provide evidence that arsenic-induced aberrant ER signaling could disrupt early life stage genetic programing in the lung leading eventually to lung tumor formation much later in adulthood.
Keywords: inorganic arsenic, transplacental exposure, lung cancer, estrogen signaling
Arsenic is an environmental pollutant toxic metalloid and a known human carcinogen (IARC, 2004; NRC, 1999). Tens of millions of people throughout the world are chronically exposed to potentially toxic levels of inorganic arsenic, mainly through ingestion of contaminated water (IARC, 2004; NRC, 1999). Epidemiological data provide evidence that ingestion of arsenic causes cancer of the urinary bladder, lung, liver, and skin among other potential target sites (IARC, 2004; NRC, 1999). Although there is strong evidence for the carcinogenicity of inorganic arsenic compounds in humans, the molecular mechanism remains incompletely defined. Because arsenicals can be carcinogenic in many diverse tissues in both humans and animals, it is reasonable to suspect that more than one mechanism may be involved (Kitchin, 2001; Rossman, 2003). Defining these mechanisms is critical to determining the nature and extent of the human health hazard presented by environmental arsenic exposure and to the development of intervention and prevention strategies. In this regard, important progress has been made in the development of mouse models of inorganic arsenic-induced skin carcinogenesis (Germolec et al., 1998; Rossman et al., 2004). In addition, dimethylarsenic acid, a methylated metabolite of inorganic arsenic, can act as a complete urinary bladder carcinogen in rat or as a multisite tumor promoter in mouse or rat (Wanibuchi et al., 2004). However, animal models for many of the internal tumors associated with inorganic arsenic exposure, such as lung tumors, have not been available until recently. We have recently shown that short-term transplacental inorganic arsenic exposure in mice produces a variety of internal tumors in the offspring when they become adults (Waalkes et al., 2003, 2004b). For instance, in female offspring, lung adenocarcinoma incidence increased from 0% in controls to over 20% in C3H mice exposed to arsenic in utero (Waalkes et al., 2003). Another study showed that gestational inorganic arsenic exposure in C3H mice could initiate pulmonary carcinogenesis that was promoted by the phorbol ester, 12-O-tetradecanoyl phorbol-13-acetate (TPA) in male or female offspring (Waalkes et al., 2004b). These results are in accord with human studies that show the lung is a target of arsenic carcinogenesis (IARC, 2004; NRC, 1999).
Gestation is a period of high sensitivity to chemical carcinogenesis in rodents and humans (Anderson, 2004), and the lung is a common target of transplacental carcinogens (Anderson et al., 2000). Since inorganic arsenic readily crosses the human and rodent placenta and enters the fetal system (Devesa et al., 2006; NRC, 1999), in utero exposure to arsenic likely occurs in human populations exposed to elevated environmental arsenic and is a plausible mode of human exposure (Smith et al., 2006). Accumulating evidence indicates that lung cancer can result from multiple changes in the genome of susceptible pulmonary cells caused by carcinogenic insult (Sekido et al., 2003). There are a variety of ways arsenic can alter gene expression (Kitchin, 2001; Rossman, 2003; Waalkes et al., 2004a), which can be associated with acquisition of aberrant cellular phenotypes (Achanzar et al., 2002; Benbrahim-Tallaa et al., 2005). We recently assessed aberrant gene expression in liver samples from male mice bearing hepatocellular carcinoma induced by in utero arsenic exposure (Liu et al., 2004, 2006; Waalkes et al., 2004a). Remarkably, disruption of estrogen signaling was observed as a key event in transplacental arsenic-induced hepatocarcinogenesis (Liu et al., 2006; Waalkes et al., 2004a). Interestingly, arsenic acted as a complete transplacental lung carcinogen only in female mice (Waalkes et al., 2003), although it was an effective lung tumor initiator in both males and females (Waalkes et al., 2004b).
In recent years, lung cancer has become increasingly common in women and evidence has potentially implicated estrogen in lung carcinogenesis (Gasperino and Rom, 2004; Stabile and Siegfried, 2004). For instance, data that indicate that the female gender carries a significantly elevated risk for lung cancer in cigarette smokers are emerging (Gasperino and Rom, 2004). It is suspected that the combination of carcinogen exposure, genetics, and endocrine-related factors, including potentially estrogen receptor-α (ER-α) (Stabile and Siegfried, 2004; Stabile et al., 2005), contribute to the female disparity in exposure-adjusted lung cancer risk (Gasperino and Rom, 2004). Thus, the present work examined early molecular changes, focusing on estrogen signaling, in fetal lung and subsequent lung tumors developing after in utero arsenic exposure in female mice.
MATERIALS AND METHODS
Animal treatment and fetal tissue collection
The current study was performed, in part, using lung samples from gestation day 18 female fetal C3H mice collected after transplacental arsenic exposure to a dose (85 ppm) shown to either produce lung adenocarcinoma formation in adulthood (Waalkes et al., 2003) or initiate lung tumor formation promotable by TPA (Waalkes et al., 2004b). Animal care was provided in accordance with the U.S. Public Health Policy on the Care and Use of Animals, and the study proposal was approved by Institutional Animal Care and Use Committee of National Cancer Institute (NCI). Pregnant C3H mice (n = 6) were obtained from the Animal Production Area, NCI-Frederick, Animal Program (Frederick, MD). A basal diet (NIH-31 Open Formula, 6% Modified; Teklad Standard Diets, Madison, WI) and water containing 85 ppm arsenic as sodium arsenite (Sigma Chemical Co, St Louis, MO) or unaltered water (control) were provided ad libitum from days 8 to 18 of gestation. The dose of arsenic used did not affect water consumption or the body weight of dams. On gestation day 18, dams and fetuses were killed and the lungs were collected from female fetuses. Samples were frozen in liquid nitrogen and stored at - 80°C until analysis. Samples of fully formed lung adenoma or adenocarcinoma induced by in utero arsenic exposure in adult female C3H were obtained from tissues taken in our prior study (Waalkes et al., 2003) and were used for immunohistochemistry (see below).
