

Abstract
Initial Ca2+-dependent contraction of intestinal smooth muscle is inhibited upon IL-1β treatment. The decrease in contraction reflects the upregulation of regulator of G protein signaling-4 (RGS4) via the canonical inhibitor of NF-κB kinase-2 (IKK2)/IκB-α/NF-κB pathway. Here, we show that the activation of various protein kinases, including ERK1/2, p38 MAPK, and phosphoinositide 3-kinase (PI3K), differentially modulates IL-1β-induced upregulation of RGS4 in rabbit colonic muscle cells. IL-1β treatment caused a transient phosphorylation of ERK1/2 and p38 MAPK. It also caused the phosphorylation of Akt and glycogen synthase kinase-3β (GSK3β), sequential downstream effectors of PI3K. Pretreatment with PD-98059 (an ERK inhibitor) and SB-203580 (a p38 MAPK inhibitor) significantly inhibited IL-1β-induced RGS4 expression. In contrast, LY-294002 (a PI3K inhibitor) augmented, whereas GSK3β inhibitors inhibited, IL-1β-induced RGS4 expression. PD-98059 blocked IL-1β-induced phosphorylation of IKK2, degradation of IκB-α, and phosphorylation and nuclear translocation of NF-κB subunit p65, whereas SB-203580 had a marginal effect, implying that the effect of ERK1/2 is exerted on the canonical IKK2/IκB-α/p65 pathway of NF-κB activation but that the effect of p38 MAPK may not predominantly involve NF-κB signaling. The increase in RGS4 expression enhanced by LY-294002 was accompanied by an increase in the phosphorylation of IKK2/IκB-α/p65 and blocked by pretreatment with inhibitors of IKK2 (IKK2-IV) and IκB-α (MG-132). Inhibition of GSK3β abolished IL-1β-induced phosphorylation of IKK2/p65. These findings suggest that ERK1/2 and p38 MAPK enhance IL-1β-induced upregulation of RGS4; the effect of ERK1/2 reflects its ability to promote IKK2 phosphorylation and increase NF-κB activity. GSK3β acts normally to augment the activation of the canonical NF-κB signaling. The PI3K/Akt/GSK3β pathway attenuates IL-1β-induced upregulation of RGS4 expression by inhibiting NF-κB activation.
Keywords: smooth muscle cells, rabbit, short interfering RNA, nuclear factor-κB, signal transduction, regulator of G protein signaling, interleukin-1β, extracellular signal-regulated kinase 1/2, mitogen-activated protein kinase, phosphoinositide 3-kinase
inflammatory mediators, including cytokines, chemokines, growth factors, and cell adhesion molecules, contribute to the maintenance and resolution of the inflammatory responses in patients with either inflammatory bowel diseases (IBD) or irritable bowel syndrome (IBS). Different patterns of inflammatory cytokines have been identified that distinctly regulate the motility of gastrointestinal smooth muscle (51, 69, 81). A pattern involving the time-dependent release of IL-1β, TNF-α, IL-6, and IL-8 is accompanied by decreases in the response of smooth muscle to excitatory neurotransmitters (acetylcholine, neurokinin A, etc.) (70, 77, 81, 85), whereas the pattern of T helper cell (Th)2 cytokines observed with helminth infection involves the transient activation of IL-4 and IL-13, resulting in initial hypercontractility followed by sustained expression of transforming growth factor-β1 and cyclooxygenase (COX)-2, leading to persistent hypercontractility (2–5, 51, 106). Recently, an additional pattern of Th17 cytokines, including IL-17 and IL-23, has been shown to play an important role in the pathophysiology of IBD and IBS (84, 86, 100, 105). The specific steps in the signaling pathways mediating the contraction or relaxation of smooth muscle that are affected by these cytokine patterns have not been identified.
IL-1β has been well known to inhibit the contractile response of intestinal smooth muscle (8, 9, 15, 18, 31, 48, 49, 62, 67, 95). The mechanisms for such inhibition may involve inhibitory neural regulation on the release of excitatory neurotransmitters (8, 9, 14, 67) and/or a reduction in muscular contractile responses (15, 18, 70, 85). The cellular mechanisms are becoming increasingly identified. H2O2, formed in colonic and esophageal sphincter smooth muscle in response to IL-1β (16, 18), inhibits contraction by interfering with Ca2+ mobilization. IL-1β treatment of rat ileal smooth muscle strip decreases the phosphorylation of myosin light chain phosphatase-targeting subunit 1 and protein expression of 17-kDa PKC-potentiated myosin phosphatase inhibitor (CPI-17), the two key signaling components in mediating agonist-induced sustained (tonic) contraction of smooth muscle (70). By screening the signaling targets mediating IL-1β-induced inhibition on acetylcholine-stimulated initial and sustained contraction in isolated or cultured colonic smooth muscle cells (SMCs), we have previously demonstrated that IL-1β upregulates regulator of G protein signaling-4 (RGS4) expression, which contributes to the inhibitory effect of IL-1β on the initial contraction, and confirmed that IL-1β downregulates CPI-17 expression, which is associated with IL-1β-induced inhibition on the sustained contraction (37).
RGS4 is one of seven members of the classic R4 RGS protein family that accelerates the intrinsic GTPase activity of Gαi/o and Gαq/11 family members. RGS4 is well known to regulate the strength and duration of Gαq signaling and plays an important role in regulating smooth muscle contraction, cardiomyocyte development, neural plasticity, and psychiatric disorders (36, 37, 59, 99). However, the regulatory mechanisms of RGS4 expression remain elusive. At the protein level, RGS4 is regulated by the NH2-end rule pathway (10). At the mRNA level, RGS4 is regulated by the neural type-specific transcription factor Phox2b (28). Our recent study (36) provided the first evidence that IL-1β-induced upregulation of RGS4 is transcription dependent and mediated by the canonical inhibitor of NF-κB kinase-2 (IKK2)/IκB-α pathway of NF-κB activation (36).
MAPKs are a family of serine/threonine kinases and are activated upon dual phosphorylation at threonine and tyrosine by upstream kinases in response to diverse extracellular stimuli. Recent evidence has suggested that both ERK1/2 and p38 MAPK are implicated in the Ca2+ sensitization (39) and PKC-dependent contraction of gastrointestinal smooth muscle (17, 39). However, the mechanisms by which these MAPKs modulate smooth muscle contraction are not well understood. Phosphorylation of caldesmon and/or calponin may contribute to the effect of ERK1/2 (41, 54), whereas p38 MAPK may regulate muscle contraction through the sequential phosphorylation and activation of MAPK-activated protein kinase-2 (94) and heat shock protein 27 (102).
Phosphoinositide 3-kinases (PI3K) are heterodimeric proteins and linked to an extraordinarily diverse group of cellular functions. The product of PI3K, phosphotidylinositol-3,4,5-triphosphate, triggers the accumulation of pleckstrin homology domain-containing proteins such as the serine-threonine kinase Akt. Activated Akt inactivates glycogen synthase kinase-3 (GSK3) through phosphorylation (55, 61). IL-1β is known to activate the PI3K/Akt/GSK3β pathway in several types of cells, such as epithelia cells, hepatocytes, sertolic cells, and airway SMCs (23). Our previous study (38) has shown that Gi-coupled receptors activate PI3K in gut SMCs. However, the effect of the PI3K/Akt/GSK3β pathway on gut smooth muscle contraction remains to be elucidated.
Here, we show that IL-1β-induced activation of either ERK1/2 or p38 MAPK enhances the upregulation of RGS4 expression, which reflects the ability to inhibit initial contraction, whereas the PI3K/Akt/GSK3β pathway attenuates IL-1β-induced upregulation of RGS4 expression. The effect of the ERK1/2 and PI3K pathway is dependent on the signaling of NF-κB activation, whereas p38 MAPK exerts its role independently of NF-κB signaling.
MATERIALS AND METHODS
Reagents and antibodies.
IL-1β was obtained from Alexis Biochemicals (San Diego, CA). 2′-Amino-3′-methoxyflavone (PD-98059), 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole (SB-203580), 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY-294002), triciribine [Akt/PKB signaling inhibitor-2 (API-2)], 3-[1-(3-hydroxypropyl)-1H-pyrrolo-(2,3-b)pyridin-3-yl]-4-pyrazin-2-yl-pyrrole-2,5-dione [a specific GSK3β inhibitor (GSK3β-XI)], 4-(4′-phenoxyanilino)-6,7-dimethoxyquinazoline (Src kinase inhibitor I), [5-(p-fluorophenyl)-2-ureido]thiophene-3-carboxamide [IKK2 inhibitor IV (IKK2-IV)], and carbobenzoxy-l-leucyl-l-leucyl-l-leucinal Z-LLL-CHO (MG-132) were from EMD Chemicals (San Diego, CA) and dissolved in DMSO. Antibodies against p65, IKK2, IκB-α, GAPDH, and β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Affinity-purified anti-RGS4 antibody was kindly provided by Dr. Susanne M. Mumby (University of Texas Southwest Medical Center). Antibodies against ERK1/2, phospho-ERK1/2 (Thr202/Tyr204), phospho-p38 MAPK (Thr180/Tyr182), phosphor-Akt (Ser473), phospho-GSK3β (Ser9), phospho-IKK2 (Ser177/Ser181), phospho-IκB-α (Ser32/Ser36), and phospho-p65 (Ser546) were from Cell Signal Technology (Danvers, MA). All other reagents were from Sigma (St. Louis, MO).
