

Abstract
The small GTPase proteins RhoA and RhoC are essential for tumor invasion and/or metastasis in breast carcinomas. However, it is poorly understood how RhoA and RhoC are activated in breast cancer cells. Here we describe the role of MyoGEF in regulating RhoA and RhoC activation as well as cell polarity and invasion in an invasive breast cancer cell line MDA-MB-231. RNA-interference (RNAi)-mediated depletion of MyoGEF in MDA-MB-231 cells not only suppresses the activation of RhoA and RhoC, but also decreases cell polarity and invasion activity. The dominant negative mutants of RhoA and RhoC, but not Rac1 and Cdc42, dramatically decrease actin polymerization induced by MyoGEF. In addition, MyoGEF colocalizes with nonmuscle myosin IIA (NMIIA) to the front of migrating cells, and depletion of NMIIA by RNAi disrupts the polarized localization of MyoGEF at the cell leading edge, suggesting a role for NMIIA in regulating MyoGEF localization and function. Moreover, MyoGEF protein levels significantly increase in infiltrating ductal carcinomas as well as in invasive breast cancer cell lines. Taken together, our results suggest that MyoGEF cooperates with NMIIA to regulate the polarity and invasion activity of breast cancer cells through activation of RhoA and RhoC.
Keywords: MyoGEF, RhoA, RhoC, MDA-MB-231, cell motility, cell polarity, nonmuscle myosin II
INTRODUCTION
Cell migration plays a critical role in normal physiological processes such as embryogenesis, immune surveillance, and angiogenesis, as well as in pathological processes such as tumor invasion and metastasis (Burridge and Wennerberg, 2004; Jaffe and Hall, 2005). Cell migration involves lamellipodia formation and membrane protrusion at the front and retraction of the rear part of migrating cells. Actin polymerization at the front of migrating cells is critically important for membrane protrusion while myosin-based contractility is required for the retraction of the rear of migrating cells (Lauffenburger and Horwitz, 1996; Raftopoulou and Hall, 2004; Ridley et al., 2003; Webb et al., 2002).
The small GTPase proteins such as Rac1, Cdc42, and RhoA are key regulators of the actin cytoskeleton. RhoA stimulates the assembly of contractile actomyosin filaments and associated focal adhesion complexes (Ridley and Hall, 1992). Rac1 induces the formation of lamellipodia and membrane ruffles (Ridley et al., 1992), whereas Cdc42 induces filopodia (Kozma et al., 1995). Both lamellipodia and filopodia are actin-rich membrane protrusions, which define the leading edge of a migrating cell. Earlier studies suggest that Rac1 and Cdc42 are restricted to the front of migrating cells and are responsible for membrane protrusion, whereas RhoA is localized to the rear of migrating cells and is responsible for tail retraction (Kraynov et al., 2000; Nalbant et al., 2004; Raftopoulou and Hall, 2004; Sastry et al., 2006).
However, studies using fluorescence resonance energy transfer (FRET) to monitor the distribution of active RhoA in randomly migrating cells concluded that active RhoA also localizes to the front of migrating cells (Kurokawa and Matsuda, 2005; Pertz et al., 2006), consistent with findings that RhoA can induce membrane protrusion and ruffling in epithelial cells (Kawano et al., 1999; O'Connor et al., 2000). It is now believed that active RhoA can localize to the front of migrating cells in a cell type and/or signal specific manner. In randomly migrating cells, high levels of active RhoA are found at the cell leading edge. In contrast, low levels of active RhoA are found in platelet-derived growth factor (PDGF)-induced membrane protrusions (Pertz et al., 2006).
Tumor invasion and metastasis involve uncontrolled cell migration. Accumulating evidence suggests that activation of small GTPase proteins RhoA and RhoC is critical for tumor invasion and/or metastasis in breast carcinoma (Clark et al., 2000; Hakem et al., 2005; Kleer et al., 2005; Kleer et al., 2002; Kleer et al., 2004; Kusama et al., 2006; Pille et al., 2005; Simpson et al., 2004; van Golen et al., 1999; van Golen et al., 2000). RhoA and RhoC are activated by guanine nucleotide exchange factors (GEFs) and inactivated by GTPase-activating proteins (GAPs). However, the GEFs that are responsible for RhoA/RhoC activation in breast cancer cells remain to be identified. In this article, we have demonstrated that MyoGEF can activate RhoA and RhoC in an invasive breast cancer cell line MDA-MB-231 cells. MyoGEF colocalizes with nonmuscle myosin IIA (NMIIA) to the front of migrating cells. In addition, depletion of MyoGEF in MDA-MB-231 cells by RNAi impairs cell polarity and invasion. Further, MyoGEF is highly expressed in invasive breast cancer cell lines as well as in infiltrating ductal carcinomas. Our results indicate that MyoGEF plays an important role in regulating the polarity and invasion activity of invasive breast cancer cells by activating RhoA/RhoC.
RESULTS
MyoGEF is required for the invasion activity of MDA-MB-231 cells
To correlate the expression level of MyoGEF with the invasive potential of breast cancer cells, immunoblot analysis with an antibody specific for MyoGEF was carried out to examine MyoGEF protein levels in breast cancer cell lines. As shown in Figure 1A, MyoGEF is highly expressed in invasive breast cancer cells MDA-MB-231 and MDA-MB-435S, but is not detectable in noninvasive (MDA-MB-361 and MCF-7) or poorly invasive (MDA-MB-468) breast cancer cells. This finding indicates that MyoGEF may play a critical role in regulating the invasion activity of breast cancer cells. Therefore, we used RNAi to deplete MyoGEF in MDA-MD-231 cells and then examined the effect of MyoGEF depletion on the invasion activity of MDA-MB-231 cells. At 48 h after transfection with control or MyoGEF siRNA, transfected MDA-MB-231 cells were subjected to Matrigel invasion assays. As shown in Figures 1C and 1D, depletion of MyoGEF dramatically inhibited MDA-MB-231 cell invasion activity. Nonmuscle myosin II (NMII) has also been implicated as an important regulator of cell invasion and migration (Conti and Adelstein, 2008; Lo et al., 2004; Meshel et al., 2005; Vicente-Manzanares et al., 2007). In addition, MyoGEF can interact with NMIIA (Wu et al., 2006). Therefore, we also depleted both NMIIA and MyoGEF to examine whether NMIIA and MyoGEF synergistically regulate cell invasion. As shown in Figures 1C and 1D, RNAi-mediated depletion of both NMIIA and MyoGEF further decreased the invasion activity of MDA-MB-231 cells, suggesting that MyoGEF and NMIIA may cooperate in regulating breast cancer cell invasion.
Figure 1. MyoGEF is required for the invasion activity of MDA-MB-231 cells.