Gene expression analysis
Total RNA was isolated from frozen fetal lung samples using TRIzol reagent (Invitrogen, Carlsbad, CA) followed by purification on RNeasy columns (Qiagen, Palo Alto, CA). RNA was then reverse transcribed with MuLV reverse transcriptase and Oligo-dT primers. The forward and reverse primers for selected genes were designed using Primer Express software (Applied Biosystems, Foster City, CA). Primers for select genes were listed in Table 1 and synthesized by Sigma-Genosys (Woodlands, TX). The SYBR green DNA PCR kit (Applied Biosystem) was used for realtime polymerase chain reaction analysis. The relative differences in expression between groups were expressed using cycle time (Ct) values as follows: the Ct values of the interested genes were first normalized with β-actin of the same sample, and then the relative transcript levels are expressed as percent control with control being set at 100%.
TABLE 1.
Primer Sequences for Real-Time Reverse Transcriptase-Polymerase Chain Reaction Analysis
| Gene name | Accession number | Forward | Reverse |
|---|---|---|---|
| AFP | V00743 | AGCTCAGCGAGGAGAAATGGT | GTTCACAGGGCTTGCTTCATTC |
| Agr2 | NM_011783 | CCTTGCGGCTCACACAAAG | ATGGCCACAAGAAGCAGGAT |
| Aromatase | D00659 | CCCTGGTCTTGTTCGAATGG | TGAGGGTCAACACATCCACGTA |
| β-Actin | M12481 | GGCCAACCGTGAAAAGATGA | CAGCCTGGATGGCTACGTACA |
| EGFR | X78987 | CCCTTGGGATTGGCCTATTC | TCCACGAGCTCTCTCTCTTGAA |
| ER-α | M38651 | TCTCTGGAAGAGAAGGACCACATC | TGCAGAGTCAGGCCAGCTTT |
| ER-β | NM_207707 | CAGGTGCATTGCCTGAACAA | CAGCTGCTCGGGACTCAGA |
| 17βHSD5 | NM_030611 | GCCATGGAGAAATGCAAGGA | GTTGCACACAGGCTTGTACTTGAG |
| IGF-1 | X04480 | TTGCTTCCGGAGCTGTGATC | AGAGCGGGCTGCTTTTGTAG |
| IGF-2 | M14951 | AGAGTTCAGAGAGGCCAAACGT | TTGCTGGACATCTCCGAAGAG |
| IGFBP-1 | X81579 | TGGACAGCTTCCACCTGATG | TGATGGCGTTCCACAGGAT |
| IGFBP-5 | X81583 | CCCAAGCACACTCGCATTT | TGGACTGGGTCAGCTTCTTTCT |
| IGFR2 | U04710 | TTTGGGCGCCTTGCAT | AGGGCAAGGATCACCATTCA |
| L-myc, lung-specific myc oncogene | X13945 | CCCATCACCTCCCCATTTTC | GGGCTCCAATCACCATCAGA |
| MT-1 | BC027262 | AATGTGCCCAGGGCTGTGT | GCTGGGTTGGTCCGATACTATT |
| TFF3 | NM_011575 | CTCTGTCACATCGGAGCAGTGT | TGAAGCACCAGGGCACATT |
Western blot analysis
Fetal lung samples were homogenized (1:20, wt/vol) in PER-Tissue protein extraction buffer (Pierce, Rockford, IL) and freshly added protease inhibitors cocktail (CalBiochem, La Jolla, CA) and 500μM phenylmethylsulfonyl fluoride. Cytosols were prepared by centrifugation at 12,000 × g for 10 min at 4°C. Protein concentrations were determined using the dye-binding assay (Bio-Rad, Hercules, CA). Total protein (30 μg) was subjected to electrophoresis on Nupage gels (4-12%) (Invitrogen), followed by electrophoretic transfer to nitrocellulose membranes at 30 V for 1 h. Membranes were blocked in 5% dried milk in TBST (15mM Tris-HCl, pH 7.4, 150mM NaCl, and 0.05% Tween 20) for 2 h at room temperature, followed by incubation with the first antibody (1:200 to 1:1000) in Blotto (Pierce) overnight at 4°C. After three washes with TBST, the membranes were incubated in secondary antibody (1:4000 to 1:10,000) for 45 min. After four washes with TBST, proteins were visualized using SuperSignal substrate (Pierce).
Immunohistochemical localization for ER-α
Representative lung adenocarcinomas or adenomas induced by transplacental arsenic exposure in female mice (Waalkes et al., 2003) were compared with normal lung or with lung carcinomas induced by diethylnitrosamine (DEN) from female mice. A polyclonal antibody against ER-α (Santa Cruz Biotechnology, Santa Cruz, CA) was used as the primary antibody (diluted 1:1000) together with a streptavidinconjugated secondary antibody (Santa Cruz Biotechnology). Antibody binding was visualized with an avidin-biotin-peroxidase kit (VECTASTAIN Elite ABC Kit; Vector Laboratories, Burlingame, CA) with diaminobenzidine as the chromagen and hematoxylin as a nuclear counterstain.
