

Abstract
Berkeley sickle cell mice are used as an animal model of human sickle cell disease but there are no reports of platelet studies in this model. Since humans with sickle cell disease have platelet abnormalities, we studied platelet morphology and function in Berkeley mice (SS). We observed elevated mean platelet forward angle light scatter (FSC) values (an indirect measure of platelet volume) in SS compared to wild type (WT) (37 ± 3.2 vs. 27 ± 1.4, mean ± SD; p <0.001), in association with moderate thrombocytopenia (505 ± 49 × 103/μl vs. 1151 ± 162 × 103/μl; p <0.001). Despite having marked splenomegaly, SS mice had elevated levels of Howell-Jolly bodies and “pocked” erythrocytes (p <0.001 for both) suggesting splenic dysfunction. SS mice also had elevated numbers of thiazole orange positive platelets (5 ± 1 % vs. 1 ± 1%; p <0.001), normal to low plasma thrombopoietin levels, normal plasma glycocalicin levels, normal levels of platelet recovery, and near normal platelet life spans. Platelets from SS mice bound more fibrinogen and antibody to P-selectin following activation with a threshold concentration of a protease activated receptor (PAR)-4 peptide compared to WT mice. Enlarged platelets are associated with a predisposition to arterial thrombosis in humans and some humans with SCD have been reported to have large platelets. Thus, additional studies are needed to assess whether large platelets contribute either to pulmonary hypertension or the large vessel arterial occlusion that produces stroke in some children with sickle cell disease.
Keywords: Sickle cell, Berkeley mouse model, Platelet size
Introduction
Murine models of sickle cell disease (SCD) offer the potential to elucidate the pathophysiology of the disorder and improve its therapy. It is important however, to fully characterize the models to assess their similarities and differences from human SCD. Berkeley sickle cell (SS) mice were generated by injecting a transgenic construct containing human HBA1 (hemoglobin, alpha 1), HBG2 (hemoglobin, gamma G), HBG1 (hemoglobin, gamma A), HBD (hemoglobin, delta), and HBBS (hemoglobin, beta, sickle allele) genes and the locus control region into fertilized FVB/N mouse eggs and then breeding the resultant offspring carrying the transgene to mice bearing targeted mutations in the endogenous mouse Hba and Hbb genes (1). This model has many similarities to human SCD, including anemia, reticulocytosis, sickled erythrocytes in the peripheral blood (1), and evidence of both systemic oxidant stress (2;3) and inflammation (4). The mice differ from humans with SCD most notably in that mice develop splenomegaly rather than splenic atrophy; more subtle differences also exist in the histopathology of affected organs (5). In addition, the erythrocytes of Berkeley mice have an excess of α-globin chain synthesis (α/βS =1.26) indicating mild β-thalassemia (1).
Thrombocytosis and several qualitative platelet abnormalities, including defects in platelet aggregation and evidence for in vivo platelet activation, have been described in patients with SCD (6–14). While some studies have suggested that platelets contribute to vasoocclusive crisis (15–18), there is considerable doubt about their role (19) because antiplatelet agents have not significantly altered the frequency or severity of pain crisis (20–22;22–24). A potential role of platelets in large vessel occlusion leading to thrombotic stroke in children, however, remains to be assessed critically (25–29). Since we could not identify any reports of platelet studies in Berkeley SS mice, we studied platelets and related parameters in these mice.
Materials
Mouse Studies
A colony of SS mice was established with approximately tenth generation breeding pairs kindly provided by Dr Cheryl Hillery (Blood Center of Southeastern Wisconsin, Milwaukee, WI). The initial background of this strain was a mixture of FVB/N, 129X1/SvJ, DBA/2, Black Swiss, and C57BL/6 (1). The studies described below included SS mice that have targeted mutations of both the endogenous murine α (30) and β (31) globin genes and carry the transgene Tg(HBA–HBBS)41Paz (1), genotype Hbatm1Paz/Hbatm1PazHbbtm1Tow/Hbbtm1Tow Tg(HBA–HBBS)41Paz/0. C57BL/6J and 129X1/SvJ mice, the two major background strains present in SS mice, were purchased from Jackson Laboratories (Bar Harbor, ME) and crossbred to generate wild type (WT) controls and a colony was established by interbreeding their progeny for up to 10 generations. SS mice from the same original colony, but which were backcrossed onto the C57BL/6 background over eight generations (kindly supplied by Dr. Mary Fabry, Albert Einstein College of Medicine, New York, NY) were also analyzed in some experiments. β-thalassemia mice homozygous for a deletion of the murine β globin gene Hbb (Hbbd3th/Hbbd3th) (32), also kindly supplied by Dr. Fabry, served as controls for anemia. WT and C57BL/6J mice were obtained from Jackson Laboratories for the experiments involving transplantation of SS bone marrow. All of the animal experiments performed in these studies were approved by the Laboratory Animal Research Committee at the Rockefeller University.
Antibodies
Antibody 1B5 (hamster anti-mouse αIIbβ3) (33) was produced at the National Cell Culture Center (Minneapolis, MN) and rat anti-mouse CD45 and ter119 were from BD Biosciences (San Diego, CA). Rat anti-mouse GPIb antibodies p0p 3 and HRP-conjugated p0p 4 were a kind gift of Dr. Bernard Nieswandt (Rudolf-Virchow-Zentrum für Experimentelle Biomedizin, Würzburg, Germany) (34) and rat anti-mouse GPIbβ antibody Xia.C3 was obtained from Emfret (Würzburg, Germany). Labeling of antibody 1B5 with NHS-LC-Biotin (Pierce, Rockford, IL), Alexa Fluor488 or Alexa Fluor647 (Molecular Probes, Eugene, OR) was performed according to the manufacturer’s instructions.
Blood collection
Mice were anesthetized with isoflurane (Baxter Healthcare Corp, Deerfield, IL) and blood obtained from the retrobulbar venous plexus using 12–15 mm long glass capillary tubes (Fischer Scientific, Pittsburg, PA); blood was dripped directly into tubes containing sufficient ethylenediaminetetraaceticacid (EDTA) to produce a final concentration of 10 mM and gently mixed by pipetting. For studies of erythropoietin, blood was collected into tubes containing heparin (American Pharmaceutical Partners Inc, Los Angeles, CA; 15Units/ml) and for platelet function and intravascular platelet recovery studies, blood was collected into 3.8 % sodium citrate (Fischer Scientific, Pittsburg, PA). All other assays were conducted using EDTA-anticoagulated blood.
Hematological data
Blood smears were prepared from every sample and stained with Wright’s stain (Sigma-Aldrich Inc, St Louis, MO). Smears were evaluated for overall erythrocyte, leukocyte and platelet morphology, as well as the presence of nucleated RBCs (NRBCs) and sickled erythrocytes, Howell-Jolly bodies, erythrocyte fragments, and platelet aggregates. When a sample was found to contain platelet aggregates, the hematologic data obtained from that animal was excluded from the analysis.
Hematological parameters were performed on EDTA-anticoagulated blood 30–60 min after blood collection using an automated dual angle light scatter instrument (ADVIA120, Bayer Diagnostics, Tarrytown, NY) with software designed to measure murine blood (35). Samples (20 μl) were diluted 1:10 in phosphate-buffered saline (PBS) containing 5% bovine serum albumin (BSA) and then analyzed immediately. Preliminary studies demonstrated that diluting the blood before testing did not affect the hematocrit or reticulocyte values, and hematocrit values obtained by centrifugation of whole blood in microhematocrit tubes (Fischer Scientific Inc, Pittsburg, PA) were concordant with hematocrit values obtained with the automated instrument. Leukocyte and platelet count assessments by the ADVIA120 in WT mice demonstrated concordance with manual assessments using a hemocytometer. In SS mice, however, platelet count and mean platelet volume (MPV) determinations were confounded by the presence of erythrocyte fragments and leukocyte counts were confounded by the presence of NRBCs.
