

Abstract
There is evidence that NO can regulate CO production, however less is known about CO regulation of NO synthesis. Our studies were undertaken to define how CO regulates iNOS in cultured hepatocytes. CO (250 ppm) exposure resulted in a significant decrease in iNOS protein, nitrite production, level of active iNOS dimer and cytosolic iNOS activity in cells stimulated with cytokines (IL-1β) or transfected with the human iNOS gene. However, IL-1β-stimulated iNOS mRNA expression was unaffected by CO. These effects of CO on iNOS protein levels were inhibited when CO was scavenged using hemoglobin. HO-1 induction with an adenoviral vector carrying HO-1 showed a decrease in total iNOS protein, nitrite production, and iNOS dimer level from cells stimulated by IL-1β. iNOS protein level was significantly higher in lung endothelial cells isolated from HO-1 knockout mice compared to wild type cultures stimulated with cytokines mixture. CO was found to increase p38 phosphorylation and p38 inhibition using SB203580 increased iNOS protein levels in response to IL-1β. Interestingly, proteasome inhibitors (MG132 and Lactacystin) and an autophagy inhibitor (3-Methyladenine) reversed CO influence iNOS levels. Our results imply that CO exposure decreases NO production by suppressing dimer formation and increasing iNOS degradation through a process involving p38 activation.
Keywords: Heme oxygenase, Carbon monoxide, Inducible nitric oxide synthase, Nitric oxide, proteosome
Introduction
The inducible nitric oxide synthase (iNOS) gene is expressed by hepatocytes in a number of physiologic and pathophysiological conditions affecting the liver including sepsis and hemorrhagic shock (1–3). iNOS protein and the CO productof its enzymatic activity, nitric oxide (NO) modulates several aspects of liver injury. iNOS expression potentiates the hepatic oxidative injury in warm ischemia/reperfusion (4), while NO protects against hepatic apoptotic cell death seen in models of sepsis and hepatitis. Anti-apoptotic actions are either cyclic nucleotide dependent or independent, and include the induction of heat shock protein 70 expression, prevention of mitochondrial dysfunction, and inhibition of caspase activity (5–8).
Heme oxygenase (HO) catalyzes the breakdown of heme into equimolar concentrations of carbon monoxide (CO), biliverdin, and iron. The inducible isoform, HO-1, is potentially involved in the control of intracellular heme levels and is believed to play an important role in attenuating tissue injury caused by inflammatory stimuli (9). Notably, up-regulation of HO-1 has been shown to protect against lipopolysaccharide (LPS)-induced cardiovascular collapse, and minimizes ischemic liver damage (10).
CO, like NO, is a second messenger gas involved in a number of physiological processes (11). Both CO and NO activate soluble guanylate cyclase to increase cyclic GMP (cGMP) levels. It is becoming increasingly clear that iNOS/NO and HO-1/CO can modulate each other’s activity. These two system are linked in that NO can up-regulate HO-1 expression leading to the formation of endogenous CO (10; 12; 13), and CO can bind to the heme group in the iNOS protein and influence the production of NO (14; 15). Although much is known about the HO-1/CO and iNOS/NO pathways, how these two important systems interact is less understood.
It has been postulated that CO serves as an intracellular signal to modulate the tissue stress response and interacts with iNOS to control its expression and activity (13; 16–19). Specifically, CO could act as a feedback inhibitor of iNOS when the concentrations of NO exceed a critical threshold.
Interleukin-1beta (IL-1β) is a key mediator in a variety of inflammatory responses and the most effective cytokine for the induction of the iNOS gene expression (20; 21). In this report we investigated the effect of CO on iNOS expression and NO production in IL-1β-treated hepatocytes. Exogenous CO decreased iNOS expression associated with suppressed iNOS dimer formation. Our findings suggest CO is a feedback inhibitor of iNOS expression and activity.