(A) Immunoblot analysis with anti-MyoGEF antibody shows that MyoGEF is expressed in MDA-MB-231 and MDA-MB-435S cells, but not in MDA-MB-361, MDA-MB-468, and MCF-7 cells. (B) Immunoblot analysis confirms the depletion of MyoGEF in MDA-MB-231 cells by RNAi. (C) MDA-MB-231 cells depleted of MyoGEF and/or NMIIA were subjected to Matrigel invasion assays. (D) Images in (C) were quantitated by using the NIH ImageJ program. (E) Immunohistochemical analysis of a breast cancer tissue array with MyoGEF antibody. Three arrays were analyzed independently and similar results were obtained. Immunohistochemistry with preimmune serum shows light, background straining (data not shown). Images in (C) and (E) were taken by using a 20x objective (Leica DMI 6000 B microscope).
We then asked whether MyoGEF protein levels increase in invasive breast carcinomas. To this end, we carried out immunohistochemistry with MyoGEF antibody to examine MyoGEF protein levels in a human breast cancer tissue array (US Biomax, Inc.). This tissue array contains 21 cases of infiltrating ductal carcinomas with normal and adjacent tissues. As shown in Figure 1E, MyoGEF protein levels significantly increase in infiltrating ductal carcinomas as compared to normal or adjacent breast tissues (compare panels a and c with panel b). We found that 17 out of the 21 cases of infiltrating ductal carcinomas (n=17/21 cases) show a dramatic increase in MyoGEF protein levels.
Activation of RhoA and RhoC by MyoGEF
To determine whether MyoGEF can activate RhoA and RhoC in breast cancer cells, MDA-MB-231 cells treated with control or MyoGEF siRNAs were subjected to RBD (rhotekin Rho-binding domain) or PBD (PAK1/p21 binding domain) pull-down assays to estimate the activity of the small GTPase proteins, including RhoA, RhoC, Rac1, and Cdc42. RBD binds to activated (GTP-bound) RhoA and RhoC, whereas PBD binds to activated (GTP-bound) Rac1 and Cdc42 (Abe et al., 2000; Arthur et al., 2002; Liu and Burridge, 2000). As shown in Figures 2C and 2D, RBD-conjugated agarose beads could precipitate a significant amount of GTP-RhoA and RhoC from MDA-MB-231 cells treated with control siRNA (lane 1 in Figures 2C and 2D), whereas depletion of MyoGEF by RNAi dramatically decreased the amount of active RhoA and RhoC precipitated by RBD-beads (lane 2 in Figures 2C and 2D). In contrast, MyoGEF depletion did not dramatically affect the amount of active Rac1 and Cdc42 pulled down by PBD-conjugated agarose beads (Figures 2E and 2F). These results suggest that MyoGEF can activate RhoA and RhoC, but not Rac1 and Cdc42, in MDA-MB-231 cells.
Figure 2. Depletion of MyoGEF represses RhoA and RhoC activation in MDA-MB-231 cells.

(A) Immunoblot analysis confirms the depletion of MyoGEF in MDA-MB-231 cells by RNAi. (B) The image in (A) was quantitated by using the NIH ImageJ program to estimate the efficiency of MyoGEF depletion in MDA-MB-231 cells by RNAi. (C-F) Depletion of MyoGEF decreases the amount of active RhoA (C) and RhoC (D), but not Rac1 (E) and CDc42 (F), in MDA-MB-231 cells. ~6% of transfected cell lysates were used as control to estimate the amount of total RhoA, RhoC, Rac1, and Cdc42. (G) The images in (C), (D), (E), and (F) were quantitated by using the NIH ImageJ program.
To further characterize the in vitro activity of MyoGEF towards RhoA, RhoC, Rac1, and Cdc42, HeLa cells expressing Myc-MyoGEF was subjected to immunoprecipitation with anti-Myc-antibody, and then the immunoprecipitated Myc-MyoGEF was used for the analysis of MyoGEF activity towards RhoA, RhoC, Rac1, or Cdc42 with a fluorescence-based GTPase in vitro assay. As shown in Figure 3, Myc-MyoGEF could activate RhoA (3A), RhoC (3B), and Rac1 (3C), but not Cdc42 (3D). Consistent with these findings, in vitro pull-down assays demonstrated that full-length MyoGEF (ThioHis-MyoGEF) or a truncated MyoGEF fragment 71-388 (GST-71-388; containing the DH domain) could bind to RhoA and RhoC that were preloaded with GTP or GDP (Figures 3E and 3F), indicating that MyoGEF could bind to GTP- and GDP-bound RhoA and RhoC. In contrast, MyoGEF only bound to Rac1 that was preloaded with GTP (Figure 3G), suggesting that MyoGEF differentially bound to the active form of Rac1 (GTP-Rac1). However, RNAi-mediated depletion of MyoGEF in HeLa and MDA-MB-231 cells decreased the amount of activated RhoA/RhoC (but not Rac1and Cdc42) (Figure 2) (Wu et al., 2006). Therefore, our results indicate that RhoA and RhoC are most likely the physiological effectors of MyoGEF.
Figure 3. In vitro activation of RhoA, RhoC, and Rac1, but not Cdc42, by MyoGEF.

(A-D) The immunoprecipitated Myc-MyoGEF from transfected HeLa cells could activate RhoA (A), RhoC( B), and Rac1 (C), but not Cdc42 (D) in a fluorescence-based GEF assay. (E) ThioHis-MyoGEF (full-length) could bind both GDP-RhoA (lane 4) and GTP-RhoA (lane 5). (F) A MyoGEF fragment (amino acids 71-388) that contain the DH domain could bind both GDP-RhoC (lane 4) and GTP-RhoC (lane 5). (G) ThioHis-MyoGEF could bind GTP-Rac1 (lane 5) but not GDP-Rac1 (lane 4). D, preloaded with GDP; T, preloaded with GTP.
MyoGEF colocalizes with actin-myosin filaments at the cell leading edge
To determine the localization of MyoGEF in migrating MDA-MB-231 cells, immunofluorescence with anti-MyoGEF antibody was carried out in fixed MDA-MB-231 cells after ~6 h of culture on fibronectin-coated coverslips. As shown in Figure 4A, endogenous MyoGEF colocalized with actin filaments at the front of migrating cells (arrowheads in panels a-c and a'-c'). Consistent with these observations, exogenously expressed GFP-MyoGEF also colocalized with actin filaments at the cell leading edge (Figure 4C, panels a-c and a'-c'). It should be noted that expression of exogenous GFP-MyoGEF could induce the formation of thick actin bundles (panels b' and c'). In addition, GFP-MyoGEF formed filament-like structures that overlap with these thick actin bundles (arrowheads in panel c'). These results indicate that MyoGEF can localize to the cell leading edge, where it induces actin filament formation.
Figure 4. MyoGEF colocalizes with actin-myosin filaments at the cell leading edge.