Platelets were therefore enumerated by flow cytometry (FACS Calibur, BD Biosciences, San Diego, CA) (36). Briefly, 5 μl of 10-fold diluted EDTA-anticoagulated blood was labeled with antibody Alexa647-1B5 (2.5 μg/ml) in 45 μl of HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid)-saline (NaCl 165 mM, HEPES 10 mM, pH 7.4, 0.22 μm filtered, degassed for 1 h) for 15 min at 22 °C in the dark. Samples were then further diluted by adding 300 μl of HEPES-saline. Immediately before flow cytometry analysis, 50 μl of a suspension containing 5.5 × 103 latex beads /μl of 7.32 μm diameter (Bangs Laboratories Inc, Fischers, IN) was added to each sample to serve as an internal standard to quantify the volume of sample analyzed, thus permitting the calculation of the whole blood platelet count from the flow cytometry data. The concentration of latex beads was determined using the automated instrument. The platelet count, in platelets/μl, was calculated by dividing the number of particles in the platelet gate by the volume of sample analyzed [(number of beads counted/final concentration of beads in sample analyzed)] and then multiplying the value by the final dilution of the original blood sample. Platelet size was estimated by determining the geometric mean value of the forward angle light scatter (FSC) of particles that exhibited fluorescence with Alexa647-1B5, after excluding particles that reflected platelet-erythrocyte coincidence or platelet-erythrocyte aggregates. Previous studies performed by Holme et al. on fixed human platelets establish that platelet size determined by mean FSC correlates well with MPV determined by resistive particle sizing (r=0.83) (37;38). As a size reference, we tested a series of nonfluorescent microspheres of 1, 2, 4, and 6 μm diameter (Molecular Probes) and found that they gave geometric mean FSC values of 17.4, 37.5, 170.4, and 514.3. When the FSC values of the beads were plotted on a log scale versus bead size, a line was obtained with r2=0.996. WT mouse platelets had FSC values of 26.6 ± 1.4, corresponding to microspheres of ~1.4 μm diameter and two normal human donor platelets had FSC of 59 and 85 corresponding to microspheres of ~2.5–3 μm diameter.
Since leukocyte counts in SS mice using the automated instrument were confounded by the presence of NRBCs, WBC and NRBC counts were measured by flow cytometry (39;40) using an antibody to the pan-leukocyte antigen CD45 and propidium iodide (PI) to stain nucleated cells. Briefly, 25 μl of EDTA-anticoagulated blood was incubated with FITC-labeled anti-CD45 (5 μg/ml final concentration) for 30 min at 22°C in the dark, and then permeabilized and fixed for 30 min by adding an equal volume of a solution containing 2% paraformaldehyde-0.1% saponin. Erythrocytes were lysed by the addition of 400 μl of a murine erythrocyte lysing reagent (R&D Systems, Minneapolis, MN) and the nucleated cells were stained with PI (Sigma Aldrich Inc; 5 μg/ml final concentration). The samples were finally incubated with 50 μl RNAse (Sigma Aldrich Inc, 10 mg/ml) at 37°C for 10 min to eliminate PI staining of reticulocytes and platelets, both of which contain RNA. Subsequently, 25 μl of 10 μm fluorescent beads (Flow-Count Fluorospheres; Beckman Coulter, Fullerton, CA) containing 972 beads/μl were added to the sample and flow cytometry was performed. Particles exhibiting fluorescence for CD45 were defined as leukocytes and particles that were negative for CD45 and positive for PI (CD45−/PI+ve) were defined as NRBCs. In separate experiments, we observed that CD45−/PI− cells labeled with a murine erythrocyte-specific marker (ter119)(41), confirming their erythrocyte lineage. The numbers of whole blood leukocytes and NRBCs per μl were calculated as above using the manufacturer’s formula.
Erythrocytes with irregular indentations (“pocked” erythrocytes) were enumerated by immediately fixing EDTA-anticoagulated blood with 0.1% glutaraldehyde in saline (0.15 M NaCl, 310 mOsm) and then examining wet mounts by differential interference contrast microscopy using a 100x oil immersion objective (Olympus Inc, Mahwah, NJ) (42).
Analysis of Platelet RNA
Blood (5 μl) was incubated with a combination of thiazole orange (5 μg/ml final concentration) and Alexa647-1B5 (5 μg/ml) in PBS (final volume 50 μl) for 30 min at 22°C in the dark and analyzed by flow cytometry. Platelets were gated using a combination of light scatter and 1B5 staining and considered positive for RNA (“reticulated platelets”) based on thiazole orange staining as previously described (43;44). In particular, a boundary was constructed at the upper edge of the thiazole orange fluorescence intensity of the WT platelet population as a function of side angle scatter such that ~1% of gated WT platelets were thiazole orange positive (44).
Immunofluorescent Detection of Platelets on Blood Smears
Blood samples were incubated with Alexa488-1B5 (5 μg/ml final concentration) for 30 min at 22°C. Blood smears were prepared, heat fixed, mounted with an antifade reagent (Prolong, Molecular Probes) and a cover slip, and viewed with a 100x oil immersion objective (Olympus Inc). Images of platelets were acquired from 10 consecutive random fields per slide for SS and WT mice (n = 5 each). Using image analysis software (Slide Book; Intelligent Imaging, Baltimore, MD), the individual platelet areas occupied by 100 ± 40 platelets and the percentage of platelets with mean platelet areas ≥ 3.5 μm2 were determined.
Splenic Sequestration Studies
The size of the splenic platelet pool was estimated by assessing the recovery of injected platelets labeled with the fluorescent dye 5-chloromethyl fluorescein diacetate (CMFDA; Cell Tracker Green, Molecular Probes, Eugene, OR)(45). Platelet-rich plasma (PRP) from SS and WT mice was prepared by centrifugation (250 x g at 22°C for 2.5 min), treated with PGE1 (1 μM), and labeled with green CMFDA in DMSO (0.04 vol. 200 μM stock solution in DMSO) at 37°C for 15 min. Labeled platelets were then washed with 0.35% BSA in HEPES-saline with PGE1, resuspended in 200 μl of HEPES-saline, counted, and injected into the left ventricle under ultrasound guidance using a 40–70 mHz probe (VisualSonics, Toronto, Canada). Intravascular platelet recovery was measured 30 min later on retrobulbar venous blood using flow cytometry and calculated by multiplying the number of fluorescent platelets/μl in the circulation by the estimated blood volume (7.2 ml/100 gm weight (46)) and dividing the product by the number of labeled platelets injected.
Platelet Survival Studies
Survival of biotin-labeled platelets was determined by injecting NHS-LC-biotin (Pierce; 100 μl of 3 mg/ml in 0.13 M NaCl, 330 mOsm) via the tail vein into SS (n = 5), and WT (n = 7) mice (47). Pre-injection and 2 hr post-injection blood samples were obtained by retrobulbar blood sampling to determine the background and day 0 platelet labeling, respectively; subsequent 5 μl samples were obtained daily from the tail using EDTA-coated capillary tubes and anticoagulated by mixing with 45 μl of 10 mM EDTA in HEPES -saline. Samples (5 μl) were incubated with FITC-avidin (10 μg/ml final concentration; Leinco Technologies Inc, St Louis, MO) and Alexa647-1B5 for 30 min at 22°C and then the samples were diluted with PBS and analyzed by flow cytometry. Platelets were identified by a combination of light scatter properties and 1B5 labeling, and then the percentage of biotinylated platelets was determined from the intensity of avidin labeling. The percentage of platelets labeled 2 hr post-injection on day 0 was considered 100% labeling and subsequent data were normalized to this value. Platelet survival was determined using a linear best-fit line (48).
Effect of Splenectomy on Hematological Parameters
Twenty week-old WT mice (n = 4) had baseline hematologic evaluations for cell counts and indices of splenic function (Howell-Jolly bodies and “pocked” erythrocytes), and then were anesthetized with isoflurane and splenectomized. Repeat blood samples were obtained and analyzed nine weeks later under isoflurane anesthesia.
Plasma Glycocalicin, Thrombopoietin, and Erythropoietin assays
Plasma glycocalicin was measured using the rat anti-mouse GPIb antibodies p0p 3 and HRP-conjugated p0p 4 as previously reported (34). Thrombopoietin and erythropoietin levels were measured by ELISA with commercially available kits (R&D Diagnostics Inc, Minneapolis, MN).