Statistics
Means and SEMs were calculated, and comparisons between groups were made by the Student's t-test. Fischer's exact test was used to analyze the incidence of lungs immunohistochemically positive for ER-α expression. A two-sided probability level of p< 0.05 was considered significant.
RESULTS
Estrogen Receptor and Related Gene Expression in Female Fetal Lung After In Utero Arsenic Exposure
Prior work showed that transplacental arsenic exposure in C3H mice induces a marked increase in lung adenocarcinoma incidence in female offspring as adults (Waalkes et al., 2003). Thus, the exposure protocol producing lung cancers (85 ppm arsenic in drinking water from days 8 to 18 of gestation) was duplicated, and female fetal lungs were collected for analysis. Genes were selected based on prior work showing aberrant estrogen signaling associated with transplacental arsenic hepatocarcinogenesis (Waalkes et al., 2004a) and based on relevance to pulmonary carcinogenesis. Figure 1 shows the expression of ER-α and ER-β, as well as the ER-linked genes trefoil factor-3 (Tff3) and anterior gradient-2 (Agr2). In addition, genes encoding for enzymes important in steroid metabolism such as 17-β-hydroxysteroid dehydrogenase type 5 (17β-HSD5) and aromatase were assessed. Compared with control, a dramatic 530% increase occurred in pulmonary ER-α transcript in female fetuses after gestational exposure to arsenic. The expression of ER-β in arsenic-treated fetal lung was unchanged. The expression of various estrogen-related genes was markedly increased, including Tff3 (966%), Agr2 (321%), 17β-HSD5 (355%), and aromatase (253%). In marked contrast, the expression of ER-α or these ER-linked genes was unchanged in male fetal lung as compared with control (data not shown).
FIG. 1.
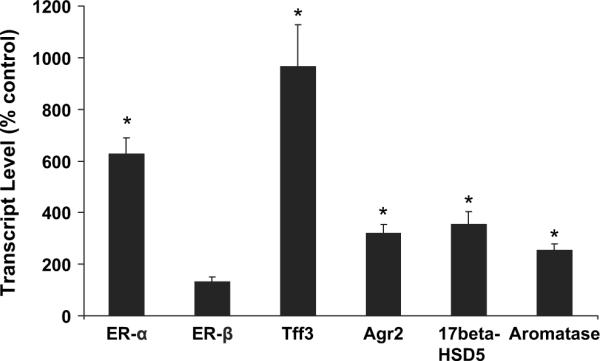
Expressions of ER-α, ER-β, Tff3, Agr2, 17β-HSD5 and aromatase transcripts in gestation day 18 female fetal mouse lung after in utero arsenic exposure. Data are expressed as mean ± SEM (n = 6). *Significantly difference (p ≤ 0.05) from control.
To help confirm the results of transcript analysis, western blot analysis was performed in control and arsenic-treated fetal lung on selected proteins (Fig. 2). ER-α protein was increased in fetal lung by arsenic exposure, which is in accord with increases seen in the transcript. The potential oncogene and tumor cell marker, α-fetoprotein (AFP), was also markedly increased at the protein level in arsenic-treated fetal lung.
FIG. 2.
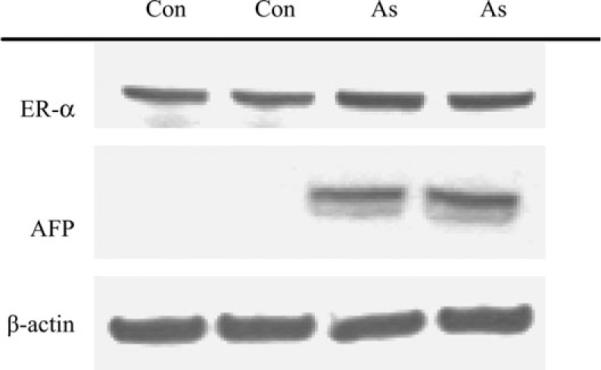
Western blot analysis of selected proteins in control fetal lung (lanes 1 and 2) and arsenic-exposed fetal lung (lanes 3 and 4). The kilodalton sizes are ER-α (≈ 70 kDa), AFP (≈ 75 kDa), and β-actin (≈ 43 kDa).
Oncogenesis-Related Gene Expression in Female Fetal Lung Exposed to Arsenic In Utero
Figure 3 shows the impact of in utero arsenic exposure on the expression of selected genes related to the pulmonary oncogenic process. Various transcriptional products were increased including those for the epidermal growth factor receptor (EGFR, 3.2-fold), AFP (6.9-fold), L-myc (1.9-fold), and metallothionein-1 (MT-1, 2.1-fold) genes. EGFR, AFP, and L-myc are all potentially important in lung cancer (Cappuzzo et al., 2005; Hiroshima et al., 2002; Shih et al., 2002). In fact, overexpression of EGFR and AFP can be associated with particularly aggressive lung malignancies (Cappuzzo et al., 2005; Hiroshima et al., 2002). These very early expression changes in lung oncogenesis-related genes may be relevant to the arsenic-induced carcinogenic process that results in lung cancer formation much later in adulthood.
FIG. 3.
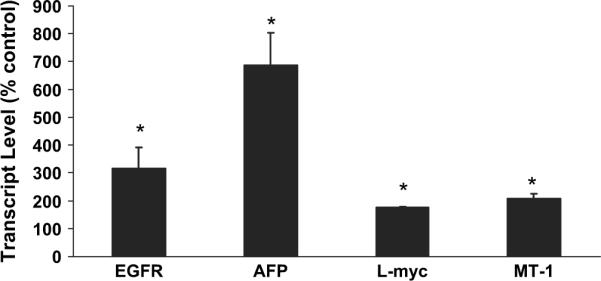
Expressions of EGFR, AFP, L-myc, and MT-1 transcripts in gestation day 18 female fetal mouse lung after in utero arsenic exposure. Data are expressed as mean ± SEM (n = 6). *Significantly difference (p ≤ 0.05) from control.