(A) MDA-MB-231 cells were subjected to immunofluorescence with anti-MyoGEF antibody (green) and rhodaminephalloidin (red). (B) Immunoblot analysis of total cell lysates from MDA-MB-231 with anti-MyoGEF antibody. Note that a single band was recognized by MyoGEF antibody in MDA-MB-231 cell lysates. (C) Exogenously expressed GFP-MyoGEF (green) colocalizes with actin filaments (red) in transfected MDA-MB-231 cells. (D) Exogenously expressed GFP-IIA (green) colocalizes with endogenous MyoGEF (red) at the cell leading edge of transfected MDA-MB-231 cells. (E) Exogenously expressed GFP-MyoGEF (green) colocalizes with endogenous NMIIA (red) at the cell leading edge of transfected MDA-MB-231 cells. Bars, 10 μm.
NMII has been shown to localize to the cell leading edge, and it plays a critical role in regulating cell polarity and motility (Conti and Adelstein, 2008; Kolega, 2006; Sandquist et al., 2006; Vicente-Manzanares et al., 2007). We also reported previously that MyoGEF interacts with NMIIA (Wu et al., 2006). Therefore, we also examined whether MyoGEF and NMIIA colocalized in randomly migrating MDA-MB-231 cells. MDA-MB-231 cells expressing GFP-tagged nonmuscle myosin heavy chain IIA (GFP-IIA) were subjected to immunofluorescence with anti-MyoGEF antibody. As shown in Figure 4D, MyoGEF colocalized with GFP-IIA at the cell leading edge (arrowheads in panels a'-d'). Consistently, exogenously expressed GFP-MyoGEF also colocalized with endogenous NMIIA in transfected MDA-MB-231 cells as indicated in Figure 4E (arrowheads in panels a'-d').
MyoGEF preferentially interacts with NMIIA in MDA-MB-231 cells
We previously showed that MyoGEF interacts with NMIIA in vitro (Wu et al., 2006). Three isoforms of the nonmuscle myosin heavy chain (NMHC), IIA, IIB, and IIC, have been identified in humans and mice (Bresnick, 1999; Conti and Adelstein, 2008; Golomb et al., 2004; Krendel and Mooseker, 2005; Sellers, 2000). MDA-MB-231 cells express IIA and IIB, but not IIC (Betapudi et al., 2006). To confirm the interaction between MyoGEF and NMII, MDA-MB-231 cells expressing Myc-MyoGEF were subjected to immunoprecipitation with anti-Myc antibody followed by immunoblot analysis with anti-IIA or anti-IIB antibodies. As shown in Figure 5A, endogenous NMIIA, but not NMIIB, could be co-immunoprecipitated with Myc-MyoGEF from total cell lysates of transfected MDA-MB-231 cells, indicating that MyoGEF can preferentially interact with NMIIA in MDA-MB-231 cells. To further characterize the interaction between MyoGEF and NMIIA, we generated different truncated versions of MyoGEF (Figure 5B). Plasmids encoding these Myc-tagged MyoGEF fragments were transfected into a HeLa cell line that only expresses NMIIA, but not NMIIB and NMIIC (Wei and Adelstein, 2000). As shown in Figure 5C, Myc-MyoGEF full-length, Myc-PH, Myc-1-409, and Myc-1-500 could immunoprecipitate a significant amount of NMIIA, suggesting that the PH domain as well as the 351-409 region that is C-terminal to the DH domain are required for interaction with NMIIA.
Figure 5. MyoGEF interacts with NMIIA.

(A) MDA-MB-231 cells expressing Myc-MyoGEF were subjected to immunoprecipitation with anti-Myc antibody followed by immunoblot analysis with anti-IIA or anti-IIB antibodies. Note that Myc-MyoGEF binds to NMIIA but not NMIIB. (B) Schematic diagram of MyoGEF fragments that were used in (C). (C) Interactions between Myc-tagged MyoGEF fragments and endogenous NMIIA. Full-length MyoGEF (lane 3) as well as MyoGEF fragments Myc-PH (lane 5), Myc-1-409 (lane 8), and Myc-1-500 (lane 9) could pull down a significant amount of endogenous NMIIA. Note that cell lysate from lane 3 was also used for immunoprecipitation with normal IgG (lane 2). ~5% of cell lysates were loaded.
Depletion of MyoGEF by RNAi impairs cell polarity
Rho GTPase signaling is essential for cell polarity and cell migration (Ridley et al., 2003). Localization of MyoGEF to the cell leading edge (Figure 4) suggests that MyoGEF may play a role in regulating cell polarity. Therefore, we examined whether depletion of MyoGEF had an impact on the polarity of MDA-MB-231 cells. As shown in Figures 6A and 6B, only ~25% of MDA-MB-231 cells treated with control siRNA (n=200 cells) remain unpolarized after 6 h of culture on fibronectin-coated coverslips. However, ~60% of MDA-MB-231 cells (n=250 cells) did not polarize following MyoGEF depletion (Figures 6A and 6B). Consistent with these findings, MyoGEF localized to the cell leading edge in MDA-MB-231 cells transfected with control siRNA, and these cells showed polarized actin polymerization (Figure 6C; arrowheads in panels a-c). In contrast, depletion of MyoGEF by RNAi decreased MyoGEF protein levels (Figure 6C; compare panel b with panel e), and MyoGEF-depleted cells did not show polarized actin organization (panels d and f). Notably, actin filaments were predominantly assembled at the periphery of MyoGEF-depleted cells (Figure 6C; panels d and f). These results suggest that MyoGEF is required for cell polarization in MDA-MB-231 cells, even though MyoGEF-depletion did not dramatically decrease actin filament formation (based on the intensity of actin filament staining; data not shown).
Figure 6. Depletion of MyoGEF by RNAi impairs MDA-MB-231 cell polarity.

(A) MDA-MB-231 cells treated with control siRNA (siCont) or MyoGEF siRNA (siMyoGEF) for 48 h were trypsinized, replated on fibronectin-coated coverslips, and cultured for an additional 6 h. Note that cells depleted of MyoGEF did not polarize. (B) Quantitation of nonpolarized MDA-MB-231 cells treated with control or MyoGEF siRNAs. (C) MDA-MB-231 cells treated with siCont or siMyoGEF were subjected to immunofluorescence with MyoGEF antibody (red) and FITC-phalloidin (green). (D) MDA-MB-231 cells treated with siCont or siMyoGEF were stained with antibodies specific for p-MRLC (green) and NMIIA (red). (E) MDA-MB-231 cells treated with siCont or siMyoGEF were stained with antibodies specific for p-MRLC (green) and NMIIB (red). Bar in (A), 80 μm; Bars in (C), (D), and (E), 10 μm
We then examined the effect of MyoGEF-depletion on myosin filament organization. MDA-MB-231 cells treated with control siRNA (siCont) were able to polarize, and a significant amount of p-MRLC (phosphorylated myosin regulatory light chain), NMIIA, and NMIIB localized at the cell leading edge of randomly migrating cells (arrowheads in Figure 6D, a-c and Figure 6E, a-c). In contrast, cells treated with MyoGEF siRNA did not polarize (Figure 6D, d-f and Figure 6E, d-f). NMIIA and NMIIB filaments were predominantly assembled at the periphery of MyoGEF-depleted cells (arrowheads in Figure 6De and Figure 6Ee). Consistently, p-MRLC staining was also found predominantly at the cell periphery (arrowheads in Figure 6Dd and Figure 6Ed). However, we did not observe a dramatic decrease in NMIIA, NMIIB, and p-MRLC staining. These results suggest that MyoGEF depletion impairs cell polarity as well as polarized actin-myosin organization without having an obvious effect on overall actin-myosin filament formation.