Material and Methods
Materials
Williams Medium E, penicillin, streptomycin, L-glutamine, HEPES were purchased from Life Technologies, Inc. Mouse recombinant IL-1β was obtained from R & D Systems. Polyclonal antibodies towards the following: phospho-ERK 1/2 (Thr202/Tyr204), phospho-Akt (Ser473), phospho-p38, p38, phospho-JNK and IκB were purchased from Cell Signaling. Polyclonal anti-HO-1 anti-NF-κB antibodies were purchased from StressGen Biotechnologies and SantaCruze. KT5823, LY294002, PD 98059, SP600125 and wortmannin are from Calbiochem. [γ-32P]ATP was obtained from NEN Life Science Products. 1H-(1,2,4)-oxadiazole [4,3-a] quinoxalon-1-one (ODQ) was purchased from Promega. Cobalt protoporphyrin (CoPPIX), and tin protoporphyrin (SnPPIX) were acquired from Porphyrin Products. All other reagents are from Sigma, unless otherwise indicated.
Cell Culture
Primary hepatocytes were isolated and purified from rat and cultured as described previously (22). Highly purified hepatocytes (>98% purity and >98% viability by trypan blue exclusion) were suspended in Williams medium E supplemented with 10% calf serum, 1 μM insulin, 2 mM L-glutamine, 15 mM HEPES (pH 7.4), 100 units/ml penicillin, and 100 μg/ml streptomycin. The cells were plated on collagen-coated tissue culture plates at a density of 2×105 cells/well in 12-well plates for cell viability analysis or 3 × 106 cells/60-mm dish for Western blot and EMSA. After 18 h fresh medium containing 5% calf serum was added including designated treatments.
Preparation of Whole Cell Lysate
Cells were cultured in medium with or without serum for 24 h and then stimulated with CO or other specified reagent. Monolayers were rinsed twice with ice-cold PBS, and crude cell lysates were isolated with 300 μl of whole cell lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Nonidet P-40, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1μg/ml leupeptin, and 1mM PMSF) was then added to each plate. The cells were lysed by three freeze and thaw cycles. Cytosolic fractions were obtained by centrifugation of the ruptured crude lysates at 12,000×g for 20 min at 4°C and aliquots were stored at −80 °C until use. Protein concentration was determined by BCA (Pierce).
Western blot Analysis
Proteins were separated on 10% SDS-PAGE in denaturing or non-denaturing conditions and transferred to nitrocellulose membranes. The membranes were hybridized with antibodies for iNOS (1:1000), HO-1 (1:2000), p-Akt (1:1000), p-ERK (1:1000), p-p38 (1:500), p-JNK (1:500), beta-actin (1:5000) in PBS-Tween (0.01%) (PBS-T) containing 1% nonfat milk for 1 h. The immunoblots were developed following a 2 min incubation with enhanced chemiluminescent (ECL) solution (Pierce), and exposure to x-ray film.
Luciferase Reporter Assays
Evaluation of NF-κB activation was performed using a luciferase reporter assay. In brief, hepatocytes were co-transfected with NF-κ B reporter constructs (100 ng/well; pGL3-NF-κ B luciferase; provided by D. Golenbock, University of Massachusetts, Worcester, MA) and pIEP-Lac-z (0.5 μg/well) using Lipofectin (Invitrogen) according to the manufacturer’s instructions (23). Evaluation of iNOS expression was performed using a luciferase reporter assay with transient transfection of hepatocytes being accomplished with an iNOS promoter reporter constructs (1 μg/well; pXP2; provided by C. Lowenstein, Johns Hopkins University, Baltimore, MD) or pIEP-LacZ (0.5 μg/well) described in published work (24). Hepatocytes were treated with various stimuli after transfection for 24 h. Luciferase activity (reported as arbitrary units [A.U.]) was assayed 6 h post treatment using a luciferase assay kit (Promega) as per the manufacturer’s instructions and measured on a Berthold Luminometer. Results were corrected for transfection efficiency and protein concentration.
Northern Blot
Total RNA isolated from cultured hepatocytes using the method of RNAzol adapted as described by Chomoczynski and Sacchi (25). RNA (20 μg) was loaded onto a 1% agarose gel containing 3% formaldehyde, transferred onto a nylon membrane and UV auto-crosslinked (UV Stratalinker 1800, Stratagene). The membranes were pre-hybridized at 43°C overnight as previously described and hybridized by random priming (Boehringer Mannheim) with [32P} dCTP-iNOS probe (~2×106 cpm/ml), a 2.7 kb Not I fragment of the murine macrophage cDNA effective for identifying iNOS mRNA in rat hepatocytes (26). The hybridized filters were washed three times, and exposed to autoradiography film.