Isolation and culture of SMCs.
Institutional Animal Care and Use Committee (IACUC) approval was obtained from Virginia Commonwealth University. Rabbit colonic circular muscle cells were isolated and cultured as previously described (37, 66). Briefly, the distal colon from euthanized New Zealand White rabbits (2∼2.5 kg) was placed in HEPES-buffered smooth muscle media. The circular smooth muscle layer was dissected from the mucosa and the longitudinal muscle layer under a stereomicroscope and treated with 0.1% collagenase (type II) and 0.1% soybean trypsin inhibitor for 30 min at 31°C. The isolated single muscle cells after several rounds of spontaneous dispersion were harvested by filtration through 500-μm Nitex and centrifuged twice at 350 g for 10 min. Aliquots of freshly isolated SMCs in HEPES-buffered smooth muscle media without serum and antibiotics were placed in six-well plates and incubated at 37°C for 30 min before treatment with various inhibitors and cytokines. For cultures, isolated SMCs were placed in a 100-mm dish with DMEM containing 10% FBS and 1% antibiotics and antimycotics. After 10–14 days, SMCs attained confluence and were then passaged once for use in various experiments. Full confluent muscle cells were deprived of serum for 24 h before experiments.
Conventional and real-time RT-PCR.
Freshly dispersed or cultured colonic SMCs were treated with the TRIzol reagent (Invitrogen, Carlsbad, CA) for total RNA extraction. The potentially contaminated genomic DNA was removed by treating 10 μg of the RNA sample at 37°C for 30 min with 1 μl of TURBO DNase (Ambion, Austin, TX) followed by an extraction with phenol-chloroform-isoamylalcohol (25:24:1). RNA (2 μg) was used to synthesize cDNA using SuperScript II reverse transcriptase (Invitrogen) with random hexanucleotide primers. Conventional PCR was performed on cDNA using the HotMaster Taq DNA polymerase kit (Eppendorf). The primer sequences for rabbit RGS4 (GenBank Accession No. DQ120011) were forward 5′-ATGTGCAAAGGACTTGCAGGTC-3′ and reverse 5′-GTGAGAATTAGGCACACTGGG-3′, generating a fragment of 624 bp. The primer sequences for rabbit GAPDH (GenBank Accession No. DQ403051) were forward 5′-TCACCATCTTCCAGGAGCGA-3′ and reverse 5′-CACAATGCCGAAGTGGTCGT-3′, generating a fragment of 292 bp. The PCR product was purified and cloned into the T-A vector for confirmation by sequencing.
Real-time PCR analysis was carried out on the ABI Prism 7300 Sequence Detection System (Applied Biosystems, Foster, CA). Expression of RGS4 was analyzed using the TaqMan PCR Master Mix Reagent Kit (Applied Biosystems). The TaqMan probe and primers for rabbit RGS4 designed using Primer Express (version 2.0) were as follows: forward (nucleotides 232–252, exon 2) 5′-TCCCACAGCAAGAAGGACAAA-3′; reverse (nucleotides 303–284, exon 3) 5′-TTCGGCCCATTTCTTGACTT-3′; and probe (nucleotides 254–279, across exons 2 and 3 with 321 bp of intron 2) 5′-TTGACTCACCCTCTGGCAAACAACCA-3′. cDNA was synthesized from 500 ng RNA using the TaqMan RT Reagent Kit (Applied Biosystems). The optimized concentrations for real-time PCR were 0.4 μM for both primers and 0.2 μM for probe and 5 ng cDNA in a 20-μl reaction volume. Rabbit GAPDH primers (forward 5′-CGCCTGGAGAAAGCTGCTAA-3′ and reverse 5′-CGACCTGGTCCTCGGTGTAG-3′) were used as internal controls. Each sample was tested in triplicate. Cycle threshold (Ct) values were obtained graphically for RGS4 and GAPDH. The difference in Ct values between GAPDH and RGS4 were represented as ΔCt values. ΔΔCt values were obtained by subtracting ΔCt values of control samples from those of treated samples. The relative fold change in gene expression was calculated as 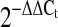 .
.
Immunofluorescent cytochemistry and semiquantitative analysis.
SMCs were seeded on eight-well glass chamber slides (Nalge Nunc, Lab-Teck, Rochester, NY) and cultured untill full confluence. After 24 h of serum starvation, cells were treated with IL-1β for different time periods followed by a fixation with 4% paraformaldehyde-PBS for 30 min. After being washed with PBS, cells were treated with 0.5% Triton X-100 for 30 min, blocked with 10% normal donkey serum for 1 h, and incubated with the primary anti-p65 polyclonal antibody (1:200) for 2 h. After cells had been washed, the Alexa fluor-488 (green)-linked donkey anti-rabbit secondary antibody (1:200, Molecular Probes, Eugene, OR) was applied for 1 h. The staining specificity was determined by omitting the primary antibody. Hoechst 33258 was used for counterstaining of nuclei. Slides were coverslipped with antifading aqueous mounting media (Biomeda, Foster City, CA). Fluorescent images were taken under the fluorescent invert microscope using NIS Elements F (version 2.10) software (Nikon, Japan). Fluorescences of p65 and Hoechst staining were captured using sequential acquisition. Image analysis of p65 nuclear translocation was performed using NIH ImageJ software according to previously described methods (22, 26, 68). Briefly, the nuclear mask was generated by applying a median filter (3 × 3-pixel radius) and automatic thresholding of nuclear staining followed by a 1-pixel erosion to avoid cytoplasmic contamination. The cytoplasmic mask was generated using a 3-pixel dilation of the nuclear mask and subtraction. Each mask was then applied to the original p65 staining image to obtain the integrated density, and the ratio of the nuclear and cytoplasmic integral was compared between different groups (see Fig. 3C).
Western blot analysis.
Freshly isolated and cultured SMCs were solubilized for 30 min in Triton X-100-based lysis buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 100 μg/ml PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 30 mM sodium fluoride, and 3 mM sodium vanadate. After centrifugation of the lysates at 20,000 g for 10 min at 4°C, protein concentrations of the supernatant were determined with the DC Protein Assay kit from Bio-Rad (Hercules, CA). Equal amount of proteins were fractionated by SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad). Blots were blocked in 5% nonfat dry milk with Tris-buffered saline (TBS; pH 7.6) plus 0.1% Tween 20 (TBS-T) for 1 h and then incubated overnight at 4°C with various primary antibodies in TBS-T plus 1% milk. The dilution of 1:1,000 was used for most primary antibodies except for anti-RGS4 (1:10,000) and β-actin (1:100,000). After incubation for 1 h with horseradish peroxidase-conjugated corresponding secondary antibody (1/2,000, 10 μg/ml, Pierce) in TBS-T plus 1% milk, immunoreactive proteins were visualized using the SuperSignal Femto maximum sensitivity substrate kit (Pierce, Rocjford, IL). All washing steps were performed with TBS-T.
Statistical analysis.
Images from Western blots and RT-PCR were scanned and analyzed with NIH ImageJ (version 1.41) densitometric measurements. Data are expressed as integrated densities and presented as relative fold changes compared with the corresponding control. Quantitative data are expressed as means ± SE of n experiments, and statistical significance was determined using Student's t-test for unpaired values. The ratio of nuclear to cytoplasmic fluorescence of p65 was analyzed by ANOVA and Newman-Keuls comparison.
RESULTS
Activation of MAPK by IL-1β in rabbit colonic SMCs.
The activation of ERK1/2 and p38 MAPK in rabbit colonic SMCs was examined by determining the level of phosphorylation of ERK1/2 at Thr202/Tyr204 and p38 MAPK at Thr180/Tyr182. As shown in Fig. 1, exposure of cultured and serum-starved rabbit circular colonic muscle cells to IL-1β (10 ng/ml) led to a rapid and transient phosphorylation of ERK1/2 and p38 MAPK. Parallel experiments using freshly isolated colonic SMCs were performed. As shown in Fig. 1B, IL-1β treatment of freshly isolated SMCs induced a similar pattern of activation to p38 MAPK. In addition, the constitutive phosphorylation of MAPK was detectable in freshly isolated muscle cells, which may result from residual serum. A similar pattern of activation occurred with ERK1/2 and PI3K (data not shown).
Fig. 1.