NMII filaments are required for polarized distribution of MyoGEF
NMII plays a central role in regulating cell polarity and motility (Conti and Adelstein, 2008; Lo et al., 2004; Meshel et al., 2005; Vicente-Manzanares et al., 2007). In addition, MyoGEF can bind to NMIIA (Figure 5) (Wu et al., 2006). Further, MyoGEF and NMIIA colocalize to the cell leading edge in MDA-MB-231 cells (Figure 4). Therefore, we used RNAi to deplete NMIIA in MDA-MB-231 cells and then carried out immunofluorescence with anti-MyoGEF antibody to examine the effect of NMIIA depletion on MyoGEF localization. As shown in Figure 7A, depletion of NMIIA by RNAi led to cell spreading indiscriminately and the disruption of polarized MyoGEF localization (arrowheads in Figure 7Ab). In contrast, depletion of NMIIB did not impair cell polarity and the polarized distribution of MyoGEF in MDA-MB-231 cells (data not shown), even though it has been demonstrated that depletion of NMIIB also decreases MDA-MB-231 cell migration (Betapudi et al., 2006). These results indicate that NMIIA is required for polarized localization of MyoGEF during cell migration.
Figure 7. NMIIA is required for polarized localization of MyoGEF as well as the formation of MyoGEF-induced actin bundles.

(A) MDA-MB-231 cells treated with control siRNA (siCont; panel a) or NMIIA siRNA (siIIA; panel b) were subjected to immunofluorescence with anti-MyoGEF antibody. (B) MDA-MB-231 cells treated with siCont or siIIA were subjected to immunoblot analysis with antibodies specific for NMIIA or β-tubulin. (C-D) A plasmid encoding GFP-MyoGEF was cotransfected into HeLa cells with siCont or siIIA. The transfected cells were subjected to immunofluorescence with anti-IIA antibody (C) or phalloidin (D). Bars, 10 μm.
To further confirm that NMIIA is required for MyoGEF function in other epithelial cells, we transfected NMIIA siRNA and a plasmid encoding GFP-MyoGEF into a HeLa cell line that expresses only NMIIA (Golomb et al., 2004; Wei and Adelstein, 2000), and then examined the formation of actin-myosin bundles induced by exogenously expressed GFP-MyoGEF. As shown in Figure 7C, GFP-MyoGEF induced the formation of massive myosin bundles in the presence of control siRNA (arrowheads in panels a-c), and GFP-MyoGEF colocalized with these NMIIA bundles (arrowheads in panel c). In contrast, RNAi-mediated depletion of NMIIA completely abrogated the formation of massive myosin bundles induced by GFP-MyoGEF (Figure 7C; panels d-f). We also examined the effect of NMIIA depletion on the formation of actin bundles induced by GFP-MyoGEF. As shown in Figure 7D, depletion of NMIIA by siRNA dramatically reduced the formation of massive actin bundles induced by GFP-MyoGEF (compare panels a-c with panels d-f). These results suggest that the presence of NMIIA and MyoGEF-myosin II interaction may be critical for MyoGEF localization and function.
Expression of dominant negative mutants N19RhoA or N19RhoC interferes with MyoGEF-induced actin polymerization
To determine whether RhoA and/or RhoC are required for MyoGEF-induced actin bundles, a plasmid encoding GFP-MyoGEF was co-transfected into HeLa cells with plasmids encoding the dominant negative mutants of the small GTPase proteins RhoA, RhoC, Rac1, or Cdc42. Actin filaments were visualized by staining with rhodamine-conjugated phalloidin. Co-transfection of an empty vector did not affect the formation of massive actin bundles induced by GFP-MyoGEF (Figure 8; arrowheads in panel b). In contrast, transfection with plasmids encoding N19RhoA (d-f) or N19RhoC (g-i) significantly decreased the formation of massive actin bundles induced by GFP-MyoGEF (compare panels e and h with panel b). These findings indicate that RhoA or RhoC is required for MyoGEF-induced actin bundles. However, expression of dominant negative mutants N17Rac1 or N17Cdc42 did not significantly decrease the formation of actin bundles induced by GFP-MyoGEF (Figure 8; panels j-o), even though expression of N17Rac1 induced the formation of filopodia in GFP-MyoGEF-expressing cells, and it appeared that GFP-MyoGEF predominantly localized to cell periphery in the presence of N17Rac1 (compare panels a-c with panels j-i). In addition, MyoGEF can specifically bind to GTP-Rac1 (see Figure 3G). Therefore, our results suggest that MyoGEF may act as a downstream effector of Rac1 or as a carrier of GTP-Rac1 to potential target sites. Interestingly, it has been reported previously that a Rho-specific GEF, DBL's big sister (DBS), can bind to GTP-Rac1 and act as a downstream effector of GTP-Rac1 (Cheng et al., 2004). However, it is yet to be determined whether MyoGEF activity towards RhoA and RhoC can be regulated by Rac signaling.
Figure 8. Expression of dominant negative mutants N19RhoA and N19RhoC inhibits the formation of MyoGEF-induced actin bundles.
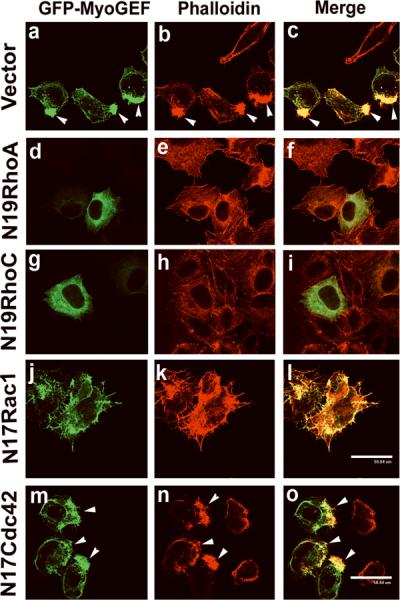
A plasmid encoding GFP-MyoGEF was cotransfected into HeLa cells with an empty vector (a-c) or plasmids encoding N19RhoA (d-f), N19RhoC (g-i), N17Rac1 (j-l), or N17Cdc42 (m-o). Note that co-transfection of N19RhoA or N19RhoC decreases the formation of massive actin bundles induced by GFP-MyoGEF. Bars, 60 μm.