Platelet P-selectin expression and fibrinogen binding
Citrate-anticoagulated blood samples from SS (n = 4) and WT (n = 4) mice were diluted in HEPES-buffered modified Tyrodes solution containing 100 μM Ca++ and 50 μM Mg++ and stimulated with a protease activated receptor 4 (PAR-4) peptide (AYPGKF)(49) at 75 and 200 μM in the presence of an antibody to P-selectin (BD Biosciences; 5 μg/ml final concentration) or Alexa647-labeled human fibrinogen (Molecular Probes, 200 μg/ml final concentration) for 5 min at 37°C. Platelets were then immediately fixed by the addition of paraformaldehyde (0.5% final concentration) and labeled with either fluorescently-labeled antibody 1B5 for P-selectin studies or an antibody to GPIb (Xia.C3) for fibrinogen studies. Platelets identified by either antibody were examined for baseline and agonist-induced surface P-selectin expression and fibrinogen binding using flow cytometry. Negative controls included samples with 10 mM EDTA for the fibrinogen studies and excess unlabeled antibody for the P-selectin studies.
Statistical analysis
Statistical comparisons between groups were analyzed using the one-way analysis of variance (ANOVA) for gaussian distributed data and the Kruskal Wallis test for non-gaussian data. When multiple group comparisons using these tests were statistically significant (p <0.05, 2- tailed), pairwise comparisons were conducted using the Bonferroni method or the Mann Whitney test as appropriate. Statistical analysis was performed using SPSS, version 10.0 for Windows (Chicago, IL).
Results
Hematological data
SS mice had severe anemia with marked reticulocytosis and high levels of circulating NRBCs (Table 1). Their blood smears revealed NRBCs, Howell-Jolly bodies, and erythrocyte fragments, as well as extensive polychromasia (Fig 1). Quantitative assessments of NRBCs by smear confirmed the presence of large numbers of NRBCs, although the number obtained by smear determination (43 ± 18 per100 WBCs; mean ± SD; n = 15) was approximately one half of the value obtained by flow cytometry, perhaps reflecting inhomogeneities in the distribution of NRBCs and WBCs on the smear. β-thalassemia mice had similar hematocrit and reticulocyte values, but much lower NRBC values (Table 1).
Table 1.
Hematologic parameters of mice studied
| N | WT | N | SS | N | β-Thal | N | WT Splx§ | |
|---|---|---|---|---|---|---|---|---|
| Hematocrit (%) | 16 | 40 ± 2 | 13 | 26 ± 4 B,E | 4 | 29 ± 7 A | 4 | 44 ± 2 G |
| Reticulocyte (x10−3/μl) | 16 | 233 ± 117 | 13 | 2171 ± 438 B,E | 4 | 2273 ± 784 B | 4 | 212 ± 24 G |
| WBC count (x10−3/μl) | 13 | 8 ± 2 | 12 | 17 ± 3 B,E | 4 | 12 ± 7 A | 4 | 11 ± 2† G |
| Platelets (x10−3/μl) | 16 | 1151 ± 162 | 13 | 505 ± 49 B,D | 4 | 1400 ± 251 A | 4 | 1195 ± 126† G |
| Platelet forward angle light scatter (FSC) | 16 | 26.6 ± 1.4 | 13 | 37.2 ± 3.2 B,E | 4 | 36.5 ± 1.9 B | 4 | 23.1 ± 1.7 G |
| Thiazole orange positive platelets (%) | 12 | 1 ± 1 | 15 | 5 ± 1B, D | 3 | 2 ± 1 A | 4 | 2 ± 1 G |
| Nucleated RBCs (x10−3/μl) | 13 | 0 | 12 | 17 ± 5 B,C | 4 | 2.1 ± 1.4 A | 4 | ND |
| Howell-Jolly bodies (%) | 5 | 0.2 ± 0.2 | 10 | 4.7 ± 1.4 B,C | 4 | 1.0 ± 0.2 A | 4 | 0.3 ± 0.2 G |
| “Pocked” RBC (%) | 15 | 0.3 ± 0.4 | 5 | 40.4 ± 3.0 B,C | 4 | 34.6 ± 4.0 B | 4 | 0.4 ± 0.7 G |
Data are shown as mean ± SD. WT, wild type; SS, Berkeley SS mice; β-thal, β-thalassemia mice; Splx, surgically splenectomized; WBC, white blood count; FSC, foward angle light scatter; RBC, red blood cell.
P < 0.05 vs. WT.
P < .001 vs. WT.
P < 0.05 vs. β-thal mice.
P < .001 vs.β-thal mice.
P > 0.05 vs. β-thal mice.
P < 0.05 vs. pre splenectomy mice.
P >0.05 vs pre-splenectomy mice.
Figure 1. SS mice have markedly abnormal blood smears.
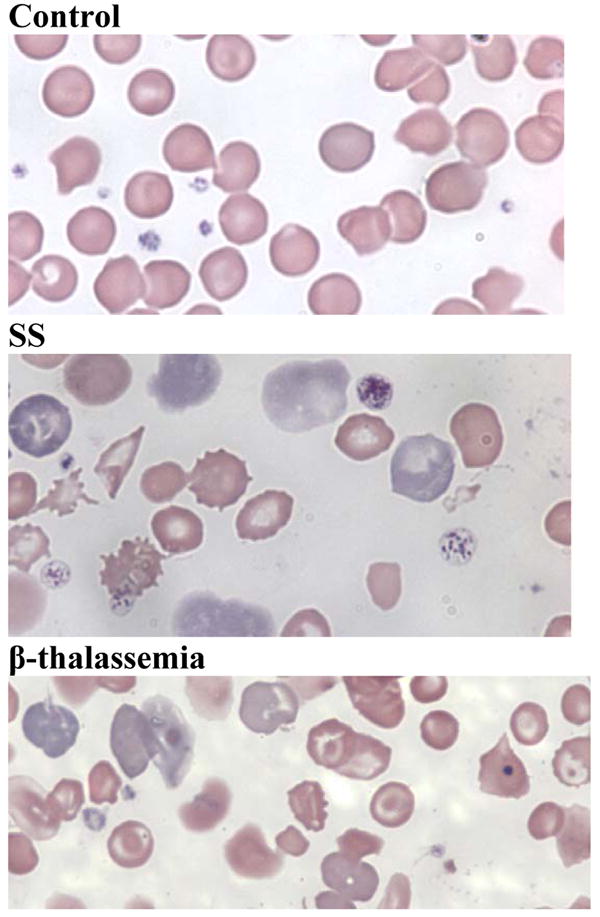
Peripheral blood smears of EDTA-anticoagulated blood obtained by retrobulbar venous plexus puncture from WT, SS and β-thalassemia mice. Blood smears from SS animals show anisocytosis, poikilocytosis, sickled erythrocytes, target cells, erythrocyte fragments of varying sizes, Howell-Jolly bodies, and an increased percentage of polychromatophilic erythrocytes. In sharp contrast, smears from WT animals do not show any of these findings. Blood smears from β-thalassemia mice show anisocytosis, microcytosis, targets cells, erythrocyte fragments, and an increased percentage of polychromatophilic erythrocytes.
SS mice were moderately thrombocytopenic, with platelet counts of 505 ± 40 × 10−3 per μl compared to 1152 ± 162 × 10−3 per μl in WT mice (p<0.001). The platelets of SS mice had FSC values that were almost 40% higher than those of WT mice [SS = 37 ± 3.2 (mean ± SD); WT = 27 ± 1.4; p<0.001] (Figure 2). SS mice that were backcrossed onto the C57BL/6J background also demonstrated higher FSC values than WT animals (39 ± 0.9; n=3). β-thalassemia mice had significantly elevated platelet counts compared to both SS and WT mice and the platelet FSC values of β-thalassemia mice were similar to those of SS mice (Table 1).
Fig 2. Platelets from SS mice have increased forward light scatter properties.

Histogram of forward light scatter (abscissa, log scale) and number of particles positive for anti-murine αIIbβ3 (1B5) staining (ordinate) from blood of wild type and SS mice. Blood samples were reacted with Alexa647-1B5 (platelet specific mAb to murine αIIbβ3) and then the platelets were identified on the basis of light scatter properties and 1B5 staining. In the SS mice, there is a rightward shift of the entire platelet population, contributing to an elevation in the mean forward scatter value.