Insulin Growth Factor-Related Gene Expression in Female Fetal Lung Exposed to Arsenic In Utero
Transcript levels of genes encoding for the insulin growth factor (IGF) system, which can be influenced by ER and have also been implicated in the lung oncogenic process (Pavelic et al., 2005), are shown in Figure 4. The expression of both IGF-1 and -2 was increased approximately 2-fold by in utero arsenic exposure. In addition, IGF receptor-1 (IGFR1; 2.4-fold) and IGFR2 (1.8-fold) were similarly increased. Expression of IGF-binding protein 1 (IGFBP-1; 2.5-fold) and IGFBP-5 (1.6-fold) was also increased by in utero arsenic exposure.
FIG. 4.
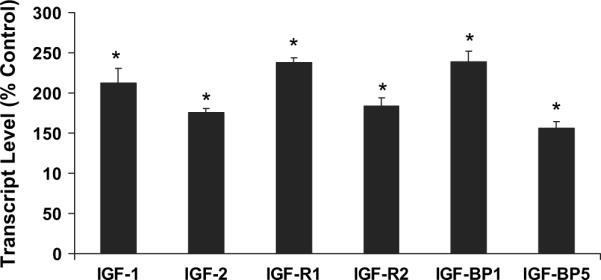
Expression of IGF-1, IGF-2, IGFR1, IGFBP-1, and IGFBP-5 transcripts in gestation day 18 female fetal mouse lung after in utero arsenic exposure. Data are expressed as mean ± SEM (n = 6). *Significant difference (p ≤ 0.05) from control.
ER-α Expression in Lung Adenocarcinoma From Adult Female Mice Exposed to Arsenic in utero
To determine if ER-α overexpression was related to arsenic-induced lung cancer in female offspring, lung adenocarcinomas resulting from gestational arsenic exposure were studied immunohistochemically. A typical arsenic-induced lung adenocarcinoma stained with hematoxylin and eosin (H&E) is shown for reference (Fig. 5A). When stained for ER-α, the lung adenocarcinomas induced by in utero arsenic exposure showed intense and widespread expression of ER-α that was particularly pronounced in tumor cell nuclei (Fig. 5B). A nuclear localization is thought to represent an active form of this receptor. ER-α overexpression appeared to be common at all stages of lung tumor development induced by in utero arsenic exposure in female mice as evidenced by the intense and widespread staining for ER-α in a typical arsenic-induced lung adenoma (Fig. 6A). To see if ER-α expression was specific to arsenic, a lung adenocarcinoma induced by DEN was also examined (Fig. 6B). There was little immunohistochemical evidence of ER-α expression in the DEN-induced pulmonary adenocarcinoma.
FIG. 5.

Immunohistochemical analysis of mouse ER-α expression in lung adenocarcinoma in adult female mice induced by in utero arsenic exposure. (A) Morphology of arsenic-induced lung adenocarcinoma (hematoxylin and eosin, ×400). (B) Immunohistochemical analysis for ER-α expression in a typical arsenic-induced lung adenocarcinoma (×400). The lung adenocarcinoma shows widespread and intense expression for ER-α (brown staining), particularly in the tumor cell nuclei.
FIG. 6.

Immunohistochemical analysis of ER-α expression of mouse lung tumors induced by in utero arsenic exposure or DEN treatment in adulthood. (A) ER-α expression in a typical lung adenoma (×100) induced by in utero arsenic exposure showing widespread and intense expression for ER-α (brown staining), particularly in the tumor cell nuclei. (B) ER-α expression in a DEN-induced lung adenocarcinoma (×100) showing little or no expression.
The rate of positive ER-α expression in mouse lung tumors induced by in utero arsenic exposure or DEN treatment in adulthood as assessed immunochemically is shown in Table 2. Most of arsenic-induced lung tumors were strongly positive for ER-α (83%), while all DEN-induced lung tumors examined were negative.
TABLE 2.
Immunohistochemical Assessment of ER-a Expression in Mouse Lung Tumors Induced by In Utero Arsenic Exposure or by DEN Treatment in Adulthood
| Group | n | Strongly positive | Variably positive | Negative |
|---|---|---|---|---|
| Arsenic induced | 6 | 5* | 1 | 0 |
| DEN induced | 3 | 0 | 0 | 3 |
Note. The asterisk (*) indicates a significant (p < 0.05) difference from incidence rates for DEN-induced tumors (Fischer's exact test, two sided).
DISCUSSION
The present study clearly showed that in utero arsenic exposure at a level that affects pulmonary tumor development in adulthood increased expression of ER-α and genes related to estrogen signaling in the fetal lung of female mice. In lung adenoma or adenocarcinoma occurring in adult mice exposed to arsenic in utero, ER-α protein overexpression was widespread and intense, indicating that in utero arsenic induced aberrant ER signaling in the lung in a fashion similar to that seen during transplacental arsenic hepatocarcinogenesis (Waalkes et al., 2004a). Thus, this study further fortifies the hypothesis that aberrant estrogen signaling is associated with transplacental arsenic carcinogenesis, at least in some tissues (Liu et al., 2004, 2006; Waalkes et al., 2004a, 2006a,b). Our recent results in CD1 mice indicate that urinary bladder proliferative lesions, including tumors, associated with prenatal arsenic exposure are greatly enhanced by postnatal exposure to estrogenic agents (Waalkes et al., 2006a,b). As a corollary in humans, increased mortality occurs from lung cancers in young adults following in utero exposure to elevated levels of arsenic in the drinking water (Smith et al., 2006). Thus, the developing fetus appears to be quite sensitive to arsenic-induced carcinogenesis.