Measurement of Nitrite
Nitrite was assayed by the Griess reaction. Briefly, 100μl of Griess reagent was added to 100μl of culture supernatant in a 96-well plate. Absorbance was measured at 550 nm using a 96-well plate reader, and nitrite concentration was calculated from a standard curve of sodium nitrite.
Transfection of Adenovirus
Adenoviral vectors carrying the genes for bacterial β-galactosidase (Ad-LacZ), human iNOS (Ad-iNOS) and recombinant HO-1 (Ad-HO-1) were prepared as described previously (27; 28). Following overnight culture, hepatocytes were washed with Hanks’ buffered saline twice prior to transduction at multiplicity of infection of 1 in a volume of 2 ml of Opti-MEM (Life Technology). Following a 6 h infection, the medium was changed to fresh Williams E containing 5% calf serum. After 24 h, cells infected with adenovirus were treated with CO or IL-1β. Samples were isolated at the indicated time points.
Statistical Analysis
Data are presented as means ± S.D. of the mean of at least three separate experiments.
Results
CO decreased the level of iNOS protein in hepatocytes stimulated by cytokines
To examine the effects of CO on hepatocyte iNOS expression and activity we first measured the consequences of exogenous CO exposure on iNOS protein levels and NO production in cytokine-stimulated hepatocytes (29). We have previously shown that IL-1β is the most effective cytokine stimulator of hepatocyte iNOS expression (30). Consistent with this previous report, Fig. 1A shows that iNOS protein levels at 24 hrs following cytokine exposure are highest with IL-1β alone with weak induction by TNF or IFNγ. A combination of the three cytokines shows the expected synergy with greater iNOS expression. CO exposure for 24 hr markedly suppresses iNOS protein levels and decreases nitrite release by over 50% (Fig. 1B). The reduction in iNOS protein expression for IL-1β stimulated hepatocytes was confirmed by immunohistochemistry (Fig. 1C).
Figure 1.
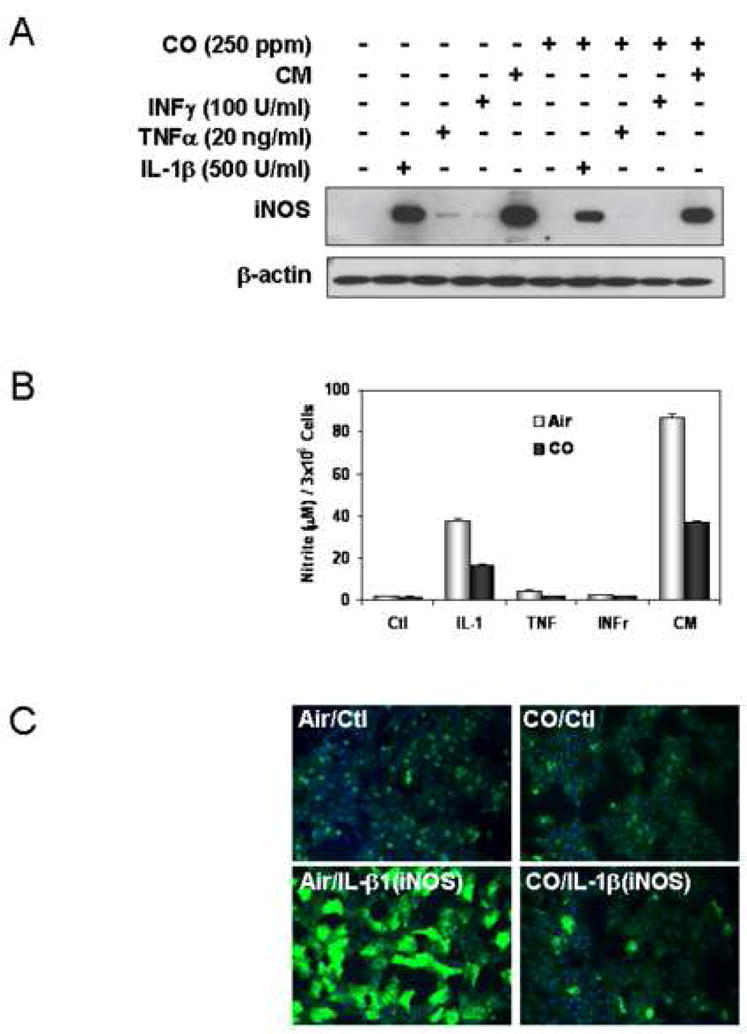
CO decreases hepatic iNOS protein and nitrite production in response to IL-1β and cytokine mixture (CM). Rat hepatocytes were cultured for 24h in the presence of CM (IL-1β;500 U/ml, INFγ;100U/ml, TNFα;10ng/ml) with or without a 2h pre-exposure of CO (250 ppm), (A) Western blot was performed using denaturing SDS-PAGE. (B) Accumulated nitrite for 24h was measured in the culture medium using Griess reagent. Data are means±S.D of three separate