Independent assessment of platelet size by direct immunofluorescent microscopic evaluation of blood smears demonstrated that blood from SS mice contained larger platelets than those from WT. For example, the percentage of platelets occupying areas ≥3.5 μm2 was 35 ± 12% for SS vs. 7 ± 8% for WT mice; n=5 ; p< 0.01).
SS mice had leukocytosis, with leukocyte counts approximately double those of WT mice (Table 1). β-thalassemia mice had leukocyte counts that were intermediate in value between SS and WT mice.
Thiazole orange staining
SS mice had a significantly higher percentage of platelets staining strongly with the RNA stain thiazole orange compared with WT (p<0.001, Table 1). Platelets staining strongly for thiazole orange varied in forward scatter properties over a wide distribution (data not shown), indicating that both large and small platelets in SS mice had increased amounts of RNA. β-thalassemia mice had values closer to those of WT than SS mice.
Splenic size and indicators of splenic function
Splenic weight (either as a percentage of body weight or as an absolute value) was dramatically increased in SS mice (4.9 ± 0.8%; 1.4 ± 0.3 g, n=7) compared with WT mice (0.3 ± 0.1%; 0.08 ± 0.02 g, n=12; p<0.001) for both determinations. The percentage of erythrocytes containing Howell-Jolly bodies was more than twenty-fold higher in the blood of SS mice than in the blood of WT mice (Table 1; p<0.001). In addition, more than 40% of the erythrocytes in SS mice were “pocked” compared with 0.3% of the erythrocytes in WT mice (Table 1; p<0.001). β-thalassemia mice had values for both Howell-Jolly bodies and “pocked” erythrocytes that were in between those of SS than WT mice. Surprisingly, surgically splenectomized WT mice, which had essentially normal hematologic values, did not have elevated values for Howell-Jolly bodies or “pocked” erythrocytes compared to WT mice (Table 1).
Platelet recovery and survival
Intravascular recovery of injected fluorescent platelets in SS mice was 55 ± 17% (n=5), which was similar to that in WT mice (55 ± 14%; n=14; p=0.98), although there was considerable variability in results in both groups. Injection of NHS-LC-biotin into SS and WT mice led to labeling of ~60 and 95% of circulating platelets, respectively, at 2 h. The subsequent decreases in circulating biotinylated platelets over time indicated that the mean platelet survival in WT mice was 5.7 ± 0.7 days (n=7), which is similar to values reported by others (48). Platelet survival was 5.4 ± 0.4 days in SS (n=5) mice (p>0.05 for SS vs. WT).
Plasma thrombopoietin, erythropoietin, and glycocalicin
The mean plasma thrombopoietin value in SS mice (268 ± 81 pg/ml, n=12) was 28% lower than the value in WT mice (370 ± 93 pg/ml; n = 16; p<0.05). Erythropoietin levels were markedly elevated in SS mice (3020 ± 2380 pg/ml; n=8), compared with WT mice (290 ± 290 pg/ml; n=6; p<0.001). Plasma glycocalicin values (expressed as a percentage of pooled mouse plasma) in the SS mice (86 ± 17%; n=7) were similar to those in WT mice (80 ± 40 %; n=6; p>0.05).
Binding of fibrinogen and surface expression of P-selectin by activated platelets
Antibodies to P-selectin demonstrated similar binding (as judged by mean fluorescent intensity) to platelets from WT and SS mice in both the resting state and after maximal stimulation with a PAR-4 peptide (200 μM), with the modestly higher values in SS mice presumably reflecting differences in platelet size (Table 2). In contrast, submaximal platelet stimulation with the PAR-4 peptide (75 μM) resulted in 5-fold greater anti-P-selectin antibody binding to SS platelets compared with WT platelets (p=0.03). The baseline binding of fibrinogen was low to both WT and SS platelets and binding increased dramatically to both types of platelets with maximal stimulation with the PAR-4 peptide (200 μM) (Table 2). Once again, the SS platelets bound modestly higher amounts of fibrinogen under both conditions, presumably reflecting the differences in platelet size. Upon submaximal stimulation with PAR-4 peptide (75 μM), however, fibrinogen binding to SS platelets was nearly three-fold greater than that to WT platelets (p=0.03). Thus, both the surface expression of the α-granule protein P-selectin and activation of the αIIbβ3 receptor to bind fibrinogen showed increased sensitivity to submaximal PAR-4 activation, with changes that could not be accounted for based exclusively on the differences in platelet size.
Table 2.
P-selectin and Fibrinogen binding to Platelets at baseline and after stimulation with PAR4 peptide
| Platelet | Wild Type n = 4 |
SS n = 4 |
P value |
|---|---|---|---|
| Geometric MFI† |
|||
| P-selectin expression | |||
| Baseline | 0.3 ± 0.1 | 0.6 ± 0.7 | 0.89 |
| 75μM PAR4 | 0.8 ± 0.9 | 4.0 ± 2.6 | 0.03 |
| 200μM PAR4 | 18.9 ± 4.6 | 26.6 ± 4.1 | 0.11 |
| Fibrinogen binding | |||
| Baseline | 1.7 ± 0.3 | 3 ± 0.6 | 0.03 |
| 75μM PAR4 | 38 ± 14 | 110 ± 31 | 0.03 |
| 200μM PAR4 | 161 ± 23 | 192 ± 30 | 0.20 |
Expressed as mean ± SD of the geometric mean fluorescent intensities (MFI) after subtraction of negative controls
Discussion
The major new findings in this study are that compared to WT mice on a combined C57BL/6J and 129X1/SvJ background (2 of the 5 mouse background strains for the Berkeley SS mouse), SS mice have: 1) an ~50 % lower platelet count, 2) increased platelet size, 3) increased sensitivity to submaximal agonist-induced granule secretion and activation of αIIbβ3 receptors 4) normal to low thrombopoietin levels, 5) an increased percentage of platelets staining with the RNA dye thiazole orange, 6) normal intravascular recovery, 7) nearly normal platelet life span, and 8) evidence of compromised splenic function (elevated levels of Howell-Jolly bodies and “pocked” cells) despite marked splenomegaly (1;5).
The most striking anatomic difference between SS mice and humans with SCD is the presence of massive splenomegaly in SS mice rather than splenic infarction and atrophy.(1;5) This results, in large part, from murine expansion of splenic hematopoiesis, including megakaryopoiesis, in response to anemia, but must also reflect differences in splenic susceptibility to vascular occlusion and tissue fibrosis mediated by sickle cell disease. Paradoxically, despite their splenomegaly, SS mice demonstrate profound compromise of what is generally considered normal splenic function in humans as judged by the increased percentages of “pocked” erythrocytes and erythrocytes with Howell-Jolly bodies. In fact, these abnormalities are more severe in SS mice than in splenectomized WT mice. One possible explanation for these data is that the extra-splenic monocyte-macrophage system in mice contributes to removing and/or remodeling “pocked” cells and cells containing Howell-Jolly bodies and that SS mice have compromise of both the splenic and extra-splenic components of the monocyte-macrophage system. The splenomegaly observed in SS mice also differs functionally from splenomegaly in humans with regard to platelet sequestration since intravascular recovery of injected platelets in SS mice with splenomegaly was essentially normal, whereas intravascular recovery of platelets in humans with splenomegaly is invariably reduced(50).
Platelet size in SS mice was consistently increased, but the extent could not be quantitated by standard laser light scattering techniques because of contributions from erythrocyte fragments. As a result, we had to employ a combination of immunodetection of platelets based on their expression of αIIbβ3 and forward angle laser light scatter as an indicator of platelet size. We calibrated the FSC measurement with beads of known spherical diameter. The mean FSC value for SS mice platelets was 40% greater than the WT value, and this translated into a 32% increase in equivalent spherical diameter when analyzed on the reference curve produced by the beads of known spherical diameter. Since the volume of a sphere is proportional to the cube of its radius, we estimate the mean platelet volume of SS mice to be ~232% of that of WT mice. Thus, even though SS mice have an ~56% decrease in platelet count [which also contrasts with the mild thrombocytosis commonly found in patients with SCD(51;52)], their approximately two-fold increase in mean platelet volume results in their having a near normal total intravascular platelet mass. These data provide an explanation for why SS mice do not have elevated thrombopoietin levels despite their thrombocytopenia, since total platelet mass rather than platelet count is the prime determinant of thrombopoietin levels(53;54).