Lung cancer is the leading cause of cancer death in the United States and, once diagnosed, often has a poor prognosis (Gasperino and Rom, 2004). Accumulating evidence indicates that females may be more sensitive to insult by lung carcinogens (Gasperino and Rom, 2004). In humans, lung cancer is associated with inorganic arsenic exposure either from drinking water or by inhalation (IARC, 2004; NRC, 1999). A recent study showed that chronic oral exposure to inorganic arsenate increased lung tumor formation in A/J mice (Cui et al., 2006). Inorganic arsenic can also act as a complete transplacental lung carcinogen or as a lung tumor initiator in female mice (Waalkes et al., 2003, 2004b). A variety of clinical and experimental data suggest that aberrant estrogen signaling plays a role in lung cancer (Gasperino and Rom, 2004; Patel, 2005; Stabile and Siegfried, 2004; Stabile et al., 2005). A study in humans found that ER-α expression occurs more often in the lungs and in lung tumors of women than men, and this potential gender-dependent difference could contribute unique phenotypic characteristics to lung cancer development and progression in women (Fasco et al., 2002). The present study shows that in utero exposure to the carcinogenic dose of inorganic arsenic (Waalkes et al., 2003, 2004b) resulted in the overexpression of pulmonary ER-α in female fetus. ER-α overexpression was clearly associated with arsenic-induced lung adenoma and adenocarcinoma in adult females treated with arsenic during prenatal life. In fact, arsenic-induced lung tumors that arise in adults after gestation exposure show widespread overexpression of ER-α which is particularly intense in the tumor cell nuclei, the site where this transcription factor is normally active (Hart and Davie, 2002). In contrast, ER-α expression was very poor in normal lung tissue or in DEN-induced lung adenocarcinoma, indicating that ER-α expression may be specific to arsenic, rather than typical for mouse pulmonary tumors in general.
The overexpression of pulmonary ER-α observed in the present work is consistent with our earlier observation of uterine (Waalkes et al., 2006b) or hepatic (Waalkes et al., 2004a) proliferative lesions, including tumors, induced by in utero arsenic exposure. In addition, in utero arsenic exposure combined with postnatal exposure to diethylstilbestrol greatly increases urogenitial cancers in female CD1 mice and enhances expression of estrogen-regulated genes in neonatal uterus (Waalkes et al., 2006b). Furthermore, prenatal arsenic plus postnatal diethylstilbestrol or tamoxifen increases urinary bladder transitional cell hyperplasia and tumors that show clear evidence of ER-α overexpression (Waalkes et al., 2006a,b). Thus, arsenic carcinogenesis may, in some cases, involve enhanced cellular responses to endogenous or exogenous steroidal growth signals. In this regard, recent evidence indicates exposure to environmental factors during mammalian development, including estrogenic compounds, can aberrantly “reprogram” physiological responses in target tissue to enhance tumor formation in later life (Cook et al., 2005). The alterations in steroid metabolism and estrogen signaling induced by in utero arsenic exposure in mouse lung, liver, urinary bladder, and uterus are consistent with this reprograming concept during early life exposure to environmental carcinogens.
The consistent finding of arsenic-induced overexpression of ER-α and ER-linked genes in mouse models of arsenic toxicity and transplacental carcinogenesis (Chen et al., 2004; Liu et al., 2004; Waalkes et al., 2000, 2004a, 2006b) may have important implications for arsenic-exposed humans. Estrogens are widely used pharmaceuticals, and a large number of environmental chemicals act as xenoestrogens. The combined results of the present and prior studies raise the possibility that people exposed to arsenic may be more sensitive to the potential oncogenic effects of estrogens, at least in certain tissues. Conversely, exposure to pharmacologic or environmental estrogens may enhance the carcinogenic effects of arsenic.
Arsenic-induced ER-α in the fetal lung could potentially be active in regulating gene expression. This conclusion is supported by our findings that the ER-regulated gene Tff3 (dos Santos Silva et al., 2000) was expressed at high levels in the lungs after arsenic exposure in utero. TFF3 is expressed and secreted in normal and inflamed human airways. Overexpression of TFF3 is often found in lung cancer patients (dos Santos Silva et al., 2000). In human breast cancer, Agr2, an important estrogen-related gene, is overexpressed (Shen et al., 2005). Agr2 gene expression is associated with ER-α positive breast carcinomas but not with those that are ER-α negative (Liu et al., 2005). In addition, 17β-HSD5 gene is related to the process of human prostate cancer (Nakamura et al., 2005). Overexpression of aromatase is found in human non-small-cell lung cancer cells (Weinberg et al., 2005). Thus, the increased expressions of these genes in arsenic-exposed fetal lung could be an important genetic event in arsenic pulmonary carcinogenesis.