experiments. (C). Immunostaining for iNOS, cells were treated with or without CO (250 ppm) for 1 h, stimulated with IL-1β (500 U/ml) for 24h in the presence or absence of CO, monolayers were washed 2× with cold PBS and then cells were fixed in 2% paraformaldehyde. Fixed cells were incubated with the primary monoclonal anti-iNOS antibody, and the primary antibody staining was detected with the FITC-conjugated anti mouse IgG. Nuclei of cells were stained with Hoechst 33342. Images were taken at a magnification of 20× using a Olympus Provis microscope.
CO decreases the level of active iNOS dimer and iNOS activity in response to IL-1β
iNOS is active only as a homodimer (31) and CO may alter iNOS activity by interfering with dimer formation. Using non-denaturing SDS PAGE and Western blotting we found that CO exposure for 24hr markedly decreased the levels of dimeric iNOS (Fig. 2A). Doubling the CO levels to 500 ppm had no additional effect. To determine how CO affects the dimer to monomer ratio we quantified levels by UN-SCAN- IT gel for Windows 5.1 (Silk Scientific Corporation) at 12 hrs following IL-1β exposure. The ratio of iNOS dimer to monomer was 1 to 1 in air treated cells, but 0.5 to 1 in CO exposed cells (Fig. 2B). Fig. 2C shows the corresponding decrease in nitrite production at 12 hrs.
Figure 2.
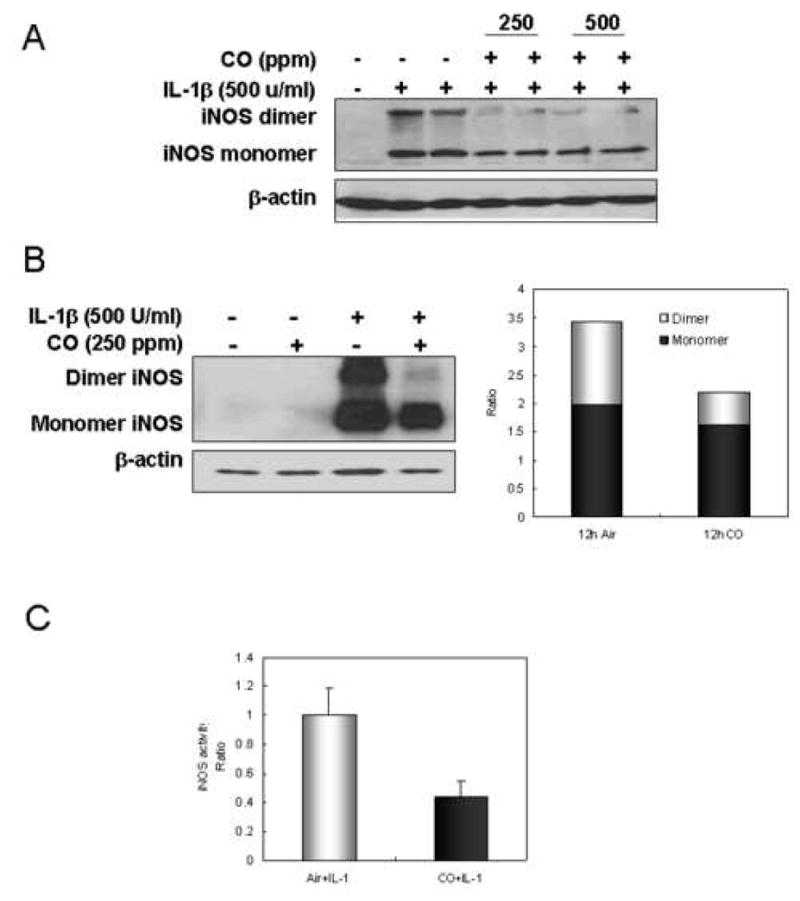
CO decreases the level of active iNOS dimer and iNOS activity in response to IL-1β. (A) Hepatocytes were stimulated with IL-1β (500 U/ml) for 24h at different concentration of CO (250 or 500 ppm). After 24h, cells were harvested, washed with ice-cold PBS, and lysed by three cycles of freeze and thaw. Cytosolic fractions were prepared by centrifugation at 12,000×g for 20 min and then subjected to western blot. (B) Hepatocytes were treated with or without CO (250 ppm) for 2h, and stimulated with IL-1β (500 U/ml) for 12h in a presence or absence of CO. After 12h, monolayers were washed with ice-cold PBS, and lysed by three cycles of freeze and thaw. Cytosolic fractions were prepared by centrifugation at 12,000×g for 20 min and then western blot was performed using non-denaturing SDS-PAGE. Graph displays the ratio of active dimer to monomer from western blot. (C) Cytosolic iNOS activity was measured in the presence of 1 mM NADPH, 20 μM FAD, 20 μM FMN, and 4 mM L-arginine, with exogenous 100 μM BH4 at 37°C for 2h. Accumulated nitrite of cytosolic protein was measured using the Griess reagent. Data represent the mean ± S.D. of more than three independent experiments.