IL-1β induces a rapid and transient phosphorylation of ERK1/2 and p38 MAPK in rabbit colonic smooth muscle cells (SMCs). Cultured and serum-starved (left; duplicate) or freshly isolated (right) SMCs were treated with IL-1β (10 ng/ml) for indicated time periods followed by Western blot analysis with anti-phosphorylated (p-)ERK1/2 (A) or anti-p-p38 (B) antibodies. Expression levels of total ERK1/2 (A) or p38 (B) were used as internal controls. The number under each blot indicates the relative fold of optical density compared with the corresponding control.
ERK1/2 and p38 MAPK enhance IL-1β-induced upregulation of RGS4 expression.
Our previous study (36) demonstrated that IL-1β induced a time-dependent upregulation of RGS4 mRNA and protein. To determine whether the activation of endogenous ERK1/2 and/or p38 MAPK is involved in IL-1β-induced upregulation of RGS4, selective inhibitors for MEK1 (PD-98059) or p38 MAPK (SB-203580) were used. The optimal concentrations of both inhibitors were based on our preliminary studies and previous reports (38, 60, 87). The efficacy and specificity of the inhibitors were validated by Western blot analysis in cultured SMCs after IL-1β exposure for 15 min using anti-phospho-specific antibody against the corresponding MAPK (Fig. 2A). Treatment with either inhibitor alone for 4 and 24 h did not effect the basal expression of RGS4 in serum-starved colonic SMCs (data not shown). Pretreatment with either PD-98059 (20 μM) or SB-203580 (1 μM) for 1 h before IL-1β exposure for 3 h significantly inhibited IL-1β-induced increases in the expression of RGS4 mRNA and protein, as determined by real-time RT-PCR (Fig. 2B) and Western blot analysis (Fig. 2, C and D), respectively. These data suggest that ERK1/2 and p38 MAPK are stimulatory for IL-1β-induced upregulation of RGS4 expression.
Fig. 2.

Inhibition of either ERK1/2 or p38 MAPK significantly blocks IL-1β-induced upregulation of RGS4 expression in rabbit colonic SMCs. Cultured SMCs were starved for 24 h and pretreated with the p38 MAPK inhibitor SB-203580 (1 μM) or MEK1 inhibitor PD-98059 (20 μM) for 1 h before exposure to IL-1β (10 ng/ml) for 15 min (A) and 3 h (B–D). The effectiveness of SB-203580 and PD-98059 to inhibit p38 MAPK and MEK1 was confirmed by Western blot analysis (A). The expression of regulator of G protein signaling-4 (RGS4) at the mRNA (B) and protein (C and D) levels was determined by real-time RT-PCR and Western blot analysis, respectively. The level of β-actin was used as a loading control. Relative levels of RGS4 mRNA expression (fold induction) are shown compared with the control after GAPDH normalization. Relative density (in %) is shown compared with the control after β-actin normalization. **P < 0.01, significant decrease after IL-1β treatment by the inhibitors compared with DMSO.
ERK1/2 enhances RGS4 upregulation via NF-κB activation, whereas p38 MAPK enhances RGS4 expression independently of NF-κB signaling.
The canonical IKK2/IκB-α pathway of NF-κB activation mediates IL-1β-induced upregulation of RGS4 (36). To address the interaction between the ERK1/2 and/or p38 MAPK pathways and the NF-κB signaling pathway, we performed Western blot analysis and immunocytochemistry in cells treated with selected MAPK inhibitors. The MEK inhibitor (PD-98059, 20 μM) almost completely blocked (from 26- to 3-fold) IL-1β-induced phosphorylation of p65 (Ser536; Fig. 3A), implying that the effect of ERK1/2 on RGS4 expression was dependent on NF-κB signaling, predominantly at the level of p65. The MEK inhibitor also inhibited the phosphorylation of IKK2 (Ser177/Ser181, from 112- to 13-fold) and the degradation of IκB-α (from 99% to 63%), suggesting that ERK1/2 also exerted its effect at least partially on IκB-α and IKK2 or its upstream signaling components. On the other hand, the p38 MAPK inhibitor (SB-203580, 1 μM) had a marginal effect on IκB-α degradation and p65 phosphorylation (Fig. 3A), implying that NF-κB activation may not be the predominant target for the effect of p38 MAPK on RGS4 expression. However, SB-203580 partially inhibited the phosphorylation of IKK2 (from 112- to 55-fold), implying that p38 MAPK may regulate IKK2 activity that involves other signaling pathways in addition to the canonical IKK2/IκB-α/NF-κB signaling pathway.
Fig. 3.

Inhibition of MEK1 blocks IL-1β-induced inhibitor of NF-κB kinase-2 (IKK2) phosphorylation, IκB-α degradation, and p65 phosphorylation and nuclear translocation but p38 MAPK inhibition has less effect. Cultured rabbit colonic SMCs after serum starvation for 24 h were pretreated with the MEK1 inhibitor PD-98059 (10 μM) or the p38 MAPK inhibitor SB-203580 (1 μM) for 1 h before treatment with or without IL-1β (10 ng/ml) for 15 min (A) or 1 h (B–D) followed by Western blot analysis using the indicated specific antibodies (A) or immunocytochemistry with anti-p65 antibody (B–D). Representative images of nuclear and cytoplasmic masks are shown in C, and the quantitative ratio of nuclear to cytoplasmic fluorescence of p65 was analyzed by ANOVA and Newman-Keuls comparison. **P < 0.01 and *P < 0.05, significant decrease after IL-1β treatment by the inhibitors compared with DMSO; ++P < 0.01, significant increase after IL-1β treatment compared with the control. The number under each blot in A indicates the relative fold of optical density compared with the corresponding control.
To further corroborate the effect of the selected MAPK inhibitors on NF-κB activation, we determined the nuclear translocation of p65 in serum-starved SMCs by immunofluorescent cytochemistry and semiquantitative analysis. As shown in Fig. 3, B and D, pretreatment with PD-98059 prevented IL-1β-induced p65 nuclear translocation, whereas SB-203580 induced a partial inhibition of IL-1β-induced p65 nuclear translocation. Treatment with PD-98059 or SB-203580 alone did not affect the constitutive cellular distribution of p65 (Fig. 3B).
IL-1β activates PI3K in rabbit colonic circular SMCs.
In multiple cell types, IL-1β has been shown to activate PI3K (23). To address whether IL-1β activates PI3K in rabbit colonic SMCs, we examined the phosphorylation of Akt at Ser473 and GSK3β at Ser9, two sequential downstream effectors of PI3K, by performing Western blot analysis with phospho-specific antibodies. As shown in Fig. 4A, IL-1β treatment caused the phosphorylation of Akt and GSK3β, implying that IL-1β activates PI3K.
Fig. 4.

Phosphoinositide 3-kinase (PI3K) activation inhibits IL-1β-induced upregulation of RGS4 in rabbit colonic SMCs. A: IL-1β induces the phosphorylation of Akt and glycogen synthase kinase-3β (GSK3β). Cultured and serum-starved SMCs were treated with IL-1β (10 ng/ml) for indicated time periods followed by Western blot analysis with anti-p-Akt or anti-p-GSK3β antibodies. B–D: PI3K inhibition enhances IL-1β-induced upregulation of RGS4 mRNA and protein expression. Confluent colonic SMCs were serum starved for 24 h and pretreated with DMSO, the PI3K inhibitor LY-294002 (LY; 10 μM), or Src kinase inhibitor I (10 μM) for 1 h before an exposure to IL-1β (10 ng/ml) for 3 h (B) and 3 days (B–D). Expression levels of RGS4 mRNA were determined by conventional RT-PCR (B) and real-time RT-PCR using GAPDH for normalization (C). The expression of RGS4 protein was measured by Western blot analysis (D). The number under each blot indicates the relative fold of optical density compared with the corresponding control.
PI3K inhibits IL-1β-induced upregulation of RGS4.
As previously shown (37), IL-1β treatment of colonic SMCs induced an acute and long-term effect on RGS4 expression (Fig. 4, B and C). Pretreatment with the selective PI3K inhibitor (LY-294002, 10 μM) enhanced IL-1β-induced upregulation of RGS4 mRNA expression, whereas Src inhibitor I (10 μM) had no effect on RGS4 expression (Fig. 4, B and C). This was confirmed by Western blot analysis of RGS4 protein expression (Fig. 4D). The affinity-purified anti-RGS4 antibody recognizes one band of endogenous RGS4 protein but two bands of overexpressed HA-RGS4 fusion protein, consistent with a previous report (36). LY-294002 treatment alone had a marginal effect on the constitutive expression of RGS4 mRNA and protein (Fig. 4, B and D). These data suggest that the activation of PI3K by IL-1β plays an inhibitory role in regulating IL-1β-induced upregulation of RGS4 expression.
PI3K inhibits RGS4 upregulation via inactivation of GSK3β signaling.