DISCUSSION
In this article, we have demonstrated that MyoGEF can activate RhoA and RhoC in a breast cancer cell line MDA-MB-231. We also show that MyoGEF plays a role in regulating the polarity and invasion activity of MDA-MB-231 cells. In addition, polarized distribution of MyoGEF at the cell leading edge is dependent on the integrity of the actin-myosin cytoskeleton. Importantly, MyoGEF is highly expressed in invasive (MDA-MB-231 and MDA-MB-435s) breast cancer cells, but is not detectable in noninvasive (MDA-MB-361 and MCF-7) or poorly invasive (MDA-MB-468) breast cancer cells. Expression of dominant negative mutants N19RhoA or N19RhoC interferes with the formation of MyoGEF-induced actin-myosin bundles. Our results suggest that MyoGEF and NMII cooperate to regulate cell polarity and motility in invasive breast cancer cells.
Rho GTPase signaling has been implicated in regulating cell polarity and motility (Arthur and Burridge, 2001; Kurokawa et al., 2005; Worthylake and Burridge, 2003; Yamana et al., 2006). Rac1 is widely considered as a key regulator of cell migration in different cell lines and organisms (Ridley, 2001; Ridley et al., 2003). However, accumulating evidence also suggests that Rac1 activity is not required for cell migration in a number of cell lines, such as colon carcinoma cells, rat fibroblast, and macrophages (Ahram et al., 2000; O'Connor et al., 2000; Wells et al., 2004). Instead, it has been demonstrated that RhoC is a key pro-metastatic protein that is essential for cell migration and invasion in breast cancer cells (Clark et al., 2000; Hakem et al., 2005; Kleer et al., 2005; Simpson et al., 2004; van Golen et al., 2000).
However, it is poorly understood whether there are specific GEFs that are responsible for RhoA and/or RhoC activation in breast cancer cells. We have shown that MyoGEF can activate both RhoA and RhoC in MDA-MB-231 cells (Figures 2 and 3). Depletion of MyoGEF by RNAi impairs cell polarity and invasion activity (Figures 1 and 6). Thus, our findings point to a mechanism by which MyoGEF regulates the polarity and invasion activity of MDA-MB-231 cells through activation of RhoA and/or RhoC. Although a number of studies suggest an essential role for RhoC in breast cancer metastasis, our results show that MyoGEF can activate both RhoA and RhoC in MDA-MB-231 cells. It remains to be determined whether both RhoA and RhoC are required for MyoGEF-mediated regulation of cell migration. Nonetheless, a line of evidence suggests that RhoA also plays a critical role in regulating the motility and invasion of MDA-MB-231 cells (Kusama et al., 2006; Pille et al., 2005; Pille et al., 2006; Sahai et al., 2007). Therefore, it is likely that both RhoA and RhoC are important for MyoGEF-mediated regulation of MDA-MB-231 cell invasion. Another possibility is that MyoGEF-RhoA and MyoGEF-RhoC may differentially function in different breast cancer cells.
RhoA and RhoC can induce actin-myosin filament formation through activation of ROCK and mDia. ROCK inhibits myosin phosphatase and directly phosphorylates myosin regulatory light chains, resulting in an increase in myosin contractile activity (Kimura et al., 1996; Matsui et al., 1996). mDia can induce actin polymerization through association with the barbed end of growing actin filaments (Chang and Peter, 2002; Higashida et al., 2004; Li and Higgs, 2003; Tominaga et al., 2000; Watanabe et al., 1999; Zigmond, 2004). We observed the reorganization of the actin-myosin cytoskeleton following MyoGEF depletion, even though depletion of MyoGEF does not dramatically reduce actinmyosin filament formation (Figure 6). It is likely that other actin-myosin promoting pathways can compensate for the loss of RhoA/RhoC activity resulting from MyoGEF depletion. For instance, in addition to ROCK, myosin light chain kinase (MLCK) can also phosphorylate MRLC, thus increasing the phosphorylation of MRLC as well as myosin contractile activity. Importantly, it has been reported that MLCK predominantly phosphorylates MRLC at the cell periphery, whereas ROCK is responsible for MRLC phosphorylation in the cell center (Totsukawa et al., 2004). Consistent with this conclusion, we found that actin-myosin filaments are predominantly assembled at the cell periphery in MyoGEF-depleted cells (Figure 6).
Although Rho-ROCK signaling has been implicated in cell motility in a number of cell lines as well as in animal models, our results indicated that treatment with Y27632 does not affect cell polarity and polarized distribution of MyoGEF in MDA-MB-231 cells (data not shown). Instead, MDA-MB-231 cells often show an elongated morphology indicating that the tail retraction is not completed in the presence of Y27632 (data not shown), whereas depletion of MyoGEF impairs cell polarity, leading to rounded cell morphology (Figure 6). These results suggest that Rho-ROCK signaling may be dispensable for MyoGEF function in regulating cell polarity and invasion activity. These observations are consistent with the finding that RhoA-ROCK is required for the invasion activity of MDA-MB-435S but not MDA-MB-231 cells (Demou et al., 2005). Furthermore, a correlation between the invasion activity of different tumor cells and the requirement of Rho-ROCK signaling has been suggested (Sahai and Marshall, 2003). One of the future challenges is to determine whether Rho-mDia signaling is required for MyoGEF function in the regulation of cell polarity and invasion.
NMII plays an essential role in the regulation of cell polarity and cell migration. MyoGEF interacts with NMIIA and both proteins colocalize at the cell leading edge (Figure 4). Disruption of NMIIA by RNAi impairs cell polarity as well as polarized distribution of MyoGEF (Figure 7). In addition, exogenously expressed GFP-MyoGEF forms filament-like structures that overlap with actinmyosin bundles (Figure 4). Depletion of NMIIA completely abrogates the formation of MyoGEF-induced massive actin bundles (Figure 7C). These findings indicate that NMIIA may act as a scaffold to anchor MyoGEF to the cell leading edge. In turn, MyoGEF locally activates RhoA and/or RhoC at the front of migrating cells, thereby forming a positive MyoGEF-NMIIA loop and promoting cell polarization and invasion.