Upregulation of HO-1 expression by CoPP or adenoviral vector decreased the level of iNOS protein and nitrite accumulation
Catabolism of cellular heme by HO-1 has been reported to limit new iNOS synthesis due to the requirement for heme to form functionally competent iNOS (16; 31–37). HO-1 expression was measured in CO-treated cells (Fig. 3A) and was found to be induced to a greater degree by CO in cytokine-treated cells. To test whether enhanced HO-1 expression reduced iNOS expression, cells were treated with cobalt protoprophyrin (CoPP) to up-regulate HO-1 expression. CoPP treatment decreased IL-1β induced iNOS protein expression in a dose dependent manner (Fig. 3B). Cells treated with CoPP (100 μM) and IL-1β for 12 hrs had both a decrease in the level of active iNOS dimer and nitrite accumulation (Fig. 3C and D). Overexpression of HO-1 using an adenoviral vector for 24 hrs followed by IL-1β stimulations also resulted in lower iNOS dimer protein levels and a significant decrease in nitrite accumulation (Fig. 4A and B) compared to control β-galactosidase-transduced cells.
Figure 3.

HO-1 induction by CoPP suppresses the level of iNOS protein and active iNOS dimer in response to IL-1β. (A) Rat hepatocytes were cultured for 24h in the presence of cytokines (IL-1β;500 U/ml, INFγ;100U/ml, TNFα;10ng/ml) with or without CO (250 ppm) following the 2h pre-exposure of CO. Western blot for iNOS or HO-1 was performed using denaturing SDS-PAGE. (B). Cells were treated with CoPP at indicated concentration (10, 25, 50, 100 μM) for 20h and further incubated with treatment of IL-1β (500 U/ml) for 24h. Cytosolic fractions were prepared by centrifugation at 12,000×g for 20 min and western blot was performed using denaturing SDS-PAGE. (C). Cells were incubated with CoPP (100 βM) for 20h prior to addition of IL-1β (500 U/ml). After 12h cells were washed with ice-cold PBS, and lysed by three cycles of freeze and thaw. Cytosolic fractions were prepared by centrifugation at 12,000×g for 20 min and western blot was performed using non-denaturing SDS-PAGE. (D). Cytosolic iNOS activity was measured in the presence of 1 mM NADPH, 20 μM FAD, 20 μM FMN, and 4 mM L-arginine, with exogenous 100 μM BH4 at 37°C for 2h. Accumulated nitrite of cytosolic protein was measured using Griess reagent. Data represent the mean ± S.D. of more than three independent experiments
Figure 4.