The overexpression of oncogenesis-related genes in arsenic-exposed fetal lung is noteworthy and includes overexpression of AFP, L-myc, and EGFR, which have been all implicated in lung carcinogenesis. For instance, elevated serum levels of AFP occur in patients with lung carcinoma, and primary lung carcinoma can overexpress AFP (Hiroshima et al., 2002). AFP is known as a growth-promoting oncoprotein often overex-pressed in embryonic tumors (Mizejewski and MacColl, 2003) that can regulate tumor growth (Mizejewski, 2002). Similarly, L-myc can act as an oncogene, and overexpression and amplification of Myc family members are frequently seen in lung cancer cells or tumors (Shih et al., 2002). Data also indicate that high levels of EGFR can occur in human lung cancer (Cappuzzo et al., 2005). There is a functional link between ER and EGFR, and EGFR can be activated by estrogens and act in estrogen signaling (Stabile et al., 2005). Both AFP and EGFR overexpression is associated with particularly aggressive forms of pulmonary cancer (Cappuzzo et al., 2005; Hiroshima et al., 2002). MT is a low-molecular-weight protein implicated in adaptive responses to arsenic toxicity and transplacental carcinogenesis (Liu et al., 2006). Thus, early life exposure to arsenic is clearly activating several genes associated with lung oncogenesis.
Fetal pulmonary overexpression of various members of the IGF system was evident following prenatal arsenic exposure in the present work. The IGF system plays a critical role in the development and progression of cancer (LeRoith and Roberts, 2003). IGF is implicated in carcinogenesis through regulation of malignant cell proliferation, differentiation, and apoptosis (Ibrahim and Yee, 2004). IGF-1 in particular is linked to malignant transformation and has been recognized in both experimental and clinical settings, suggesting that the enhancement of growth factor pathways potentially could increase the risk for cancer development (Ibrahim and Yee, 2004). A growing body of epidemiological data suggests that high levels of circulating IGF-1 constitute a risk factor for the development of lung cancers (LeRoith and Roberts, 2003). Clinical data indicate that serum levels of IGF-1 and IGF-2 are elevated in lung cancer patients (Izycki et al., 2004). Disruption of the IGF/IGFR axis is also potentially involved in lung cancer formation (Pavelic et al., 2005). A recent study showed that estrogen-induced stimulation of proliferation of cancer cells is partly achieved via IGF signaling (Gielen et al., 2005). IGFBPs are regulated by estrogens and potentially play a role in the modulation of cancer cell proliferation (Gielen et al., 2005). Thus, the fetal overexpression of the IGF/IGFRs axis seen in the present work after in utero exposure to arsenic could have important implications in eventual lung tumor formation.
In summary, this study indicates that in utero exposure to inorganic arsenic at a dose that induces lung cancer in female offspring in adulthood causes a remarkable increase in expression of fetal pulmonary ER-α and ER-linked genes. In both early (adenoma) and advanced (adenocarcinoma) lung tumors developing in adult female mice exposed to arsenic in utero, ER-α was also overexpressed. This work provides evidence that arsenic exposure may induce early life stage reprograming of key genes in growth signaling, leading to tumor formation much later in life.
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences, and National Cancer Institute, Center for Cancer Research. BA.D. was supported by the NCI Contract No. NO1-CO-12400. We thank Drs Lamia Benbrahim-Talla, Wei Qu, Jean-Francois Coppin, and Larry Keefer for critically reviewing the manuscript.
REFERENCES
- Achanzar WE, Liu J, Webber MM, Waalkes MP. Altered apoptotic gene expression and acquired apoptotic resistance in cadmium-transformed human prostate epithelial cells. Prostate. 2002;52:236–244. doi: 10.1002/pros.10106. [DOI] [PubMed] [Google Scholar]
- Anderson LM. Predictive values of traditional animal bioassay studies for human perinatal carcinogenesis risk determination. Toxicol. Appl. Pharmacol. 2004;199:162–174. doi: 10.1016/j.taap.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Anderson LM, Diwan BA, Fear NT, Roman E. Critical windows of exposure for children's health: Cancer in human epidemiological studies and neoplasms in experimental animal models. Environ. Health Perspect. 2000;108(Suppl 3):573–594. doi: 10.1289/ehp.00108s3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benbrahim-Tallaa L, Waterland RA, Styblo M, Achanzar WE, Webber MM, Waalkes MP. Molecular events associated with arsenic-induced malignant transformation of human prostatic epithelial cells: Aberrant genomic DNA methylation and K-ras oncogene activation. Toxicol. Appl. Pharmacol. 2005;206:288–298. doi: 10.1016/j.taap.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J. Natl. Cancer Inst. 2005;97:643–655. doi: 10.1093/jnci/dji112. [DOI] [PubMed] [Google Scholar]
- Chen H, Li S, Liu J, Diwan BA, Barrett JC, Waalkes MP. Chronic inorganic arsenic exposure induces hepatic global and individual gene hypomethylation: Implication for arsenic hepatocarcinogenesis. Carcinogenesis. 2004;25:1779–1786. doi: 10.1093/carcin/bgh161. [DOI] [PubMed] [Google Scholar]