PI3K phosphorylates Akt, which further induces the phosphorylation and thus inactivation of GSK3β in distinct cell types. This concept was corroborated in rabbit colonic SMCs. As shown in Fig. 5A, IL-1β-induced phosphorylation of GSK3β in rabbit SMCs was blocked by either the PI3K inhibitor (LY-294002) or Akt inhibitor (API-2), implying that the PI3K/Akt pathway mediates IL-1β-induced GSK3β phosphorylation. The effectiveness of LY-294002 to inhibit PI3K was verified by Western blot analysis by examining the phosphorylation of Akt at Ser473 (Fig. 5A). GSK3β is inactive upon phosphorylation. As predicted, IL-1β-induced GSK3β phorphorylation was augmented by LiCl (Fig. 5A), which is well known to inhibit GSK3β by increasing the phosphorylation of GSK3β at Ser9 and competing with magnesium in the ATP-binding site. Therefore, additional inhibition of GSK3β by LiCl attenuated IL-1β-induced upregulation of RGS4 mRNA expression (Fig. 5B). This was confirmed by the experiment using the highly selective ATP-competitive GSK3β inhibitor GSK3β-XI, although it exhibited less effective inhibition than LiCl (Fig. 5B). As shown above, the activation of endogenous PI3K/Akt signaling by IL-1β inhibits RGS4 expression through inactivating GSK3β signaling by increasing the phosphorylation of GSK3β. Exogenous addition of GSK3β inhibitors also inactivate GSK3β signaling by increasing the phosphorylation of GSK3β and ultimately generate a synergistic role with IL-1β-initiated activation of PI3K/Akt signaling (see the signaling model in Fig. 7).
Fig. 5.

Inhibition of PI3K and Akt activation prevents but the GSK3 inhibitor LiCl enhances IL-1β-induced phosphorylation of GSK3β (A), whereas inhibition of GSK3β with selective GSK3β inhibitor XI (GSK3β-XI) and LiCl inhibits IL-1β-induced upregulation of RGS4 expression (B). Cultured rabbit colonic SMCs after serum starvation for 24 h were pretreated with the PI3K inhibitor LY-294002 (10 μM), the Akt inhibitor Akt/PKB protein inhibitor-2 (API-2; 10 μM), the GSK3 inhibitor LiCl (30 mM), or GSK3β-XI (10 μM) for 1 h before treatment with or without IL-1β (10 ng/ml) for 15 min (A) or 3 h (B) followed by Western blot analysis using the indicated specific antibodies (A) or real-time RT-PCR for RGS4 mRNA expression (B). **P < 0.01, significant decrease after IL-1β treatment by the inhibitors compared with vehicle. The number under each blot indicates the relative fold of optical density compared with the corresponding control.
GSK3β stimulates the canonical IKK2/IκB-α/NF-κB pathway.
Phosphorylation of IKK2, IκB-α, and p65 induced by IL-1β treatment for 15 min was augmented by pretreatment with the PI3K inhibitor LY-294002 (10 μM). In addition, LY-294002 treatment alone induced the constitutive phosphorylation of IκB-α and p65 (Fig. 6A), leading to a marginal increase in the constitutive expression of RGS4 (Fig. 4, B and D). Therefore, endogenous PI3K retains a tonic inhibition on IKK2-mediated NF-κB signaling in colonic SMCs. To further confirm the role of the canonical IKK2/IκB-α pathway of NF-κB activation in mediating the enhancing effect of PI3K inhibition, the selective inhibitors for IKK2 and IκB-α were used to pretreat the cells before treatment with LY-294002 and IL-1β. Indeed, either the IKK2 inhibitor IKK2-IV or IκB-α degradation inhibitor MG-132 significantly blocked the LY-294002-induced enhancement of IL-1β-stimulated upregulation of RGS4 expression (Fig. 6B).
Fig. 6.

Inhibition of PI3K enhances but inhibition of GSK3β abolishes the IL-1β-induced activation of the canonical IKK2/IκB-α/NF-κB signaling pathway. Cultured rabbit colonic SMCs after serum starvation for 24 h were pretreated with the PI3K inhibitor LY-294002 (10 μM) plus the IKK2 inhibitor IKK2-IV (10 μM) or the IκB-α degradation inhibitor MG-132 (10 μM) or the GSK3 inhibitor LiCl (30 mM) for 1 h before treatment with or without IL-1β (10 ng/ml) for 15 min (A and C) or 3 h (B). Activation of NF-κB signaling was determined by Western blot analysis using the indicated specific antibodies (A and C). The expression of RGS4 mRNA was quantified by real-time RT-PCR with GAPDH normalization (B). **P < 0.01, significant decrease after IL-1β treatment by the inhibitors compared with DMSO. The number under each blot in A indicates the relative fold of optical density compared with the corresponding control.
Interestingly, the GSK3β inhibitor LiCl abolished the IL-1β-induced phosphorylation of IKK2 and p65 (Fig. 6C), implying that GSK3β by itself is capable of activating IKK2 through increasing the phosphorylation of the IKK2 activation loop. The inactivation of GSK3β via inhibitory phosphorylation by PI3K/Akt (Fig. 5A) leads to the inhibition of IKK2 and p65 phosphorylation. Therefore, the IL-1β-induced activation of PI3K/Akt negatively modulates IL-1β-induced NF-κB activation and subsequent RGS4 upregulation through inhibiting GSK3β activity.
DISCUSSION
RGS4 is a member of the R4 family of RGS proteins, which has been best investigated at the structural, biochemical, and functional levels (53, 59, 79, 80, 98, 99). However, the regulatory (molecular) mechanism of RGS4 expression has not been fully understood. In our previous study (36), we demonstrated that the proinflammatory cytokine IL-1β transcriptionally upregulates RGS4 expression through the canonical IKK2/IκB-α pathway of NF-κB activation in rabbit colonic SMCs. Upregulated RGS4 has been implicated in IL-1β-induced inhibition of acetylcholine-stimulated initial contraction of colonic SMCs (37). Here, we show that the ERK1/2 and p38 MAPK pathways enhance, whereas the PI3K/Akt/GSK3β pathway inhibits, IL-1β-induced upregulation of RGS4 expression. The effects of the ERK1/2 and PI3K/Akt/GSK3β pathways are exerted through the IKK2/NF-κB pathway, whereas the effect of the p38 MAPK pathway may not involve NF-κB signaling. These data are summarized in a proposed model showing the signaling pathway and regulatory mechanism for RGS4 induction by IL-1β in colonic SMCs (Fig. 7).
Fig. 7.

IL-1β-induced upregulation of RGS4 expression in colonic SMCs via canonical IKK2/IκB-α/NF-κB signaling is differentially modulated by the MAPK and PI3K/Akt/GSK3β pathways. IL-1β induces NF-κB activation involving the phosphorylation of IKK2, degradation of IκB-α, and nuclear translocation of p65/p50, leading to the upregulation of RGS4 mRNA expression. IL-1β also activates MAPK and PI3K. ERK1/2 and p38 MAPK enhance, whereas PI3K inhibits, IL-1β-induced RGS4 upregulation. The effect of ERK1/2 is exerted on the canonical IKK2/IκBα/p65 pathway of NF-κB activation, and p38 MAPK may target at the chromatin level. PI3K attenuates NF-κB activation at the level of IKK2 by inactivating GSK3β by increasing the phosphorylation of GSK3β via Akt.
IL-1β is well known to stimulate all of the three MAPKs in SMCs of the vasculature, airway, and intestine. However, the role and outcome of these MAPK pathways are different. In airway SMCs, IL-1β-induced upregulation of COX-2 and eotaxin is inhibited by either MEK1 inhibitors or p38 MAPK inhibitors (34, 87, 103), whereas IL-1β-induced release of the protein RANTES (regulated upon activation, normal T cell expressed, and secreted) is sensitive to inhibition of MEK1 (32) or JNK (71) but not to inhibition of p38 MAPK (32). IL-1β-induced upregulation of matrix metalloproteinase-9 (60) and TNF-α-induced expression of VCAM-1 (57) are sensitive to the inhibition of all the three MAPK pathways. In vascular SMCs, IL-1β-stimulated inducible nitric oxide synthase (iNOS) expression is prevented by MEK1 inhibition but potentiated by p38 MAPK inhibition (27, 30). Inhibition of MEK1 or p38 MAPK, but not PI3K, reduced IL-1β-stimulated expression of LIM domain kinase 2 and cofilin (11). However, in human vascular SMCs, IL-1β activates only p38 MAPK, which mediates IL-1β-induced IL-8 and VEGF expression (46, 47). In colonic SMCs, IL-1β-induced H2O2 production is inhibited by MEK inhibitor but not p38 MAPK inhibitor (18), whereas IL-1β-induced upregulation of IL-6, IL-8, and COX-2 is reduced by p38 MAPK inhibitor but not MEK-1 inhibitor (81). Sphingosylphosphorylcholine-induced contraction of ileal SMCs is blocked by MEK-1 inhibitor but not p38 MAPK inhibitor (58). In animal colitis induced by 2,4,6-trinitrobenzene sulfonic acid, ERK1/2 mediates the restoration of reduced muscle contractility by meloxicam, a COX-2 inhibitor (50). In the present study, IL-1β-induced upregulation of RGS4 mRNA is sensitive to inhibition of both MEK1 and p38 MAPK. Therefore, the functional significance of MAPK activation in SMCs relies on the stimuli, target genes, and cell resources.