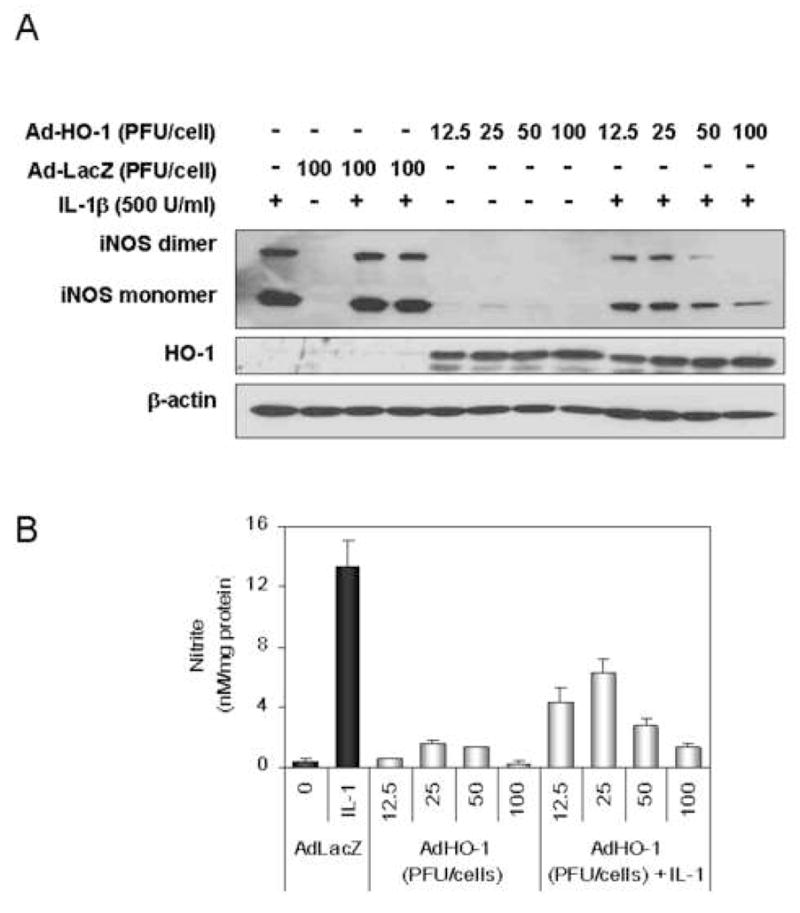
HO-1 overexpression by infection of a recombinant HO-1 adenovirus decreases iNOS protein in response to IL-1β. (A). Modified adenoviral vectors expressing HO-1 or β-galactosidase transduced in hepatocytes at the indicated multiplicity of infection (12.5, 25, 50, 100 PFU/cell). The infected hepatocytes were recovered overnight prior to changing to fresh medium and initiating iNOS expression by treatment with IL-1β (500 U/ml) for 24h. Monolayers were harvested after being washed with ice-cold PBS, and lysed by three cycles of freeze and thaw. Cytosolic fractions were prepared by centrifugation at 12,000×g for 20min and then western blot was performed using non-denaturing SDS-PAGE. (B) Accumulated nitrite was measured in the culture medium using Griess reagent. Data represent the mean ± S.D. of more than three independent experiments
Endothelial cells from HO-1 knockout mouse express higher levels of iNOS protein in response to cytokine stimulation
Lung endothelial cells isolated from HO-1 knockout animals were used to determine if iNOS dimer levels and subsequent nitrite generation are modulated by HO-1. As shown in Fig. 5, cells from the HO-1 knockout mouse expressed higher levels of iNOS protein compared with that of cells from wild type mice in response to cytokine stimulation. Exogenous CO resulted in a modest reduction in the level of active iNOS dimer in the wild type cells. These findings support the conclusion that endogenous HO-1 modulates iNOS expression.