- Cook JD, Davis BJ, Cai SL, Barrett JC, Conti CJ, Walker CL. Interaction between genetic susceptibility and early-life environmental exposure determines tumor-suppressor-gene penetrance. Proc. Natl. Acad. Sci. U.S.A. 2005;102:8644–8649. doi: 10.1073/pnas.0503218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Wakai T, Shirai Y, Hatakeyama K, Hirano S. Chronic oral exposure to inorganic arsenate interferes with methylation status of p16INK4a and RASSF1A and induces lung cancer in A/J mice. Toxicol. Sci. 2006;91:372–381. doi: 10.1093/toxsci/kfj159. [DOI] [PubMed] [Google Scholar]
- Devesa V, Adair BM, Liu J, Waalkes MP, Diwan BA, Styblo M, Thomas DJ. Speciation of arsenic in the maternal and fetal mouse tissues following gestational exposure to arsenite. Toxicology. 2006;224:147–155. doi: 10.1016/j.tox.2006.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos Silva E, Ulrich M, Doring G, Botzenhart K, Gott P. Trefoil factor family domain peptides in the human respiratory tract. J. Pathol. 2000;190:133–142. doi: 10.1002/(SICI)1096-9896(200002)190:2<133::AID-PATH518>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Fasco MJ, Hurteau GJ, Spivack SD. Gender-dependent expression of alpha and beta estrogen receptors in human nontumor and tumor lung tissue. Mol. Cell. Endocrinol. 2002;188:125–140. doi: 10.1016/s0303-7207(01)00750-x. [DOI] [PubMed] [Google Scholar]
- Gasperino J, Rom WN. Gender and lung cancer. Clin. Lung Cancer. 2004;5:353–359. doi: 10.3816/CLC.2004.n.013. [DOI] [PubMed] [Google Scholar]
- Germolec DR, Spalding J, Yu HS, Chen GS, Simeonova PP, Humble MC, Bruccoleri A, Boorman GA, Foley JF, Yoshida T, et al. Arsenic enhancement of skin neoplasia by chronic stimulation of growth factors. Am. J. Pathol. 1998;153:1775–1785. doi: 10.1016/S0002-9440(10)65692-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gielen SC, Hanekamp EE, Blok LJ, Huikeshoven FJ, Burger CW. Steroid-modulated proliferation of human endometrial carcinoma cell lines: Any role for insulin-like growth factor signaling? J. Soc. Gynecol. Investig. 2005;12:58–64. doi: 10.1016/j.jsgi.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Hart LL, Davie JR. The estrogen receptor: More than the average transcription factor. Biochem. Cell Biol. 2002;80:335–341. doi: 10.1139/o02-038. [DOI] [PubMed] [Google Scholar]
- Hiroshima K, Iyoda A, Toyozaki T, Haga Y, Baba M, Fujisawa T, Ohwada H. Alpha-fetoprotein- producing lung carcinoma: Report of three cases. Pathol. Int. 2002;52:46–53. doi: 10.1046/j.1440-1827.2002.01311.x. [DOI] [PubMed] [Google Scholar]
- Ibrahim YH, Yee D. Insulin-like growth factor-I and cancer risk. Growth Horm. IGF Res. 2004;14:261–269. doi: 10.1016/j.ghir.2004.01.005. [DOI] [PubMed] [Google Scholar]
- International Agency for Research on Cancer (IARC) Some Drinking Water Disinfectants and Contaminants, Including Arsenic. IARC; Lyon, France: 2004. IARC monographs on the evaluation of carcinogenic risks to humans, Vol. 84. Arsenic in drinking water; pp. 269–477. [PMC free article] [PubMed] [Google Scholar]
- Izycki T, Chyczewska E, Naumnik W, Talalaj J, Panek B, Ossalinska M. Serum levels of IGF-I and IGF-II in patients with lung cancer during chemotherapy. Exp. Oncol. 2004;26:316–319. [PubMed] [Google Scholar]
- Kitchin KT. Recent advances in arsenic carcinogenesis; modes of action, animal model systems, and methylated arsenic metabolites. Toxicol. Appl. Pharmacol. 2001;172:249–261. doi: 10.1006/taap.2001.9157. [DOI] [PubMed] [Google Scholar]
- LeRoith D, Roberts CT. The insulin-like growth factor system and cancer. Cancer Lett. 2003;195:127–137. doi: 10.1016/s0304-3835(03)00159-9. [DOI] [PubMed] [Google Scholar]
- Liu D, Rudland PS, Sibson DR, Platt-Higgins A, Barraclough R. Human homologue of cement gland protein, a novel metastasis inducer associated with breast carcinomas. Cancer Res. 2005;65:3796–3805. doi: 10.1158/0008-5472.CAN-04-3823. [DOI] [PubMed] [Google Scholar]
- Liu J, Xie Y, Ducharme DMK, Shen J, Diwan BA, Merrick BA, Grissom SF, Tucker CJ, Paules PS, Tennant R, et al. Global gene expression associated with hepatocarcinogenesis in adult male mice induced by in utero arsenic exposure. Environ. Health Perspect. 2006;114:404–411. doi: 10.1289/ehp.8534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xie Y, Ward JM, Diwan BA, Waalkes MP. Toxicogenomic analysis of aberrant gene expression in liver tumors and nontumorous livers of adult mice exposed in utero to inorganic arsenic. Toxicol. Sci. 2004;77:249–257. doi: 10.1093/toxsci/kfh055. [DOI] [PubMed] [Google Scholar]
- Mizejewski GJ. Biological role of alpha-fetoprotein in cancer: prospects for anticancer therapy. Expert Rev. Anticancer Ther. 2002;2:709–735. doi: 10.1586/14737140.2.6.709. [DOI] [PubMed] [Google Scholar]
- Mizejewski GJ, MacColl R. α-Fetoprotein growth inhibitory peptides: Potential leads for cancer therapeutics. Mol. Cancer Ther. 2003;2:1243–1255. [PubMed] [Google Scholar]