Large platelets in humans usually result from either “stress” thrombopoiesis, commonly due to shortened platelet survival, (55;56) or primary abnormalities in megakaryocyte maturation and fragmentation (57;58). We therefore also studied platelet kinetics in the SS mice. Platelet survival was only minimally shortened in SS mice, a finding consistent with their normal to low plasma glycocalicin levels (59), indicating that total thrombopoiesis is not increased. The one parameter suggesting thrombopoietic stress was the increased thiazole orange staining of platelets. An increase in platelets staining with thiazole-orange has been reported in humans with SCD, with the greatest increase in patients during pain crisis(52). Elevated erythropoietin levels in SS mice and patients with SCD may contribute to the increase, since erythropoietin therapy in neonates has been associated with an increase in thiazole orange-staining platelets even without producing an increase in platelet count (60).
Since humans with SCD variably demonstrate elevated markers of platelet activation, including enhanced binding of antibodies against the active conformation of the αIIbβ3 receptor and enhanced expression of platelet surface P-selectin (18;52;61), we assessed platelet surface P-selectin expression and fibrinogen binding after stimulation with a PAR-4 thrombin receptor agonist peptide. Platelets from SS mice demonstrated heightened responsiveness to activation evidenced by both increased P-selectin expression and fibrinogen binding with submaximal stimulation. The increased values could not be explained on the basis of the larger size of SS platelets because at maximal agonist stimulation, the values in SS mice were only modestly greater than those in WT mice. Our data are consistent with those of Villagra et al. who found that the platelets of humans with SCD and pulmonary hypertension had increased sensitivity to activation with ADP and a PAR-1 thrombin receptor activating peptide (62). They did not report platelet size in their patients, but data from other studies in humans have demonstrated that large platelets are functionally more active (63).
Whereas SS mice have both splenomegaly and large platelets we tried to assess whether the large platelets were coming primarily from the spleen. Splenectomy of WT mice did result in lower platelet FSC, but the differences were not significant (Table 1). In experiments not reported above, we also performed studies in which we transplanted bone marrow from SS mice into either WT or splenectomized WT mice. Eight weeks after transplantation, WT recipients had hematocrits and platelet FSC values (30 ± 4% and 42.6 ± 4.4; n=9) very similar to those of SS mice, along with the development of splenomegaly. Unfortunately, the combined splenectomy and transplantation experiments were limited by the inability of the splenectomized animals transplanted with SS bone marrow to sustain their hematocrits, demonstrating that splenic erythropoiesis is required for the survival of WT mice transplanted with SS bone marrow. Six of the seven animals did, however, survive to eight weeks post transplantation. Their hematocrits were much lower (13 ± 2%) than those in the other group, but their platelet FSC values (36.1 ± 1.0) were similar to those of the other group and those of SS mice, indicating that non-splenic murine megakaryopoiesis can produce large platelets. It is possible, however, that this response reflected the exceptional hematopoietic stress produced by the very low hematocrits and so these data may have limited relevance for interpreting the results in SS animals.
Since our subcolony of Berkeley SS mice has been isolated from the parent colony for approximately 20 generations and has not been backcrossed onto a single strain, we also studied SS mice from a colony that has been backcrossed onto the C57BL/6J background and found that their platelets also had elevated FSC values. Moreover, published data (64) and our own unpublished studies do not indicate differences in mean platelet volumes among the five different background strains of the SS mice.
There are conflicting data on platelet size in humans with SCD. Freedman and Karpatkin studied eight adults with SCD and found a 2.3-fold increase in the number of large platelets (megathrombocytes) during steady state (65), and Popescu and Martello reported qualitative data on increased platelet size in patients with SCD by blood smear assessment (66). However, a study of 16 patients with steady state sickle cell anemia in the U.K. found decreased mean platelet volume in patients compared with ethnically matched controls (7.6 ± 1.1 vs. 8.4 ± 0.9) (67). Most recently, a study of 89 Brazilian patients with SCD (including patients SC and Sβ-thalassemia) found an increase in the total number of platelets larger than 12 fl, especially in patients with pain crisis, but the percentage of large platelets was not increased(52).Genetic, environmental, and/or technical variations may account for the reported differences in platelet size in patients with SCD. Additional studies of platelet size in humans with SCD, along with appropriate controls, are needed to clarify this issue.
A broad range of abnormalities of platelets in humans with SCD have been reported, with the most consistent abnormalities being reductions in platelet number during crisis and elevated levels of platelet release products and soluble CD 40 ligand during steady state and sickle crisis (7–12;15;16;18;21;52;61;62;65;67–70). Evidence of intrinsic differences in SCD platelets comes from studies of the platelet transcriptome in SCD patients.(71) Decreased ex vivo platelet aggregation has been observed during steady state (10;12;72;73) and during sickle crisis (11;16;72), although some studies found platelet aggregation to be normal or even increased (7;12;69), and a recent study found an association between platelet activation, hemolysis, and pulmonary hypertension, suggesting decreased NO bioavailability as the link between the phenomena(62). Conducting optical platelet aggregation studies in both humans with SCD and SS mice presents technical problems because the presence of variable numbers of erythrocyte fragments in platelet-rich plasma may affect the results by decreasing the baseline and final light transmission values. It is unclear whether this was taken into consideration in the reported studies.
The role of platelets in the pathophysiology of human SCD remains unclear. While a single study found thrombocytosis to be associated with impaired cognitive dysfunction in children with SCD (74), epidemiological studies have not reported thrombocytosis to be associated with increased overall severity of SCD, (75–80), although such studies did not consider platelet size or mass. Despite the evidence for increased platelet consumption during painful crises (9), there are few data to support a role for platelets in microvascular obstruction, and small studies of aspirin, aspirin with dipyridamole, and ticlopidine failed to show a definitive benefit in reducing the frequency of pain crises (20–24). One potential link is through platelet surface expression of P-selectin and the recruitment of leukocytes since the latter have been implicated in contributing to vasoocclusion (81;82). A phase I study of the αIIbβ3 antagonist eptifibatide in four SCD patients found the therapy to be well tolerated and associated with reductions in platelet aggregation and plasma levels of myoglobin, soluble CD40 ligand, macrophage inflammatory protein-1α and TNF-α (83). It is interesting to speculate on a potential role of platelets in the development of pulmonary hypertension in patients with SCD since serotonin has been implicated in the development of pulmonary hypertension and platelets are both a rich source of serotonin and release it when activated (84). Thus, the association between platelet activation and pulmonary hypertension observed by Villagra et al. (62) may be causally related rather than two independent manifestations of hemolysis and decreased NO availability. Moreover, platelets may contribute to microvascular obstruction in the lung.
There is increasing evidence that a sizable fraction of ischemic strokes in children results from large vessel cerebrovascular occlusion due to a combination of arterial stenosis and superimposed thrombosis (28;29;85–87), raising the possibility that platelets contribute to stroke in children with SCD. Since large platelets have been associated with increased risk of cerebrovascular and cardiovascular events in humans without SCD, (88–93) it is possible that large platelets also contribute to the development of thrombotic stroke in patients with SCD. Thus, it may be appropriate to study whether antiplatelet therapy would decrease the risk of stroke in children with SCD and large vessel arterial stenosis, and currently trials using aspirin are being considered (94). It will be important, however, to exclude patients from such studies who are at high risk of cerebral hemorrhage (87;95), and to monitor the impact of aspirin therapy on vascular tone since patients with SCD have reduced responsiveness to NO and thus may be especially sensitive to inhibition of prostacyclin production (3;96;97).
Acknowledgments
We want to thank Melinda Ruiz for technical assistance and Suzanne Rivera for outstanding secretarial support.
This work was supported in part by grants HL19278 (B.S.C) and HL31579 (N.M) and contract NO-1-HB07086 (E.A.M) from the National Heart Lung and Blood Institute, a Clinical and Translational Science Award (UL1RR02414) from the national Center for Research Resources, and funds from Stony Brook University.