The cross-talk between the MAPK and NF-κB signaling pathways is not well understood. The MEK/ERK pathway has been widely shown to affect IL-1β-induced NF-κB activation. In cultured rat vascular SMCs, ERK is required for IL-1β-induced persistent NF-κB activation, whereas IL-1β induces only acute and transient NF-κB activation without ERK activation (42, 43). Inhibition of ERK does not affect IL-1β-induced phosphorylation and degradation of IκB-α but attenuates the degradation of IκB-β and, therefore, inhibits the expression of iNOS and COX-2 but has no effect on the expression of VCAM-1 and Mn-SOD (44). In the present study, we demonstrate, for the first time, that ERK activation enhances IL-1β-induced NF-κB activation and RGS4 upregulation at the levels of p65, IκB-α, and IKK2. We also show that compared with the ERK pathway, the p38 MAPK pathway has less effect on NF-κB activation in rabbit colonic SMCs. This concurs with previous reports in airway SMCs (40, 87, 96) and vascular SMCs (92, 101). In contrast, p38 MAPK has been shown preferentially to regulate the mRNA stability of IL-1β target genes such as COX-2, iNOS, IL-6, etc. (56, 76, 87, 97). Thus, the effect of MAPK on RGS4 mRNA stability is on the way of investigation.
PI3K has been shown to negatively mediate Toll-IL-1 receptor-mediated inflammatory responses (33). Early studies have shown that PI3K inhibition enhances NF-κB-dependent gene transcription induced by LPS and IL-1β through increasing NF-κB DNA binding, nuclear translocation, IκB-α degradation, p65 phosphorylation, and IKK1/2 phosphorylation (6, 20, 29, 75, 107) in distinct cell types. Here, we show that PI3K inhibition apparently enhances the IL-1β-induced phosphorylation of IKK1/2 and p65 and subsequent upregulation of RGS4 mRNA expression in rabbit colonic SMCs. The mechanisms for the PI3K-negative modulation on IL-1β-induced NF-κB activation and target gene expression are not well understood. Akt, the downstream substrate of PI3K, has been shown to increase p65 phosphorylation and NF-κB activation via IKK1 (1, 21, 63, 88, 89) or IKK2 (63, 74, 93), which argue against the inhibition of PI3K on the NF-κB pathway. However, PI3K-mediated Akt inactivates GSK3β by phosphorylating GSK3β at Ser9 (45, 104). GSK3β is known to activate NF-κB signaling in most cases (7, 24). For example, GSK3β knockout mice show a similar phenotype to that of p65 or IKK2 knockout mice (35). Inhibition of GSK3β attenuates NF-κB activation in intestinal epithelial cells (90), hepatocytes (19, 83, 90), pancreatic cancer cells (73), and leukemia B cells (72). Here, in colonic SMCs, we found that GSK3β inhibition by LiCl abolishes IL-1β-induced NF-κB activation (Fig. 6C), implying that endogenous constitutive GSK3β mediates the activation of NF-κB signaling. Therefore, the activation of PI3K/Akt by IL-1β provides an inhibitory regulation of IL-1β-induced NF-κB signaling by inactivating GSK3β via inhibitory phosphorylation. However, the specific GSK3β inhibitor XI only partially inhibited IL-1β-induced upregulation of RGS4 expression, which was abolished by LiCl. This suggests that LiCl affects other signaling components (like GSK3α) in addition to GSK3β and that these components also mediate RGS4 induction by IL-1β.
The level of GSK3β action on the NF-κB signaling pathway remains controversial. Our data show that GSK3β inhibition by LiCl abolished IL-1β-induced phosphorylation of IKK2 and p65 in rabbit colonic SMCs. This is in agreement with the previous studies in GSK3β-deficient embryonic fibroblast (91) and GSK3β-overexpressing astrocytes (82). However, several recent studies have suggested that IKK is not involved in the GSK3β-mediated regulation of NF-κB activation. GSK-3 regulates TNF-α-induced IκB-α degradation independent of IKK2 in human endothelial cells (25). GSK3β directly or indirectly induced the phosphorylation of p65 at Ser536 (107), Ser529 (91), or Ser468 (13) or between residues 354–551 (83). A gene knockout study (90) has shown that GSK-3β only affects NF-κB DNA-binding activity. In monocytes or peripheral blood mononuclear cells, GSK3β regulates the association of p65 with the nuclear coactivator cAMP response element-binding protein-binding protein (CBP) after Toll-like receptor stimulation but does not affect IκB-α degradation, p65 phosphorylaiton, nuclear translocation, and even DNA-binding activity (64).
On the other hand, several studies have suggested a negative regulation of GSK3β on NF-κB signaling in primary astrocytes (52, 82), renal medullary interstitial cells (78), the rat pheochromocytoma PC12 cell line (12), and vascular SMCs (65). Therefore, the modulatory effects and specific levels of GSK-3β on NF-κB signaling depend on the cell types, stimuli, and target genes.
In conclusion, we demonstrate, for the first time, that IL-1β-induced upregulation of RGS4 expression is differentially modulated by MAPKs and PI3K/Akt/GSK3β signaling, which directly or indirectly influence the activation of NF-κB signaling. Increased RGS4 expression by IL-1β contributes to the deactivation of Gαq signaling and inhibition of initial muscle contraction during inflammatory responses of the gut. Intervention of these signaling pathways may be a potential target for the pharmaceutical industry as well as clinical therapy.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-075964 (to W. H. Hu) and DK-015564 (to K. S. Murthy).
REFERENCES
- 1.Agarwal A, Das K, Lerner N, Sathe S, Cicek M, Casey G, Sizemore N. The AKT/IκB kinase pathway promotes angiogenic/metastatic gene expression in colorectal cancer by activating nuclear factor-κB and β-catenin. Oncogene 24: 1021–1031, 2005. [DOI] [PubMed] [Google Scholar]
- 2.Akiho H, Blennerhassett P, Deng Y, Collins SM. Role of IL-4, IL-13, and STAT6 in inflammation-induced hypercontractility of murine smooth muscle cells. Am J Physiol Gastrointest Liver Physiol 282: G226–G232, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Akiho H, Deng Y, Blennerhassett P, Kanbayashi H, Collins SM. Mechanisms underlying the maintenance of muscle hypercontractility in a model of postinfective gut dysfunction. Gastroenterology 129: 131–141, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Akiho H, Khan WI, Al-Kaabi A, Blennerhassett P, Deng Y, Collins SM. Cytokine modulation of muscarinic receptors in the murine intestine. Am J Physiol Gastrointest Liver Physiol 293: G250–G255, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Akiho H, Lovato P, Deng Y, Ceponis PJ, Blennerhassett P, Collins SM. Interleukin-4- and -13-induced hypercontractility of human intestinal muscle cells-implication for motility changes in Crohn's disease. Am J Physiol Gastrointest Liver Physiol 288: G609–G615, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Aksoy E, Vanden Berghe W, Detienne S, Amraoui Z, Fitzgerald KA, Haegeman G, Goldman M, Willems F. Inhibition of phosphoinositide 3-kinase enhances TRIF-dependent NF-kappa B activation and IFN-beta synthesis downstream of Toll-like receptor 3 and 4. Eur J Immunol 35: 2200–2209, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Ali A, Hoeflich KP, Woodgett JR. Glycogen synthase kinase-3: properties, functions, and regulation. Chem Rev 101: 2527–2540, 2001. [DOI] [PubMed] [Google Scholar]
- 8.Aube AC, Blottiere HM, Scarpignato C, Cherbut C, Roze C, Galmiche JP. Inhibition of acetylcholine induced intestinal motility by interleukin 1 beta in the rat. Gut 39: 470–474, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aube AC, Cherbut C, Roze C, Galmiche JP. Vasoactive intestinal peptide is involved in the inhibitory effect of interleukin-1 beta on the jejunal contractile response induced by acetylcholine. Gastroenterol Clin Biol 25: 1090–1095, 2001. [PubMed] [Google Scholar]