MATERIALS AND METHODS
Plasmids and cell culture
pEGFP-MyoGEF, pCS3-MyoGEF, and pEGFP-NMHC-IIA were described previously (Wei and Adelstein, 2000; Wu et al., 2006). MyoGEF fragments were cloned into XhoI/XbaI sites of pCS3+MT vector. Breast cancer cell lines MDA-MB-231, MDA-MB-435S, MDA-MB-361, MDA-MB-468, and MCF-7 were purchased from ATCC (Manassas, VA). MDA-MB-231, MDA-MB-435S, MDA-MB-361, and MDA-MB-468 were grown in Leibovitz's L-15 Medium supplemented with 10% fetal bovine serum. HeLa cells were purchased from Clontech. HeLa and MCF-7 cells were grown in DMEM supplemented with 10% fetal bovine serum. Transfection was done with Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen, Carlsbad, CA). MyoGEF siRNA has been described previously (Wu et al., 2006).
Immunoprecipitation and immunoblotting
Immunoprecipitation was carried out as described previously (Asiedu et al., 2008; Asiedu et al., 2009; Wei, 2005), excepting that 2 mM ATP and 2mM MgCl2 were included in the lysis buffer. The following primary antibodies were used: mouse anti-Myc (9E10, 1:2000; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-β-tubulin (1:2000; Santa Cruz), mouse anti-RhoA (1:200; Santa Cruz), goat anti-RhoC (1:100; Santa Cruz), mouse anti-NMIIB (1:1000; Developmental Studies Hybridoma Bank, Iowa City, IA), and rabbit anti-MyoGEF (1:200).
Immunofluorescence
Immunofluorescence was carried out as described previously (Asiedu et al., 2009). MDA-MB-231 cells transfected with plasmids or siRNA were trypsinized, cultured on coverslips for an additional 6-12 h, and then fixed with 4% paraformaldehyde. The primary antibodies used for immunofluorescence were as follows: rabbit polyclonal NMIIA antibody (1:1000), mouse monoclonal p-MRLC antibody (1:200; Cell Signaling, Beverly, MA), and rabbit polyclonal MyoGEF antibody (1:100). The secondary antibodies rhodamine goat anti-mouse IgG (1:500) and rhodamine goat anti-rabbit IgG (1:500) were purchased from Invitrogen. The nuclei were visualized by DAPI (Sigma, St. Louis, MO). Actin filaments were stained with rhodamine-phalloidin (Invitrogen). Images were taken using a Leica DMI 6000 B microscope (Leica, Deerfield, IL) and processed by blind deconvolution. To determine the cell polarity, long (L) and short (S) axes of individual cells were measured using the NIH ImageJ program. Cells were counted as polarized (L/S ratio > 2.0) or nonpolarized (L/S <2.0).
Immunohistochemistry
The breast cancer tissue arrays were purchased from US Biomax (US Biomax, Inc., Rockville, MD; Cat # BC08032). After incubation at 60°C for 2h, the paraffin-embedded sections were deparaffinized in three changes of xylene and then rehydrated using graded alcohols. Antigen retrieval were done with 10 mM citrate buffer, pH 6, in the microwave for 30 min and allowed to cool to room temperature followed by a PBS wash. The slides were then blocked with normal goat serum and incubated with anti-MyoGEF antibody (1:100) at room temperature for 1h. Preimmune serum was used on a duplicate slide in place of MyoGEF antibody as a negative control. After washing three times with PBS, the slides were incubated with biotinylated secondary goat anti-rabbit antibody at room temperature for 30 min. After washing three times with PBS, the slides were stained with the ABC/DAB Elite kit (goat IgG type; Vectorlabs, Burlingame, CA). Finally, the slides were counterstained with hematoxylin, dehydrated, cleared, and then mounted with Permount mounting medium (Fisher Scientific, Pittsburgh, PA).
Matrigel invasion assays
Transfected MDA-MB-231 cells were trypsinized and approximately 1×105 cells (in Leibovitz's L-15 medium containing 3% of BSA) were seeded on the upper wells of Biocoat Matrigel chambers (BD Biosciences, Bedford, MA). The lower wells were filled with Leibovitz's L-15 medium containing 10% FBS. The transfected cells then underwent chemoattracting across the matrigel and filter (pore size: 8 μ) to the lower surface of the transwells for 22 h. The nonmigrating cells on the upper chambers were removed by a cotton swab. The migrating cells on the lower surface were fixed in 4% paraformaldehyde, stained with 1% crystal violet, and then photographed with a 20x objective. Data were collected from three independent experiments, each done in triplicate.
RhoA, RhoC, Rac1, and Cdc42 activation assays
RhoA, RhoC, Rac1, and Cdc42 activation assays were described previously (Asiedu et al., 2008; Glaven et al., 1999; Liu and Burridge, 2000; Wu et al., 2006).
In vitro guanine nucleotide exchange analysis
The GEF exchange assay was described previously (Asiedu et al., 2008).
ACKNOWLEDGEMENTS
We thank Dr. Robert S. Adelstein and Dr. Mary Anne Conti for critical reading and comments on the manuscript. We thank Dr. Keith Burridge (University of North Carolina at Chapel Hill, Chapel Hill, NC) and Dr. Rick Cerione (Cornell University, Ithaca, NY) for providing GST-RBD and GST-PBD plasmids. This publication was made possible by NIH Grant Number P20 RR015563 from the National Center for Research Resources. This work was also supported by National Institutes of Health Grant k22 HL071542 (to QW) as well as a fund from Terry C. Johnson Center for Basic Cancer Research (to QW). This is contribution 09-136-J from the Kansas Agricultural Experiment Station, Manhattan, Kansas.