Footnotes
Presented in abstract form at the 45th annual meeting of the American Society of Hematology
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Paszty C, et al. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science. 1997;278:876–878. doi: 10.1126/science.278.5339.876. [DOI] [PubMed] [Google Scholar]
- 2.Pritchard KA, Jr, et al. Hypoxia-induced acute lung injury in murine models of sickle cell disease. Am J Physiol Lung Cell Mol Physiol. 2004;286:L705–L714. doi: 10.1152/ajplung.00288.2002. [DOI] [PubMed] [Google Scholar]
- 3.Kaul DK, Liu XD, Chang HY, Nagel RL, Fabry ME. Effect of fetal hemoglobin on microvascular regulation in sickle transgenic-knockout mice. J Clin Invest. 2004;114:1136–1145. doi: 10.1172/JCI21633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belcher JD, et al. Transgenic sickle mice have vascular inflammation. Blood. 2003;101:3953–3959. doi: 10.1182/blood-2002-10-3313. [DOI] [PubMed] [Google Scholar]
- 5.Manci EA, Hillery CA, Bodian CA, Zhang ZG, Lutty GA, Coller BS. Pathology of “Berkeley” sickle cell mice: similarities and differences with human sickle cell disease. Blood. 2006;107:1651–1658. doi: 10.1182/blood-2005-07-2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adamides S, Konstantopoulos K, Toumbis M, Douratsos D, Travlou A, Kasfiki A. A study of beta-thromboglobulin and platelet factor-4 plasma levels in steady state sickle cell patients. Blut. 1990;61:245–247. doi: 10.1007/BF01744139. [DOI] [PubMed] [Google Scholar]
- 7.Buchanan GR, Holtkamp CA. Platelet aggregation, malondialdehyde generation and production time in children with sickle cell anaemia. Thromb Haemost. 1981;46:690–693. [PubMed] [Google Scholar]
- 8.Browne PV, Mosher DF, Steinberg MH, Hebbel RP. Disturbance of plasma and platelet thrombospondin levels in sickle cell disease. Am J Hematol. 1996;51:296–301. doi: 10.1002/(SICI)1096-8652(199604)51:4<296::AID-AJH8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 9.Haut MJ, Cowan DH, Harris JW. Platelet function and survival in sickle cell disease. J Lab Clin Med. 1973;82:44–53. [PubMed] [Google Scholar]
- 10.Mehta P, Mehta J. Abnormalities of platelet aggregation in sickle cell disease. J Pediatr. 1980;96:209–213. doi: 10.1016/s0022-3476(80)80804-3. [DOI] [PubMed] [Google Scholar]
- 11.Stuart MJ, Stockman JA, Oski FA. Abnormalities of platelet aggregation in the vaso-occlusive crisis of sickle-cell anemia. J Pediatr. 1974;85:629–632. doi: 10.1016/s0022-3476(74)80504-4. [DOI] [PubMed] [Google Scholar]
- 12.Westwick J, et al. Platelet activation during steady state sickle cell disease. J Med. 1983;14:17–36. [PubMed] [Google Scholar]
- 13.Wun T, Paglieroni T, Tablin F, Welborn J, Nelson K, Cheung A. Platelet activation and platelet-erythrocyte aggregates in patients with sickle cell anemia. J Lab Clin Med. 1997;129:507–516. doi: 10.1016/s0022-2143(97)90005-6. [DOI] [PubMed] [Google Scholar]
- 14.Wun T, Cordoba M, Rangaswami A, Cheung AW, Paglieroni T. Activated monocytes and platelet-monocyte aggregates in patients with sickle cell disease. Clin Lab Haematol. 2002;24:81–88. doi: 10.1046/j.1365-2257.2002.00433.x. [DOI] [PubMed] [Google Scholar]
- 15.Mehta P, Mehta J. Circulating platelet aggregates in sickle cell disease patients with and without vaso-occlusion. Stroke. 1979;10:464–466. doi: 10.1161/01.str.10.4.464. [DOI] [PubMed] [Google Scholar]
- 16.Beurling-Harbury C, Schade SG. Platelet activation during pain crisis in sickle cell anemia patients. Am J Hematol. 1989;31:237–241. doi: 10.1002/ajh.2830310404. [DOI] [PubMed] [Google Scholar]
- 17.Francis RB. Platelets, coagulation, and fibrinolysis in sickle cell disease: their possible role in vascular occlusion. Blood Coagul Fibrinolysis. 1991;2:341–353. doi: 10.1097/00001721-199104000-00018. [DOI] [PubMed] [Google Scholar]
- 18.Tomer A, Harker LA, Kasey S, Eckman JR. Thrombogenesis in sickle cell disease. J Lab Clin Med. 2001;137:398–407. doi: 10.1067/mlc.2001.115450. [DOI] [PubMed] [Google Scholar]
- 19.Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004;364:1343–1360. doi: 10.1016/S0140-6736(04)17192-4. [DOI] [PubMed] [Google Scholar]
- 20.Greenberg J, Ohene-Frempong K, Halus J, Way C, Schwartz E. Trial of low doses of aspirin as prophylaxis in sickle cell disease. J Pediatr. 1983;102:781–784. doi: 10.1016/s0022-3476(83)80258-3. [DOI] [PubMed] [Google Scholar]
- 21.Semple MJ, Al Hasani SF, Kioy P, Savidge GF. A double-blind trial of ticlopidine in sickle cell disease. Thromb Haemost. 1984;51:303–306. [PubMed] [Google Scholar]
- 22.Cabannes R, Lonsdorfer J, Castaigne JP, Ondo A, Plassard A, Zohoun I. Clinical and biological double-blind-study of ticlopidine in preventive treatment of sickle-cell disease crises. Agents Actions Suppl. 1984;15:199–212. [PubMed] [Google Scholar]
- 23.Zago MA, Costa FF, Ismael SJ, Tone LG, Bottura C. Treatment of sickle cell diseases with aspirin. Acta Haematol. 1984;72:61–64. doi: 10.1159/000206360. [DOI] [PubMed] [Google Scholar]
- 24.Chaplin H, Jr, Alkjaersig N, Fletcher AP, Michael JM, Joist JH. Aspirin-dipyridamole prophylaxis of sickle cell disease pain crises. Thromb Haemost. 1980;43:218–221. [PubMed] [Google Scholar]
- 25.Stockman JA, Nigro MA, Mishkin MM, Oski FA. Occlusion of large cerebral vessels in sickle-cell anemia. N Engl J Med. 1972;287:846–849. doi: 10.1056/NEJM197210262871703. [DOI] [PubMed] [Google Scholar]
- 26.Merkel KH, Ginsberg PL, Parker JC, Jr, Post MJ. Cerebrovascular disease in sickle cell anemia: a clinical, pathological and radiological correlation. Stroke. 1978;9:45–52. doi: 10.1161/01.str.9.1.45. [DOI] [PubMed] [Google Scholar]
- 27.Rothman SM, Fulling KH, Nelson JS. Sickle cell anemia and central nervous system infarction: a neuropathological study. Ann Neurol. 1986;20:684–690. doi: 10.1002/ana.410200606. [DOI] [PubMed] [Google Scholar]
- 28.Steen RG, et al. Brain imaging findings in pediatric patients with sickle cell disease. Radiology. 2003;228:216–225. doi: 10.1148/radiol.2281020943. [DOI] [PubMed] [Google Scholar]
- 29.Tuohy AM, McKie V, Manci EA, Adams RJ. Internal carotid artery occlusion in a child with sickle cell disease: case report and immunohistochemical study. J Pediatr Hematol Oncol. 1997;19:455–458. doi: 10.1097/00043426-199709000-00009. [DOI] [PubMed] [Google Scholar]
- 30.Paszty C, et al. Lethal alpha-thalassaemia created by gene targeting in mice and its genetic rescue. Nat Genet. 1995;11:33–39. doi: 10.1038/ng0995-33. [DOI] [PubMed] [Google Scholar]
- 31.Ciavatta DJ, Ryan TM, Farmer SC, Townes TM. Mouse model of human beta zero thalassemia: targeted deletion of the mouse beta maj- and beta min-globin genes in embryonic stem cells. Proc Natl Acad Sci U S A. 1995;92:9259–9263. doi: 10.1073/pnas.92.20.9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skow LC, et al. A mouse model for beta-thalassemia. Cell. 1983;34:1043–1052. doi: 10.1016/0092-8674(83)90562-7. [DOI] [PubMed] [Google Scholar]