- 10.Bodenstein J, Sunahara RK, Neubig RR. N-terminal residues control proteasomal degradation of RGS2, RGS4, and RGS5 in human embryonic kidney 293 cells. Mol Pharmacol 71: 1040–1050, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Bongalon S, Dai YP, Singer CA, Yamboliev IA. PDGF and IL-1beta upregulate cofilin and LIMK2 in canine cultured pulmonary artery smooth muscle cells. J Vasc Res 41: 412–421, 2004. [DOI] [PubMed] [Google Scholar]
- 12.Bournat JC, Brown AM, Soler AP. Wnt-1 dependent activation of the survival factor NF-kappaB in PC12 cells. J Neurosci Res 61: 21–32, 2000. [DOI] [PubMed] [Google Scholar]
- 13.Buss H, Dorrie A, Schmitz ML, Frank R, Livingstone M, Resch K, Kracht M. Phosphorylation of serine 468 by GSK-3beta negatively regulates basal p65 NF-kappaB activity. J Biol Chem 279: 49571–49574, 2004. [DOI] [PubMed] [Google Scholar]
- 14.Cao W, Cheng L, Behar J, Fiocchi C, Biancani P, Harnett KM. Proinflammatory cytokines alter/reduce esophageal circular muscle contraction in experimental cat esophagitis. Am J Physiol Gastrointest Liver Physiol 287: G1131–G1139, 2004. [DOI] [PubMed] [Google Scholar]
- 15.Cao W, Fiocchi C, Pricolo VE. Production of IL-1β, hydrogen peroxide, and nitric oxide by colonic mucosa decreases sigmoid smooth muscle contractility in ulcerative colitis. Am J Physiol Cell Physiol 289: C1408–C1416, 2005. [DOI] [PubMed] [Google Scholar]
- 16.Cao W, Harnett KM, Cheng L, Kirber MT, Behar J, Biancani P. H2O2: a mediator of esophagitis-induced damage to calcium-release mechanisms in cat lower esophageal sphincter. Am J Physiol Gastrointest Liver Physiol 288: G1170–G1178, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Cao W, Sohn UD, Bitar KN, Behar J, Biancani P, Harnett KM. MAPK mediates PKC-dependent contraction of cat esophageal and lower esophageal sphincter circular smooth muscle. Am J Physiol Gastrointest Liver Physiol 285: G86–G95, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Cao W, Vrees MD, Potenti FM, Harnett KM, Fiocchi C, Pricolo VE. Interleukin 1beta-induced production of H2O2 contributes to reduced sigmoid colonic circular smooth muscle contractility in ulcerative colitis. J Pharmacol Exp Ther 311: 60–70, 2004. [DOI] [PubMed] [Google Scholar]
- 19.Chen H, Yang S, Yang Z, Ma L, Jiang D, Mao J, Jiao B, Cai Z. Inhibition of GSK-3beta decreases NF-kappaB-dependent gene expression and impairs the rat liver regeneration. J Cell Biochem 102: 1281–1289, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Choi EK, Jang HC, Kim JH, Kim HJ, Kang HC, Paek YW, Lee HC, Lee SH, Oh WM, Kang IC. Enhancement of cytokine-mediated NF-kappaB activation by phosphatidylinositol 3-kinase inhibitors in monocytic cells. Int Immunopharmacol 6: 908–915, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Dan HC, Baldwin AS. Differential Involvement of IκB kinases α and β in cytokine- and insulin-induced mammalian target of rapamycin activation determined by Akt. J Immunol 180: 7582–7589, 2008. [DOI] [PubMed] [Google Scholar]
- 22.Ding GJ, Fischer PA, Boltz RC, Schmidt JA, Colaianne JJ, Gough A, Rubin RA, Miller DK. Characterization and quantitation of NF-kappaB nuclear translocation induced by interleukin-1 and tumor necrosis factor-alpha. Development and use of a high capacity fluorescence cytometric system. J Biol Chem 273: 28897–28905, 1998. [DOI] [PubMed] [Google Scholar]
- 23.Dragon S, Rahman MS, Yang J, Unruh H, Halayko AJ, Gounni AS. IL-17 enhances IL-1β-mediated CXCL-8 release from human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 292: L1023–L1029, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Dugo L, Collin M, Thiemermann C. Glycogen synthase kinase 3beta as a target for the therapy of shock and inflammation. Shock 27: 113–123, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Eto M, Kouroedov A, Cosentino F, Luscher TF. Glycogen synthase kinase-3 mediates endothelial cell activation by tumor necrosis factor-alpha. Circulation 112: 1316–1322, 2005. [DOI] [PubMed] [Google Scholar]
- 26.George TC, Fanning SL, Fitzgeral-Bocarsly P, Medeiros RB, Highfill S, Shimizu Y, Hall BE, Frost K, Basiji D, Ortyn WE, Morrissey PJ, Lynch DH. Quantitative measurement of nuclear translocation events using similarity analysis of multispectral cellular images obtained in flow. J Immunol Methods 311: 117–129, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Ginnan R, Guikema BJ, Singer HA, Jourd'heuil D. PKC-δ mediates activation of ERK1/2 and induction of iNOS by IL-1β in vascular smooth muscle cells. Am J Physiol Cell Physiol 290: C1583–C1591, 2006. [DOI] [PubMed] [Google Scholar]
- 28.Grillet N, Dubreuil V, Dufour HD, Brunet JF. Dynamic expression of RGS4 in the developing nervous system and regulation by the neural type-specific transcription factor Phox2b. J Neurosci 23: 10613–10621, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem 277: 32124–32132, 2002. [DOI] [PubMed] [Google Scholar]
- 30.Guikema BJ, Ginnan R, Singer HA, Jourd'heuil D. Catalase potentiates interleukin-1beta-induced expression of nitric oxide synthase in rat vascular smooth muscle cells. Free Radic Biol Med 38: 597–605, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Hahn A, Huber A, Neumayer N, Allescher HD. Effect of interleukin-1beta on the ascending and descending reflex in rat small intestine. Eur J Pharmacol 359: 201–209, 1998. [DOI] [PubMed] [Google Scholar]
- 32.Hallsworth MP, Moir LM, Lai D, Hirst SJ. Inhibitors of mitogen-activated protein kinases differentially regulate eosinophil-activating cytokine release from human airway smooth muscle. Am J Respir Crit Care Med 164: 688–697, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Hazeki K, Nigorikawa K, Hazeki O. Role of phosphoinositide 3-kinase in innate immunity. Biol Pharm Bull 30: 1617–1623, 2007. [DOI] [PubMed] [Google Scholar]
- 34.Hirst SJ, Hallsworth MP, Peng Q, Lee TH. Selective induction of eotaxin release by interleukin-13 or interleukin-4 in human airway smooth muscle cells is synergistic with interleukin-1beta and is mediated by the interleukin-4 receptor alpha-chain. Am J Respir Crit Care Med 165: 1161–1171, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406: 86–90, 2000. [DOI] [PubMed] [Google Scholar]
- 36.Hu W, Li F, Mahavadi S, Murthy KS. Interleukin-1beta up-regulates RGS4 through the canonical IKK2/IkappaBalpha/NF-kappaB pathway in rabbit colonic smooth muscle. Biochem J 412: 35–43, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu W, Mahavadi S, Li F, Murthy KS. Upregulation of RGS4 and downregulation of CPI-17 mediate inhibition of colonic muscle contraction by interleukin-1β. Am J Physiol Cell Physiol 293: C1991–C2000, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang J, Mahavadi S, Sriwai W, Hu W, Murthy KS. Gi-coupled receptors mediate phosphorylation of CPI-17 and MLC20 via preferential activation of the PI3K/ILK pathway. Biochem J 396: 193–200, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ihara E, Moffat L, Ostrander J, Walsh MP, MacDonald JA. Characterization of protein kinase pathways responsible for Ca2+ sensitization in rat ileal longitudinal smooth muscle. Am J Physiol Gastrointest Liver Physiol 293: G699–G710, 2007. [DOI] [PubMed] [Google Scholar]
- 40.Issa R, Xie S, Lee KY, Stanbridge RD, Bhavsar P, Sukkar MB, Chung KF. GRO-α regulation in airway smooth muscle by IL-1β and TNF-α: role of NF-κB and MAP kinases. Am J Physiol Lung Cell Mol Physiol 291: L66–L74, 2006. [DOI] [PubMed] [Google Scholar]
- 41.Je HD, Gangopadhyay SS, Ashworth TD, Morgan KG. Calponin is required for agonist-induced signal transduction–evidence from an antisense approach in ferret smooth muscle. J Physiol 537: 567–577, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang B, Brecher P, Cohen RA. Persistent activation of nuclear factor-kappaB by interleukin-1beta and subsequent inducible NO synthase expression requires extracellular signal-regulated kinase. Arterioscler Thromb Vasc Biol 21: 1915–1920, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Jiang B, Xu S, Brecher P, Cohen RA. Growth factors enhance interleukin-1 beta-induced persistent activation of nuclear factor-kappa B in rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 22: 1811–1816, 2002. [DOI] [PubMed] [Google Scholar]