REFERENCES
- Abe K, Rossman KL, Liu B, Ritola KD, Chiang D, Campbell SL, et al. Vav2 is an activator of Cdc42, Rac1, and RhoA. J Biol Chem. 2000;275:10141–9. doi: 10.1074/jbc.275.14.10141. [DOI] [PubMed] [Google Scholar]
- Ahram M, Sameni M, Qiu RG, Linebaugh B, Kirn D, Sloane BF. Rac1-induced endocytosis is associated with intracellular proteolysis during migration through a three-dimensional matrix. Exp Cell Res. 2000;260:292–303. doi: 10.1006/excr.2000.5031. [DOI] [PubMed] [Google Scholar]
- Arthur WT, Burridge K. RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Mol Biol Cell. 2001;12:2711–20. doi: 10.1091/mbc.12.9.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur WT, Ellerbroek SM, Der CJ, Burridge K, Wennerberg K. XPLN, a guanine nucleotide exchange factor for RhoA and RhoB, but not RhoC. J Biol Chem. 2002;277:42964–72. doi: 10.1074/jbc.M207401200. [DOI] [PubMed] [Google Scholar]
- Asiedu M, Wu D, Matsumura F, Wei Q. Phosphorylation of MyoGEF on Thr-574 by Plk1 Promotes MyoGEF Localization to the Central Spindle. J Biol Chem. 2008;283:28392–400. doi: 10.1074/jbc.M801801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asiedu M, Wu D, Matsumura F, Wei Q. Centrosome/Spindle Pole-associated Protein Regulates Cytokinesis via Promoting the Recruitment of MyoGEF to the Central Spindle. Mol Biol Cell. 2009;20:1428–40. doi: 10.1091/mbc.E08-01-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betapudi V, Licate LS, Egelhoff TT. Distinct roles of nonmuscle myosin II isoforms in the regulation of MDA-MB-231 breast cancer cell spreading and migration. Cancer Res. 2006;66:4725–33. doi: 10.1158/0008-5472.CAN-05-4236. [DOI] [PubMed] [Google Scholar]
- Bresnick AR. Molecular mechanisms of nonmuscle myosin-II regulation. Curr Opin Cell Biol. 1999;11:26–33. doi: 10.1016/s0955-0674(99)80004-0. [DOI] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–79. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- Chang F, Peter M. Cell biology. Formins set the record straight. Science. 2002;297:531–2. doi: 10.1126/science.1074649. [DOI] [PubMed] [Google Scholar]
- Cheng L, Mahon GM, Kostenko EV, Whitehead IP. Pleckstrin homology domain-mediated activation of the rho-specific guanine nucleotide exchange factor Dbs by Rac1. J Biol Chem. 2004;279:12786–93. doi: 10.1074/jbc.M313099200. [DOI] [PubMed] [Google Scholar]
- Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–5. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- Conti MA, Adelstein RS. Nonmuscle myosin II moves in new directions. J Cell Sci. 2008;121:11–8. doi: 10.1242/jcs.007112. [DOI] [PubMed] [Google Scholar]
- Demou ZN, Awad M, McKee T, Perentes JY, Wang X, Munn LL, et al. Lack of telopeptides in fibrillar collagen I promotes the invasion of a metastatic breast tumor cell line. Cancer Res. 2005;65:5674–82. doi: 10.1158/0008-5472.CAN-04-1682. [DOI] [PubMed] [Google Scholar]
- Glaven JA, Whitehead I, Bagrodia S, Kay R, Cerione RA. The Dbl-related protein, Lfc, localizes to microtubules and mediates the activation of Rac signaling pathways in cells. J Biol Chem. 1999;274:2279–85. doi: 10.1074/jbc.274.4.2279. [DOI] [PubMed] [Google Scholar]
- Golomb E, Ma X, Jana SS, Preston YA, Kawamoto S, Shoham NG, et al. Identification and characterization of nonmuscle myosin II-C, a new member of the myosin II family. J Biol Chem. 2004;279:2800–8. doi: 10.1074/jbc.M309981200. [DOI] [PubMed] [Google Scholar]
- Hakem A, Sanchez-Sweatman O, You-Ten A, Duncan G, Wakeham A, Khokha R, et al. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. 2005;19:1974–9. doi: 10.1101/gad.1310805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashida C, Miyoshi T, Fujita A, Oceguera-Yanez F, Monypenny J, Andou Y, et al. Actin polymerization-driven molecular movement of mDia1 in living cells. Science. 2004;303:2007–10. doi: 10.1126/science.1093923. [DOI] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. RHO GTPases: Biochemistry and Biology. Annu Rev Cell Dev Biol. 2005 doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Kawano Y, Fukata Y, Oshiro N, Amano M, Nakamura T, Ito M, et al. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J Cell Biol. 1999;147:1023–38. doi: 10.1083/jcb.147.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–8. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Kleer CG, Griffith KA, Sabel MS, Gallagher G, van Golen KL, Wu ZF, et al. RhoC-GTPase is a novel tissue biomarker associated with biologically aggressive carcinomas of the breast. Breast Cancer Res Treat. 2005;93:101–10. doi: 10.1007/s10549-005-4170-6. [DOI] [PubMed] [Google Scholar]
- Kleer CG, van Golen KL, Zhang Y, Wu ZF, Rubin MA, Merajver SD. Characterization of RhoC expression in benign and malignant breast disease: a potential new marker for small breast carcinomas with metastatic ability. Am J Pathol. 2002;160:579–84. doi: 10.1016/S0002-9440(10)64877-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleer CG, Zhang Y, Pan Q, Gallagher G, Wu M, Wu ZF, et al. WISP3 and RhoC guanosine triphosphatase cooperate in the development of inflammatory breast cancer. Breast Cancer Res. 2004;6:R110–5. doi: 10.1186/bcr755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolega J. The role of myosin II motor activity in distributing myosin asymmetrically and coupling protrusive activity to cell translocation. Mol Biol Cell. 2006;17:4435–45. doi: 10.1091/mbc.E06-05-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma R, Ahmed S, Best A, Lim L. The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol Cell Biol. 1995;15:1942–52. doi: 10.1128/mcb.15.4.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraynov VS, Chamberlain C, Bokoch GM, Schwartz MA, Slabaugh S, Hahn KM. Localized Rac activation dynamics visualized in living cells. Science. 2000;290:333–7. doi: 10.1126/science.290.5490.333. [DOI] [PubMed] [Google Scholar]
- Krendel M, Mooseker MS. Myosins: tails (and heads) of functional diversity. Physiology (Bethesda) 2005;20:239–51. doi: 10.1152/physiol.00014.2005. [DOI] [PubMed] [Google Scholar]
- Kurokawa K, Matsuda M. Localized RhoA activation as a requirement for the induction of membrane ruffling. Mol Biol Cell. 2005;16:4294–303. doi: 10.1091/mbc.E04-12-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa K, Nakamura T, Aoki K, Matsuda M. Mechanism and role of localized activation of Rho-family GTPases in growth factor-stimulated fibroblasts and neuronal cells. Biochem Soc Trans. 2005;33:631–4. doi: 10.1042/BST0330631. [DOI] [PubMed] [Google Scholar]
- Kusama T, Mukai M, Tatsuta M, Nakamura H, Inoue M. Inhibition of transendothelial migration and invasion of human breast cancer cells by preventing geranylgeranylation of Rho. Int J Oncol. 2006;29:217–23. [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–69. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Li F, Higgs HN. The mouse Formin mDia1 is a potent actin nucleation factor regulated by autoinhibition. Curr Biol. 2003;13:1335–40. doi: 10.1016/s0960-9822(03)00540-2. [DOI] [PubMed] [Google Scholar]