Figure 5.
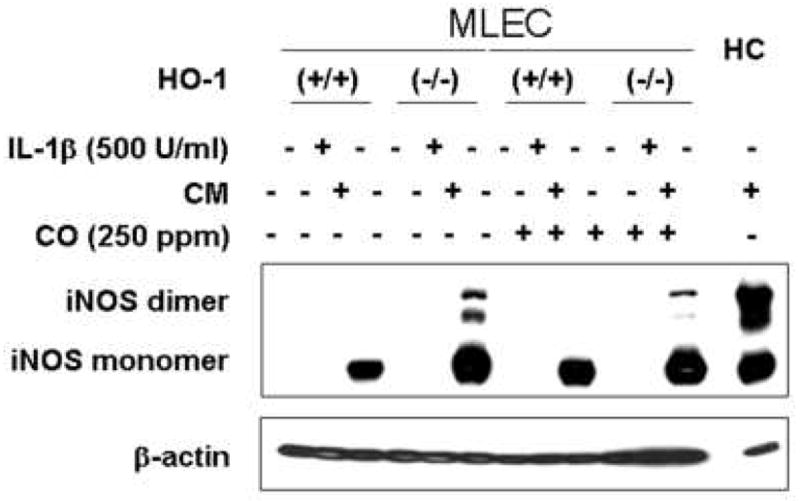
Endothelial cells from HO-1 knockout mouse increased the level of iNOS protein in response to cytokine mixture. Cells were incubated with IL-1β (500 U/ml) or cytokine mixture (CM; IL-1β (100 U/ml) INFγ (100 U/ml) and TNFα (20 ng/ml)) in the presence or absence of CO (250 ppm). After 24h, monolayers were harvested after being washed with ice-cold PBS, and lysed by three cycles of freeze and thaw. Cytosolic fractions were prepared by centrifugation at 12,000×g for 20min and then western blot was performed using non-denaturing SDS-PAGE. MLEC; mouse lung endothelial cell, HC; hepatocyte
CO regulation of iNOS protein levels occurs at the translational or post-translational level
We investigated iNOS mRNA levels from cells stimulated with IL-1β in the presence or absence of CO. Northern blot experiments showed no effect of CO on IL-1β-induced iNOS mRNA steady state levels (Fig 6A). As seen in Fig 6B, iNOS promoter activity was similarly unaffected by CO treatment.
Figure 6.
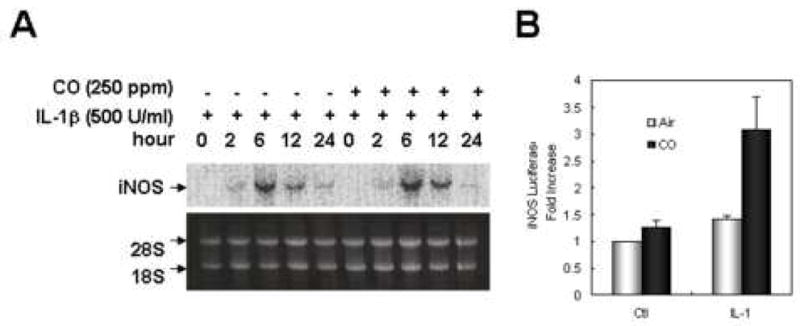
CO does not increase the level of iNOS mRNA significantly. (A) Hepatocytes were incubated with IL-1β (500 U/ml) in the presence or absence of CO (250 ppm) for the indicated times (2, 6, 12, 24h). Total RNA was extracted from hepatocytes by the RNAzol method. The RNA (20 μg) was electrophoresed on 1% agarose gel containing 1% formaldehyde, transferred onto a nylon membrane, and prehybridized with whale sperm DNA at 43°C overnight. The membrane was hybridized with 32P-labeled probes (~2×106 cpm/ml) of murine macrophage iNOS, washed three times, and exposed to autoradiography film. (B) Evaluation of iNOS promoter activity was performed using a luciferase reporter assay. Hepatocytes were transfected with human iNOS promoter reporter construct for 24 h. Cells were incubated with IL-1β (500 U/ml) in a presence or absence of CO for 6h. Luciferase activity (reported as arbitrary units [A.U.]) was assayed 6 h after initiation of treatment using a luciferase assay kit (Promega) according to the manufacturer’s instructions and measured on a Berthold Luminometer. Results were corrected for transfection efficiency and protein concentration. Data represent the mean ± S.D. of more than three independent experiments.