- Nakamura Y, Suzuki T, Nakabayashi M, Endoh M, Sakamoto K, Mikami Y, Moriya T, Ito A, Takahoshi S, Yamada S, et al. In situ androgen producing enzymes in human prostate cancer. Endocr. Relat. Cancer. 2005;12:101–107. doi: 10.1677/erc.1.00914. [DOI] [PubMed] [Google Scholar]
- National Research Council (NRC) Arsenic in Drinking Water. National Research Council; Washington, DC: 1999. pp. 1–310. [Google Scholar]
- Patel JD. Lung cancer in women. J. Clin. Oncol. 2005;23:3212–3218. doi: 10.1200/JCO.2005.11.486. [DOI] [PubMed] [Google Scholar]
- Pavelic J, Krizanac S, Kapitanovic S, Pavelic L, Samarzija M, Pavicic F, Spaventi S, Jakopovic M, Herceg-Ivanovi Z, Pavelic K. The consequences of insulin-like growth factors/receptors dysfunction in lung cancer. Am. J. Respir. Cell Mol. Biol. 2005;32:65–71. doi: 10.1165/rcmb.2004-0232OC. [DOI] [PubMed] [Google Scholar]
- Rossman TG. Mechanism of arsenic carcinogenesis: An integrated approach. Mutat. Res. 2003;533:37–65. doi: 10.1016/j.mrfmmm.2003.07.009. [DOI] [PubMed] [Google Scholar]
- Rossman TG, Uddin AN, Burns FJ. Evidence that arsenite acts as a cocarcinogen in skin cancer. Toxicol. Appl. Pharmacol. 2004;198:394–404. doi: 10.1016/j.taap.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Sekido Y, Fong KM, Minna JD. Molecular genetics of lung cancer. Annu. Rev. Med. 2003;54:73–87. doi: 10.1146/annurev.med.54.101601.152202. [DOI] [PubMed] [Google Scholar]
- Shen D, Chang HR, Chen Z, He J, Londberry V, Elshimali Y, Chia D, Seligson D, Goodglick L, Nelson SF, et al. Loss of annexin A1 expression in human breast cancer detected by multiple high-throughput analyses. Biochem. Biophys. Res. Commun. 2005;326:218–227. doi: 10.1016/j.bbrc.2004.10.214. [DOI] [PubMed] [Google Scholar]
- Shih CM, Kuo YY, Wang YC, Jian SL, Hsu YT, Wu HY, Guo MW, Wang WC. Association of L-myc polymorphism with lung cancer susceptibility and prognosis in relation to age-selected controls and stratified cases. Lung Cancer. 2002;36:125–132. doi: 10.1016/s0169-5002(01)00467-6. [DOI] [PubMed] [Google Scholar]
- Smith AH, Marshall G, Yuan Y, Ferreccio C, Liaw J, von Ehrenstein O, Steinmaus C, Bates MN, Selvin S. Increased mortality from lung cancer and bronchiectasis in young adults after exposure to arsenic in utero and in early childhood. Environ. Health Perspect. 2006;114:1293–1296. doi: 10.1289/ehp.8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabile LP, Lyker JS, Gubish CT, Zhang W, Grandis JR, Siegfried JM. Combined targeting of estrogen receptor and the epidermal growth factor receptor in non-small lung cancer shows enhanced antiproliferative effects. Cancer Res. 2005;65:1459–1470. doi: 10.1158/0008-5472.CAN-04-1872. [DOI] [PubMed] [Google Scholar]
- Stabile LP, Siegfried JM. Estrogen receptor pathways in lung cancer. Curr. Oncol. Rep. 2004;6:259–267. doi: 10.1007/s11912-004-0033-2. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Keefer LK, Diwan BA. Induction of proliferative lesions of the uterus, testes, and liver in Swiss mice given repeated injections of sodium arsenate; possible estrogenic mode of action. Toxicol. Appl. Pharmacol. 2000;166:24–35. doi: 10.1006/taap.2000.8963. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Liu J, Chen H, Xie Y, Achanzar WE, Zhou YS, Cheng ML, Diwan BA. Estrogen signaling in livers of male mice with hepatocellular carcinoma induced by exposure to arsenic in utero. J. Natl. Cancer Inst. 2004a;96:466–474. doi: 10.1093/jnci/djh070. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Liu J, Ward JM, Diwan BA. Enhanced urinary bladder and liver carcinogenesis in male CD1 mice exposed to transplacental inorganic arsenic and postnatal diethylstilbestrol or tamoxifen. Toxicol. Appl. Pharmacol. 2006a;215:295–305. doi: 10.1016/j.taap.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Liu J, Ward JM, Powell DA, Diwan BA. Urogenital carcinogenesis in female CD1 mice induced by in utero arsenic exposure is exacerbated by postnatal diethylstilbestrol treatment. Cancer Res. 2006b;66:1337–1345. doi: 10.1158/0008-5472.CAN-05-3530. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Ward JM, Diwan BA. Induction of tumors of the liver, lung, ovary and adrenal in adult mice after brief maternal gestational exposure to inorganic arsenic: Promotional effects of postnatal phorbol ester exposure on hepatic and pulmonary, but not dermal cancers. Carcinogenesis. 2004b;25:133–141. doi: 10.1093/carcin/bgg181. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Ward JM, Liu J, Diwan BA. Transplacental carcinogenicity of inorganic arsenic in the drinking water: Induction of hepatic, ovarian, pulmonary, and adrenal tumors in mice. Toxicol. Appl. Pharmacol. 2003;186:7–17. doi: 10.1016/s0041-008x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- Wanibuchi H, Salim EI, Kinoshita A, Shen J, Wei M, Morimura K, Yoshida K, Kuroda K, Endo G, Fukushima S. Understanding arsenic carcinogenesis by the use of animal models. Toxicol. Appl. Pharmacol. 2004;198:366–376. doi: 10.1016/j.taap.2003.10.032. [DOI] [PubMed] [Google Scholar]
- Weinberg OK, Marquez-Garban DC, Fishbein MC, Goodglick L, Garban HJ, Dubinett SM, Pistras RJ. Aromatase inhibitors in human lung cancer therapy. Cancer Res. 2005;65:11287–11291. doi: 10.1158/0008-5472.CAN-05-2737. [DOI] [PubMed] [Google Scholar]