- Liu BP, Burridge K. Vav2 activates Rac1, Cdc42, and RhoA downstream from growth factor receptors but not beta1 integrins. Mol Cell Biol. 2000;20:7160–9. doi: 10.1128/mcb.20.19.7160-7169.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo CM, Buxton DB, Chua GC, Dembo M, Adelstein RS, Wang YL. Nonmuscle myosin IIb is involved in the guidance of fibroblast migration. Mol Biol Cell. 2004;15:982–9. doi: 10.1091/mbc.E03-06-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, et al. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. Embo J. 1996;15:2208–16. [PMC free article] [PubMed] [Google Scholar]
- Meshel AS, Wei Q, Adelstein RS, Sheetz MP. Basic mechanism of three-dimensional collagen fibre transport by fibroblasts. Nat Cell Biol. 2005;7:157–64. doi: 10.1038/ncb1216. [DOI] [PubMed] [Google Scholar]
- Nalbant P, Hodgson L, Kraynov V, Toutchkine A, Hahn KM. Activation of endogenous Cdc42 visualized in living cells. Science. 2004;305:1615–9. doi: 10.1126/science.1100367. [DOI] [PubMed] [Google Scholar]
- O'Connor KL, Nguyen BK, Mercurio AM. RhoA function in lamellae formation and migration is regulated by the alpha6beta4 integrin and cAMP metabolism. J Cell Biol. 2000;148:253–8. doi: 10.1083/jcb.148.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertz O, Hodgson L, Klemke RL, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006;440:1069–72. doi: 10.1038/nature04665. [DOI] [PubMed] [Google Scholar]
- Pille JY, Denoyelle C, Varet J, Bertrand JR, Soria J, Opolon P, et al. Anti-RhoA and anti-RhoC siRNAs inhibit the proliferation and invasiveness of MDA-MB-231 breast cancer cells in vitro and in vivo. Mol Ther. 2005;11:267–74. doi: 10.1016/j.ymthe.2004.08.029. [DOI] [PubMed] [Google Scholar]
- Pille JY, Li H, Blot E, Bertrand JR, Pritchard LL, Opolon P, et al. Intravenous delivery of anti-RhoA small interfering RNA loaded in nanoparticles of chitosan in mice: safety and efficacy in xenografted aggressive breast cancer. Hum Gene Ther. 2006;17:1019–26. doi: 10.1089/hum.2006.17.1019. [DOI] [PubMed] [Google Scholar]
- Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Rho GTPases and cell migration. J Cell Sci. 2001;114:2713–22. doi: 10.1242/jcs.114.15.2713. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–99. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–10. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–9. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Sahai E, Garcia-Medina R, Pouyssegur J, Vial E. Smurf1 regulates tumor cell plasticity and motility through degradation of RhoA leading to localized inhibition of contractility. J Cell Biol. 2007;176:35–42. doi: 10.1083/jcb.200605135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol. 2003;5:711–9. doi: 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- Sandquist JC, Swenson KI, Demali KA, Burridge K, Means AR. Rho kinase differentially regulates phosphorylation of nonmuscle myosin II isoforms A and B during cell rounding and migration. J Biol Chem. 2006;281:35873–83. doi: 10.1074/jbc.M605343200. [DOI] [PubMed] [Google Scholar]
- Sastry SK, Rajfur Z, Liu BP, Cote JF, Tremblay ML, Burridge K. PTP-PEST couples membrane protrusion and tail retraction via VAV2 and p190RhoGAP. J Biol Chem. 2006;281:11627–36. doi: 10.1074/jbc.M600897200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers JR. Myosins: a diverse superfamily. Biochim Biophys Acta. 2000;1496:3–22. doi: 10.1016/s0167-4889(00)00005-7. [DOI] [PubMed] [Google Scholar]
- Simpson KJ, Dugan AS, Mercurio AM. Functional analysis of the contribution of RhoA and RhoC GTPases to invasive breast carcinoma. Cancer Res. 2004;64:8694–701. doi: 10.1158/0008-5472.CAN-04-2247. [DOI] [PubMed] [Google Scholar]
- Tominaga T, Sahai E, Chardin P, McCormick F, Courtneidge SA, Alberts AS. Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol Cell. 2000;5:13–25. doi: 10.1016/s1097-2765(00)80399-8. [DOI] [PubMed] [Google Scholar]
- Totsukawa G, Wu Y, Sasaki Y, Hartshorne DJ, Yamakita Y, Yamashiro S, et al. Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J Cell Biol. 2004;164:427–39. doi: 10.1083/jcb.200306172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Golen KL, Davies S, Wu ZF, Wang Y, Bucana CD, Root H, et al. A novel putative low-affinity insulin-like growth factor-binding protein, LIBC (lost in inflammatory breast cancer), and RhoC GTPase correlate with the inflammatory breast cancer phenotype. Clin Cancer Res. 1999;5:2511–9. [PubMed] [Google Scholar]
- van Golen KL, Wu ZF, Qiao XT, Bao LW, Merajver SD. RhoC GTPase, a novel transforming oncogene for human mammary epithelial cells that partially recapitulates the inflammatory breast cancer phenotype. Cancer Res. 2000;60:5832–8. [PubMed] [Google Scholar]
- Vicente-Manzanares M, Zareno J, Whitmore L, Choi CK, Horwitz AF. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J Cell Biol. 2007;176:573–80. doi: 10.1083/jcb.200612043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol. 1999;1:136–43. doi: 10.1038/11056. [DOI] [PubMed] [Google Scholar]
- Webb DJ, Parsons JT, Horwitz AF. Adhesion assembly, disassembly and turnover in migrating cells -- over and over and over again. Nat Cell Biol. 2002;4:E97–100. doi: 10.1038/ncb0402-e97. [DOI] [PubMed] [Google Scholar]
- Wei Q. Pitx2a binds to human papillomavirus type 18 E6 protein and inhibits E6-mediated P53 degradation in HeLa cells. J Biol Chem. 2005;280:37790–7. doi: 10.1074/jbc.M502974200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Adelstein RS. Conditional expression of a truncated fragment of nonmuscle myosin IIA alters cell shape but not cytokinesis in HeLa cells. Mol Biol Cell. 2000;11:3617–27. doi: 10.1091/mbc.11.10.3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells CM, Walmsley M, Ooi S, Tybulewicz V, Ridley AJ. Rac1-deficient macrophages exhibit defects in cell spreading and membrane ruffling but not migration. J Cell Sci. 2004;117:1259–68. doi: 10.1242/jcs.00997. [DOI] [PubMed] [Google Scholar]
- Worthylake RA, Burridge K. RhoA and ROCK promote migration by limiting membrane protrusions. J Biol Chem. 2003;278:13578–84. doi: 10.1074/jbc.M211584200. [DOI] [PubMed] [Google Scholar]
- Wu D, Asiedu M, Adelstein RS, Wei Q. A novel guanine nucleotide exchange factor MyoGEF is required for cytokinesis. Cell Cycle. 2006;5:1234–9. doi: 10.4161/cc.5.11.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamana N, Arakawa Y, Nishino T, Kurokawa K, Tanji M, Itoh RE, et al. The Rho-mDia1 pathway regulates cell polarity and focal adhesion turnover in migrating cells through mobilizing Apc and c-Src. Mol Cell Biol. 2006;26:6844–58. doi: 10.1128/MCB.00283-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigmond SH. Formin-induced nucleation of actin filaments. Curr Opin Cell Biol. 2004;16:99–105. doi: 10.1016/j.ceb.2003.10.019. [DOI] [PubMed] [Google Scholar]