- 44.Jiang B, Xu S, Hou X, Pimentel DR, Brecher P, Cohen RA. Temporal control of NF-kappaB activation by ERK differentially regulates interleukin-1beta-induced gene expression. J Biol Chem 279: 1323–1329, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 29: 95–102, 2004. [DOI] [PubMed] [Google Scholar]
- 46.Jung YD, Fan F, McConkey DJ, Jean ME, Liu W, Reinmuth N, Stoeltzing O, Ahmad SA, Parikh AA, Mukaida N, Ellis LM. Role of P38 MAPK, AP-1, and NF-kappaB in interleukin-1beta-induced IL-8 expression in human vascular smooth muscle cells. Cytokine 18: 206–213, 2002. [DOI] [PubMed] [Google Scholar]
- 47.Jung YD, Liu W, Reinmuth N, Ahmad SA, Fan F, Gallick GE, Ellis LM. Vascular endothelial growth factor is upregulated by interleukin-1 beta in human vascular smooth muscle cells via the p38 mitogen-activated protein kinase pathway. Angiogenesis 4: 155–162, 2001. [DOI] [PubMed] [Google Scholar]
- 48.Khan I, al-Awadi FM. Colonic muscle enhances the production of interleukin-1 beta messenger RNA in experimental colitis. Gut 40: 307–312, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khan I, Collins SM. Expression of cytokines in the longitudinal muscle myenteric plexus of the inflamed intestine of rat. Gastroenterology 107: 691–700, 1994. [DOI] [PubMed] [Google Scholar]
- 50.Khan I, Oriowo MA. Mechanism underlying the reversal of contractility dysfunction in experimental colitis by cyclooxygenase-2 inhibition. Inflammopharmacology 14: 28–35, 2006. [DOI] [PubMed] [Google Scholar]
- 51.Khan WI, Collins SM. Gut motor function: immunological control in enteric infection and inflammation. Clin Exp Immunol 143: 389–397, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim SD, Yang SI, Kim HC, Shin CY, Ko KH. Inhibition of GSK-3beta mediates expression of MMP-9 through ERK1/2 activation and translocation of NF-kappaB in rat primary astrocyte. Brain Res 1186: 12–20, 2007. [DOI] [PubMed] [Google Scholar]
- 53.Kimple AJ, Willard FS, Giguere PM, Johnston CA, Mocanu V, Siderovski DP. The RGS protein inhibitor CCG-4986 is a covalent modifier of the RGS4 Galpha-interaction face. Biochim Biophys Acta 1774: 1213–1220, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kordowska J, Huang R, Wang CL. Phosphorylation of caldesmon during smooth muscle contraction and cell migration or proliferation. J Biomed Sci 13: 159–172, 2006. [DOI] [PubMed] [Google Scholar]
- 55.Krymskaya VP Targeting the phosphatidylinositol 3-kinase pathway in airway smooth muscle: rationale and promise. BioDrugs 21: 85–95, 2007. [DOI] [PubMed] [Google Scholar]
- 56.Lasa M, Mahtani KR, Finch A, Brewer G, Saklatvala J, Clark AR. Regulation of cyclooxygenase 2 mRNA stability by the mitogen-activated protein kinase p38 signaling cascade. Mol Cell Biol 20: 4265–4274, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee CW, Lin WN, Lin CC, Luo SF, Wang JS, Pouyssegur J, Yang CM. Transcriptional regulation of VCAM-1 expression by tumor necrosis factor-alpha in human tracheal smooth muscle cells: involvement of MAPKs, NF-kappaB, p300, and histone acetylation. J Cell Physiol 207: 174–186, 2006. [DOI] [PubMed] [Google Scholar]
- 58.Lee T, Kim J, Sohn U. Sphingosylphosphorylcholine-induced contraction of feline ileal smooth muscle cells is mediated by Galphai3 protein and MAPK. Cell Signal 14: 989–997, 2002. [DOI] [PubMed] [Google Scholar]
- 59.Levitt P, Ebert P, Mirnics K, Nimgaonkar VL, Lewis DA. Making the case for a candidate vulnerability gene in schizophrenia: convergent evidence for regulator of G-protein signaling 4 (RGS4). Biol Psychiatry 60: 534–537, 2006. [DOI] [PubMed] [Google Scholar]
- 60.Liang KC, Lee CW, Lin WN, Lin CC, Wu CB, Luo SF, Yang CM. Interleukin-1beta induces MMP-9 expression via p42/p44 MAPK, p38 MAPK, JNK, and nuclear factor-kappaB signaling pathways in human tracheal smooth muscle cells. J Cell Physiol 211: 759–770, 2007. [DOI] [PubMed] [Google Scholar]
- 61.Lien SC, Usami S, Chien S, Chiu JJ. Phosphatidylinositol 3-kinase/Akt pathway is involved in transforming growth factor-beta1-induced phenotypic modulation of 10T1/2 cells to smooth muscle cells. Cell Signal 18: 1270–1278, 2006. [DOI] [PubMed] [Google Scholar]
- 62.Lodato RF, Khan AR, Zembowicz MJ, Weisbrodt NW, Pressley TA, Li YF, Lodato JA, Zembowicz A, Moody FG. Roles of IL-1 and TNF in the decreased ileal muscle contractility induced by lipopolysaccharide. Am J Physiol Gastrointest Liver Physiol 276: G1356–G1362, 1999. [DOI] [PubMed] [Google Scholar]
- 63.Madrid LV, Mayo MW, Reuther JY, Baldwin AS Jr. Akt stimulates the transactivation potential of the RelA/p65 subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J Biol Chem 276: 18934–18940, 2001. [DOI] [PubMed] [Google Scholar]
- 64.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol 6: 777–784, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Menschikowski M, Hagelgans A, Hempel U, Siegert G. Glycogen synthase kinase-3beta negatively regulates group IIA phospholipase A2 expression in human aortic smooth muscle and HepG2 hepatoma cells. FEBS Lett 577: 81–86, 2004. [DOI] [PubMed] [Google Scholar]
- 66.Murthy KS, Makhlouf GM. Differential coupling of muscarinic m2 and m3 receptors to adenylyl cyclases V/VI in smooth muscle. Concurrent M2-mediated inhibition via Galphai3 and m3-mediated stimulation via Gβγq. J Biol Chem 272: 21317–21324, 1997. [DOI] [PubMed] [Google Scholar]
- 67.Natale L, Piepoli AL, De Salvia MA, De Salvatore G, Mitolo CI, Marzullo A, Portincasa P, Moschetta A, Palasciano G, Mitolo-Chieppa D. Interleukins 1 beta and 6 induce functional alteration of rat colonic motility: an in vitro study. Eur J Clin Invest 33: 704–712, 2003. [DOI] [PubMed] [Google Scholar]
- 68.Noursadeghi M, Tsang J, Haustein T, Miller RF, Chain BM, Katz DR. Quantitative imaging assay for NF-kappaB nuclear translocation in primary human macrophages. J Immunol Methods 329: 194–200, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ohama T, Hori M, Momotani E, Elorza M, Gerthoffer WT, Ozaki H. IL-1β inhibits intestinal smooth muscle proliferation in an organ culture system: involvement of COX-2 and iNOS induction in muscularis resident macrophages. Am J Physiol Gastrointest Liver Physiol 292: G1315–G1322, 2007. [DOI] [PubMed] [Google Scholar]
- 70.Ohama T, Hori M, Sato K, Ozaki H, Karaki H. Chronic treatment with interleukin-1beta attenuates contractions by decreasing the activities of CPI-17 and MYPT-1 in intestinal smooth muscle. J Biol Chem 278: 48794–48804, 2003. [DOI] [PubMed] [Google Scholar]
- 71.Oltmanns U, Issa R, Sukkar MB, John M, Chung KF. Role of c-jun N-terminal kinase in the induced release of GM-CSF, RANTES and IL-8 from human airway smooth muscle cells. Br J Pharmacol 139: 1228–1234, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ougolkov AV, Bone ND, Fernandez-Zapico ME, Kay NE, Billadeau DD. Inhibition of glycogen synthase kinase-3 activity leads to epigenetic silencing of nuclear factor kappaB target genes and induction of apoptosis in chronic lymphocytic leukemia B cells. Blood 110: 735–742, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ougolkov AV, Fernandez-Zapico ME, Savoy DN, Urrutia RA, Billadeau DD. Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res 65: 2076–2081, 2005. [DOI] [PubMed] [Google Scholar]
- 74.Ouyang W, Li J, Ma Q, Huang C. Essential roles of PI-3K/Akt/IKKbeta/NFkappaB pathway in cyclin D1 induction by arsenite in JB6 Cl41 cells. Carcinogenesis 27: 864–873, 2006. [DOI] [PubMed] [Google Scholar]