- 33.Lengweiler S, et al. Preparation of monoclonal antibodies to murine platelet glycoprotein IIb/IIIa (alphaIIbbeta3) and other proteins from hamster-mouse interspecies hybridomas. Biochem Biophys Res Commun. 1999;262:167–173. doi: 10.1006/bbrc.1999.1172. [DOI] [PubMed] [Google Scholar]
- 34.Bergmeier W, Rackebrandt K, Schroder W, Zirngibl H, Nieswandt B. Structural and functional characterization of the mouse von Willebrand factor receptor GPIb-IX with novel monoclonal antibodies. Blood. 2000;95:886–893. [PubMed] [Google Scholar]
- 35.Moritz A, Fickenscher Y, Meyer K, Failing K, Weiss DJ. Canine and feline hematology reference values for the ADVIA 120 hematology system. Vet Clin Pathol. 2004;33:32–38. doi: 10.1111/j.1939-165x.2004.tb00347.x. [DOI] [PubMed] [Google Scholar]
- 36.Alugupalli KR, Michelson AD, Barnard MR, Leong JM. Serial determinations of platelet counts in mice by flow cytometry. Thromb Haemost. 2001;86:668–671. [PubMed] [Google Scholar]
- 37.Holme S, Heaton A, Konchuba A, Hartman P. Light scatter and total protein signal distribution of platelets by flow cytometry as parameters of size. J Lab Clin Med. 1988;112:223–231. [PubMed] [Google Scholar]
- 38.Holme S, Heaton A, Konchuba A, Hartman P. Light scatter and total protein signal distribution of platelets by flow cytometry as parameters of size. J Lab Clin Med. 1988;112:223–231. [PubMed] [Google Scholar]
- 39.Schlenke P, et al. Evaluation of a flow cytometric method for simultaneous leukocyte phenotyping and quantification by fluorescent microspheres. Cytometry. 1998;33:310–317. doi: 10.1002/(sici)1097-0320(19981101)33:3<310::aid-cyto4>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 40.Tsuji T, Sakata T, Hamaguchi Y, Wang F, Houwen B. New rapid flow cytometric method for the enumeration of nucleated red blood cells. Cytometry. 1999;37:291–301. doi: 10.1002/(sici)1097-0320(19991201)37:4<291::aid-cyto6>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 41.Kina T, et al. The monoclonal antibody TER-119 recognizes a molecule associated with glycophorin A and specifically marks the late stages of murine erythroid lineage. Br J Haematol. 2000;109:280–287. doi: 10.1046/j.1365-2141.2000.02037.x. [DOI] [PubMed] [Google Scholar]
- 42.Lane PA, et al. Functional asplenia in hemoglobin SC disease. Blood. 1995;85:2238–2244. [PubMed] [Google Scholar]
- 43.Ault KA. Flow cytometric measurement of platelet function and reticulated platelets. Ann N Y Acad Sci. 1993;677:293–308. doi: 10.1111/j.1749-6632.1993.tb38785.x. [DOI] [PubMed] [Google Scholar]
- 44.Matic GB, Chapman ES, Zaiss M, Rothe G, Schmitz G. Whole blood analysis of reticulated platelets: improvements of detection and assay stability. Cytometry. 1998;34:229–234. doi: 10.1002/(sici)1097-0320(19981015)34:5<229::aid-cyto4>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 45.Baker GR, Sullam PM, Levin J. A simple, fluorescent method to internally label platelets suitable for physiological measurements. Am J Hematol. 1997;56:17–25. doi: 10.1002/(sici)1096-8652(199709)56:1<17::aid-ajh4>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 46.Diehl KH, et al. A good practice guide to the administration of substances and removal of blood, including routes and volumes. J Appl Toxicol. 2001;21:15–23. doi: 10.1002/jat.727. [DOI] [PubMed] [Google Scholar]
- 47.Heilmann E, et al. Biotinylated platelets: a new approach to the measurement of platelet life span. Br J Haematol. 1993;85:729–735. doi: 10.1111/j.1365-2141.1993.tb03216.x. [DOI] [PubMed] [Google Scholar]
- 48.Berger G, Hartwell DW, Wagner DD. P-Selectin and platelet clearance. Blood. 1998;92:4446–4452. [PubMed] [Google Scholar]
- 49.Faruqi TR, Weiss EJ, Shapiro MJ, Huang W, Coughlin SR. Structure-function analysis of protease-activated receptor 4 tethered ligand peptides. Determinants of specificity and utility in assays of receptor function. J Biol Chem. 2000;275:19728–19734. doi: 10.1074/jbc.M909960199. [DOI] [PubMed] [Google Scholar]
- 50.McFarland JG, Anderson AJ, Slichter SJ. Factors influencing the transfusion response to HLA-selected apheresis donor platelets in patients refractory to random platelet concentrates. Br J Haematol. 1989;73:380–386. doi: 10.1111/j.1365-2141.1989.tb07757.x. [DOI] [PubMed] [Google Scholar]
- 51.Brown AK, Sleeper LA, Miller ST, Pegelow CH, Gill FM, Waclawiw MA. Reference values and hematologic changes from birth to 5 years in patients with sickle cell disease. Cooperative Study of Sickle Cell Disease. Arch Pediatr Adolesc Med. 1994;148:796–804. doi: 10.1001/archpedi.1994.02170080026005. [DOI] [PubMed] [Google Scholar]
- 52.Noronha JF, Costa FF, Saad ST, Lorand-Metze IG, Grotto HZ. Evaluation of reticulated platelets in patients with sickle cell diseases. Thromb Res. 2007 doi: 10.1016/j.thromres.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 53.Kaushansky K. Lineage-specific hematopoietic growth factors. N Engl J Med. 2006;354:2034–2045. doi: 10.1056/NEJMra052706. [DOI] [PubMed] [Google Scholar]
- 54.Kuter DJ. New thrombopoietic growth factors. Blood. 2007;109:4607–4616. doi: 10.1182/blood-2006-10-019315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang C, Smith BR, Ault KA, Rinder HM. Reticulated platelets predict platelet count recovery following chemotherapy. Transfusion. 2002;42:368–374. doi: 10.1046/j.1537-2995.2002.00040.x. [DOI] [PubMed] [Google Scholar]
- 56.Garg SK, Amorosi EL, Karpatkin S. Use of the megathrombocyte as an index of megakaryocyte number. N Engl J Med. 1971;284:11–17. doi: 10.1056/NEJM197101072840103. [DOI] [PubMed] [Google Scholar]
- 57.Tablin F, Castro M, Leven RM. Blood platelet formation in vitro. The role of the cytoskeleton in megakaryocyte fragmentation. J Cell Sci. 1990;97 (Pt 1):59–70. doi: 10.1242/jcs.97.1.59. [DOI] [PubMed] [Google Scholar]
- 58.Li J, Kuter DJ. The end is just the beginning: megakaryocyte apoptosis and platelet release. Int J Hematol. 2001;74:365–374. doi: 10.1007/BF02982078. [DOI] [PubMed] [Google Scholar]
- 59.Steinberg MH, Kelton JG, Coller BS. Plasma glycocalicin. An aid in the classification of thrombocytopenic disorders. N Engl J Med. 1987;317:1037–1042. doi: 10.1056/NEJM198710223171701. [DOI] [PubMed] [Google Scholar]
- 60.Haiden N, et al. Changes in thrombopoiesis and platelet reactivity in extremely low birth weight infants undergoing erythropoietin therapy for treatment of anaemia of prematurity. Thromb Haemost. 2005;93:118–123. doi: 10.1160/TH04-02-0093. [DOI] [PubMed] [Google Scholar]
- 61.Wun T, et al. Platelet activation in patients with sickle cell disease. Br J Haematol. 1998;100:741–749. doi: 10.1046/j.1365-2141.1998.00627.x. [DOI] [PubMed] [Google Scholar]
- 62.Villagra J, Shiva S, Hunter LA, Machado RF, Gladwin MT, Kato GJ. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood. 2007;110:2166–2172. doi: 10.1182/blood-2006-12-061697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Breimo ES, Osterud B. Studies of biological functions in blood cells from individuals with large platelets. Platelets. 2003;14:413–419. doi: 10.1080/02697450310001632597. [DOI] [PubMed] [Google Scholar]
- 64.Peters LL, et al. Large-scale, high-throughput screening for coagulation and hematologic phenotypes in mice. Physiol Genomics. 2002;11:185–193. doi: 10.1152/physiolgenomics.00077.2002. [DOI] [PubMed] [Google Scholar]