- 75.Pahan K, Raymond JR, Singh I. Inhibition of phosphatidylinositol 3-kinase induces nitric-oxide synthase in lipopolysaccharide- or cytokine-stimulated C6 glial cells. J Biol Chem 274: 7528–7536, 1999. [DOI] [PubMed] [Google Scholar]
- 76.Patil C, Zhu X, Rossa C Jr, Kim YJ, Kirkwood KL. p38 MAPK regulates IL-1beta induced IL-6 expression through mRNA stability in osteoblasts. Immunol Invest 33: 213–233, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pazdrak K, Shi XZ, Sarna SK. TNFalpha suppresses human colonic circular smooth muscle cell contractility by SP1- and NF-kappaB-mediated induction of ICAM-1. Gastroenterology 127: 1096–1109, 2004. [DOI] [PubMed] [Google Scholar]
- 78.Rao R, Hao CM, Breyer MD. Hypertonic stress activates glycogen synthase kinase 3beta-mediated apoptosis of renal medullary interstitial cells, suppressing an NFkappaB-driven cyclooxygenase-2-dependent survival pathway. J Biol Chem 279: 3949–3955, 2004. [DOI] [PubMed] [Google Scholar]
- 79.Riddle EL, Schwartzman RA, Bond M, Insel PA. Multi-tasking RGS proteins in the heart: the next therapeutic target? Circ Res 96: 401–411, 2005. [DOI] [PubMed] [Google Scholar]
- 80.Roman DL, Talbot JN, Roof RA, Sunahara RK, Traynor JR, Neubig RR. Identification of small-molecule inhibitors of RGS4 using a high-throughput flow cytometry protein interaction assay. Mol Pharmacol 71: 169–175, 2007. [DOI] [PubMed] [Google Scholar]
- 81.Salinthone S, Singer CA, Gerthoffer WT. Inflammatory gene expression by human colonic smooth muscle cells. Am J Physiol Gastrointest Liver Physiol 287: G627–G637, 2004. [DOI] [PubMed] [Google Scholar]
- 82.Sanchez JF, Sniderhan LF, Williamson AL, Fan S, Chakraborty-Sett S, Maggirwar SB. Glycogen synthase kinase 3beta-mediated apoptosis of primary cortical astrocytes involves inhibition of nuclear factor kappaB signaling. Mol Cell Biol 23: 4649–4662, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schwabe RF, Brenner DA. Role of glycogen synthase kinase-3 in TNF-α-induced NF-κB activation and apoptosis in hepatocytes. Am J Physiol Gastrointest Liver Physiol 283: G204–G211, 2002. [DOI] [PubMed] [Google Scholar]
- 84.Sheibanie AF, Yen JH, Khayrullina T, Emig F, Zhang M, Tuma R, Ganea D. The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23→IL-17 axis. J Immunol 178: 8138–8147, 2007. [DOI] [PubMed] [Google Scholar]
- 85.Shi XZ, Sarna SK. Transcriptional regulation of inflammatory mediators secreted by human colonic circular smooth muscle cells. Am J Physiol Gastrointest Liver Physiol 289: G274–G284, 2005. [DOI] [PubMed] [Google Scholar]
- 86.Shih DQ, Targan SR. Immunopathogenesis of inflammatory bowel disease. World J Gastroenterol 14: 390–400, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Singer CA, Baker KJ, McCaffrey A, AuCoin DP, Dechert MA, Gerthoffer WT. p38 MAPK and NF-κB mediate COX-2 expression in human airway myocytes. Am J Physiol Lung Cell Mol Physiol 285: L1087–L1098, 2003. [DOI] [PubMed] [Google Scholar]
- 88.Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark GR. Distinct roles of the Ikappa B kinase alpha and beta subunits in liberating nuclear factor kappa B (NF-kappa B) from Ikappa B and in phosphorylating the p65 subunit of NF-kappa B. J Biol Chem 277: 3863–3869, 2002. [DOI] [PubMed] [Google Scholar]
- 89.Sizemore N, Leung S, Stark GR. Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-kappaB p65/RelA subunit. Mol Cell Biol 19: 4798–4805, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Steinbrecher KA, Wilson W 3rd, Cogswell PC, Baldwin AS. Glycogen synthase kinase 3beta functions to specify gene-specific, NF-kappaB- dependent transcription. Mol Cell Biol 25: 8444–8455, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Takada Y, Fang X, Jamaluddin MS, Boyd DD, Aggarwal BB. Genetic deletion of glycogen synthase kinase-3beta abrogates activation of IkappaBalpha kinase, JNK, Akt, and p44/p42 MAPK but potentiates apoptosis induced by tumor necrosis factor. J Biol Chem 279: 39541–39554, 2004. [DOI] [PubMed] [Google Scholar]
- 92.Takahashi M, Suzuki E, Takeda R, Oba S, Nishimatsu H, Kimura K, Nagano T, Nagai R, Hirata Y. Angiotensin II and tumor necrosis factor-α synergistically promote monocyte chemoattractant protein-1 expression: roles of NF-κB, p38, and reactive oxygen species. Am J Physiol Heart Circ Physiol 294: H2879–H2888, 2008. [DOI] [PubMed] [Google Scholar]
- 93.Tanaka H, Fujita N, Tsuruo T. 3-Phosphoinositide-dependent protein kinase-1-mediated IkappaB kinase beta (IkkB) phosphorylation activates NF-kappaB signaling. J Biol Chem 280: 40965–40973, 2005. [DOI] [PubMed] [Google Scholar]
- 94.Taniyama Y, Ushio-Fukai M, Hitomi H, Rocic P, Kingsley MJ, Pfahnl C, Weber DS, Alexander RW, Griendling KK. Role of p38 MAPK and MAPKAPK-2 in angiotensin II-induced Akt activation in vascular smooth muscle cells. Am J Physiol Cell Physiol 287: C494–C499, 2004. [DOI] [PubMed] [Google Scholar]
- 95.Vrees MD, Pricolo VE, Potenti FM, Cao W. Abnormal motility in patients with ulcerative colitis: the role of inflammatory cytokines. Arch Surg 137: 436–445, 2002. [DOI] [PubMed] [Google Scholar]
- 96.Wang CC, Lin WN, Lee CW, Lin CC, Luo SF, Wang JS, Yang CM. Involvement of p42/p44 MAPK, p38 MAPK, JNK, and NF-κB in IL-1β-induced VCAM-1 expression in human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 288: L227–L237, 2005. [DOI] [PubMed] [Google Scholar]
- 97.Wang S, Zhang J, Zhang Y, Kern S, Danner RL. Nitric oxide-p38 MAPK signaling stabilizes mRNA through AU-rich element-dependent and -independent mechanisms. J Leukoc Biol 83: 982–990, 2008. [DOI] [PubMed] [Google Scholar]
- 98.Willars GB Mammalian RGS proteins: multifunctional regulators of cellular signalling. Semin Cell Dev Biol 17: 363–376, 2006. [DOI] [PubMed] [Google Scholar]
- 99.Xie GX, Palmer PP. How regulators of G protein signaling achieve selective regulation. J Mol Biol 366: 349–365, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yagi Y, Andoh A, Inatomi O, Tsujikawa T, Fujiyama Y. Inflammatory responses induced by interleukin-17 family members in human colonic subepithelial myofibroblasts. J Gastroenterol 42: 746–753, 2007. [DOI] [PubMed] [Google Scholar]
- 101.Yamakawa T, Eguchi S, Matsumoto T, Yamakawa Y, Numaguchi K, Miyata I, Reynolds CM, Motley ED, Inagami T. Intracellular signaling in rat cultured vascular smooth muscle cells: roles of nuclear factor-kappaB and p38 mitogen-activated protein kinase on tumor necrosis factor-alpha production. Endocrinology 140: 3562–3572, 1999. [DOI] [PubMed] [Google Scholar]
- 102.Yamboliev IA, Hedges JC, Mutnick JL, Adam LP, Gerthoffer WT. Evidence for modulation of smooth muscle force by the p38 MAP kinase/HSP27 pathway. Am J Physiol Heart Circ Physiol 278: H1899–H1907, 2000. [DOI] [PubMed] [Google Scholar]
- 103.Yang CM, Chien CS, Hsiao LD, Luo SF, Wang CC. Interleukin-1beta-induced cyclooxygenase-2 expression is mediated through activation of p42/44 and p38 MAPKS, and NF-kappaB pathways in canine tracheal smooth muscle cells. Cell Signal 14: 899–911, 2002. [DOI] [PubMed] [Google Scholar]
- 104.Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem 278: 33067–33077, 2003. [DOI] [PubMed] [Google Scholar]
- 105.Zhang Z, Zheng M, Bindas J, Schwarzenberger P, Kolls JK. Critical role of IL-17 receptor signaling in acute TNBS-induced colitis. Inflamm Bowel Dis 12: 382–388, 2006. [DOI] [PubMed] [Google Scholar]
- 106.Zhao A, Morimoto M, Dawson H, Elfrey JE, Madden KB, Gause WC, Min B, Finkelman FD, Urban JF Jr, Shea-Donohue T. Immune regulation of protease-activated receptor-1 expression in murine small intestine during Nippostrongylus brasiliensis infection. J Immunol 175: 2563–2569, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhao L, Lee JY, Hwang DH. The phosphatidylinositol 3-kinase/Akt pathway negatively regulates Nod2-mediated NF-kappaB pathway. Biochem Pharmacol 75: 1515–1525, 2008. [DOI] [PubMed] [Google Scholar]