- 65.Freedman ML, Karpatkin S. Elevated platelet count and megathrombocyte number in sickle cell anemia. Blood. 1975;46:579–582. [PubMed] [Google Scholar]
- 66.Popescu ER, Martelo OJ. Megathrombocytes and sickle cell anemia. Blood. 1977;49:490–491. [PubMed] [Google Scholar]
- 67.Mohan JS, Lip GY, Bareford D, Blann AD. Platelet P-selectin and platelet mass, volume and component in sickle cell disease: Relationship to genotype. Thromb Res. 2005 doi: 10.1016/j.thromres.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 68.Inwald DP, et al. Platelet and leucocyte activation in childhood sickle cell disease: association with nocturnal hypoxaemia. Br J Haematol. 2000;111:474–481. doi: 10.1046/j.1365-2141.2000.02353.x. [DOI] [PubMed] [Google Scholar]
- 69.Kenny MW, George AJ, Stuart J. Platelet hyperactivity in sickle-cell disease: a consequence of hyposplenism. J Clin Pathol. 1980;33:622–625. doi: 10.1136/jcp.33.7.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee SP, Ataga KI, Orringer EP, Phillips DR, Parise LV. Biologically active CD40 ligand is elevated in sickle cell anemia: potential role for platelet-mediated inflammation. Arterioscler Thromb Vasc Biol. 2006;26:1626–1631. doi: 10.1161/01.ATV.0000220374.00602.a2. [DOI] [PubMed] [Google Scholar]
- 71.Raghavachari N, et al. Amplified expression profiling of platelet transcriptome reveals changes in arginine metabolic pathways in patients with sickle cell disease. Circulation. 2007;115:1551–1562. doi: 10.1161/CIRCULATIONAHA.106.658641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gruppo RA, Glueck HI, Granger SM, Miller MA. Platelet function in sickle cell anemia. Thromb Res. 1977;10:235. doi: 10.1016/0049-3848(77)90145-1. [DOI] [PubMed] [Google Scholar]
- 73.Sarji KE, Eurenius K, Fullwood CO, Schraibman HB, Colwell JA. Abnormalities of platelet aggregation in sickle cell anemia. Presence of a plasma factor inhibiting aggregation by ristocetin. Thromb Res. 1979;14:283–297. doi: 10.1016/0049-3848(79)90238-x. [DOI] [PubMed] [Google Scholar]
- 74.Bernaudin F, et al. Multicenter prospective study of children with sickle cell disease: radiographic and psychometric correlation. J Child Neurol. 2000;15:333–343. doi: 10.1177/088307380001500510. [DOI] [PubMed] [Google Scholar]
- 75.Hayes RJ, Beckford M, Grandison Y, Mason K, Serjeant BE, Serjeant GR. The haematology of steady state homozygous sickle cell disease: frequency distributions, variation with age and sex, longitudinal observations. Br J Haematol. 1985;59:369–382. doi: 10.1111/j.1365-2141.1985.tb03002.x. [DOI] [PubMed] [Google Scholar]
- 76.Miller ST, et al. Prediction of adverse outcomes in children with sickle cell disease. N Engl J Med. 2000;342:83–89. doi: 10.1056/NEJM200001133420203. [DOI] [PubMed] [Google Scholar]
- 77.Morris J, et al. The haematology of homozygous sickle cell disease after the age of 40 years. Br J Haematol. 1991;77:382–385. doi: 10.1111/j.1365-2141.1991.tb08588.x. [DOI] [PubMed] [Google Scholar]
- 78.McKerrell TD, Cohen HW, Billett HH. The older sickle cell patient. Am J Hematol. 2004;76:101–106. doi: 10.1002/ajh.20075. [DOI] [PubMed] [Google Scholar]
- 79.Platt OS, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 80.Sebastiani P, et al. A network model to predict the risk of death in sickle cell disease. Blood. 2007;110:2727–2735. doi: 10.1182/blood-2007-04-084921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S A. 2002;99:3047–3051. doi: 10.1073/pnas.052522799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Smyth SS, Reis ED, Zhang W, Fallon JT, Gordon RE, Coller BS. Beta(3)-integrin-deficient mice but not P-selectin-deficient mice develop intimal hyperplasia after vascular injury: correlation with leukocyte recruitment to adherent platelets 1 hour after injury. Circulation. 2001;103:2501–2507. doi: 10.1161/01.cir.103.20.2501. [DOI] [PubMed] [Google Scholar]
- 83.Lee SP, et al. Phase I study of eptifibatide in patients with sickle cell anaemia. Br J Haematol. 2007;139:612–620. doi: 10.1111/j.1365-2141.2007.06787.x. [DOI] [PubMed] [Google Scholar]
- 84.de Caestecker M. Serotonin signaling in pulmonary hypertension. Circ Res. 2006;98:1229–1231. doi: 10.1161/01.RES.0000225927.04710.33. [DOI] [PubMed] [Google Scholar]
- 85.Hoppe C. Defining stroke risk in children with sickle cell anaemia. Br J Haematol. 2005;128:751–766. doi: 10.1111/j.1365-2141.2004.05310.x. [DOI] [PubMed] [Google Scholar]
- 86.Koshy M, Thomas C, Goodwin J. Vascular lesions in the central nervous system in sickle cell disease (neuropathology) J Assoc Acad Minor Phys. 1990;1:71–78. [PubMed] [Google Scholar]
- 87.Kirkham FJ. Therapy insight: stroke risk and its management in patients with sickle cell disease. Nat Clin Pract Neurol. 2007;3:264–278. doi: 10.1038/ncpneuro0495. [DOI] [PubMed] [Google Scholar]
- 88.O’Malley T, Langhorne P, Elton RA, Stewart C. Platelet size in stroke patients. Stroke. 1995;26:995–999. doi: 10.1161/01.str.26.6.995. [DOI] [PubMed] [Google Scholar]
- 89.Martin JF, Bath PM, Burr ML. Influence of platelet size on outcome after myocardial infarction. Lancet. 1991;338:1409–1411. doi: 10.1016/0140-6736(91)92719-i. [DOI] [PubMed] [Google Scholar]
- 90.Greisenegger S, Endler G, Hsieh K, Tentschert S, Mannhalter C, Lalouschek W. Is elevated mean platelet volume associated with a worse outcome in patients with acute ischemic cerebrovascular events? Stroke. 2004;35:1688–1691. doi: 10.1161/01.STR.0000130512.81212.a2. [DOI] [PubMed] [Google Scholar]
- 91.Bath P, Algert C, Chapman N, Neal B. Association of mean platelet volume with risk of stroke among 3134 individuals with history of cerebrovascular disease. Stroke. 2004;35:622–626. doi: 10.1161/01.STR.0000116105.26237.EC. [DOI] [PubMed] [Google Scholar]
- 92.Pizzulli L, Yang A, Martin JF, Luderitz B. Changes in platelet size and count in unstable angina compared to stable angina or non-cardiac chest pain. Eur Heart J. 1998;19:80–84. doi: 10.1053/euhj.1997.0747. [DOI] [PubMed] [Google Scholar]
- 93.Huczek Z, et al. Mean platelet volume on admission predicts impaired reperfusion and long-term mortality in acute myocardial infarction treated with primary percutaneous coronary intervention. J Am Coll Cardiol. 2005;46:284–290. doi: 10.1016/j.jacc.2005.03.065. [DOI] [PubMed] [Google Scholar]
- 94.Kirkham FJ, et al. Trials in sickle cell disease. Pediatr Neurol. 2006;34:450–458. doi: 10.1016/j.pediatrneurol.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 95.Dobson SR, et al. Moyamoya syndrome in childhood sickle cell disease: a predictive factor for recurrent cerebrovascular events. Blood. 2002;99:3144–3150. doi: 10.1182/blood.v99.9.3144. [DOI] [PubMed] [Google Scholar]
- 96.Reiter CD, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 97.Eberhardt RT, et al. Sickle cell anemia is associated with reduced nitric oxide bioactivity in peripheral conduit and resistance vessels. Am J Hematol. 2003;74:104–111. doi: 10.1002/ajh.10387. [DOI] [PubMed] [Google Scholar]