To express iNOS independent of cytokine stimulation, hepatocytes were transduced using an adenoviral vector carrying iNOS. As shown in Fig 7A and B, CO exposure resulted in lower levels of iNOS protein and nitrite production in cells transfected with adenoviral vector carrying the human iNOS gene compared to cells not exposed to CO.
Figure 7.
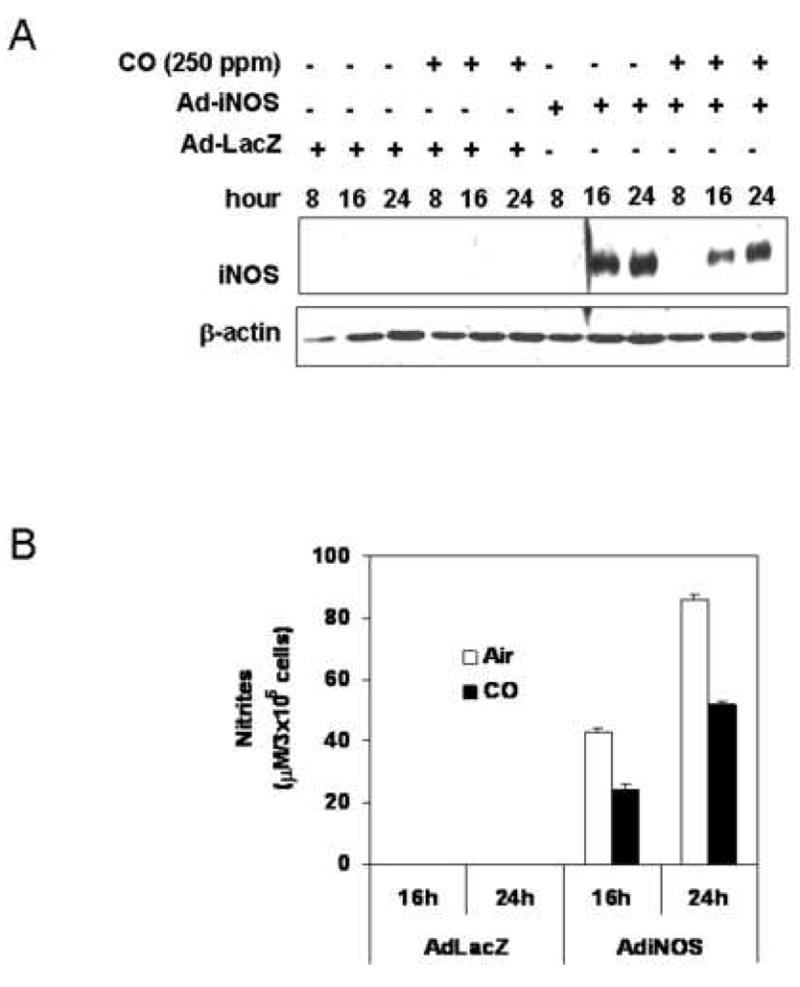
CO decreases nitrite production and iNOS protein from hepatocytes transfected with adenoviral vector carrying a human iNOS gene. Modified adenoviral vectors (MOI of 3) carrying a human iNOS gene or β-galactosidase were prepared. (A). The hepatocytes were incubated with CO (250 ppm) for 4h prior to infection of adenoviral vector. The cells were transfected with adenoviral vector in a serum-free medium for 6h, and recovered for indicated time (8, 16, 24h) in fresh medium with 5 % serum in the absence or presence of CO. Monolayers were washed with ice-cold PBS, and lysed by three cycles of freeze and thaw. Western blot was performed from cytosolic fractions collected by centrifugation at 12,000×g for 20min. (B). Accumulated nitrite was measured in the culture medium using Griess reagent. Data represent the mean ± S.D. of two independent experiments.
CO suppressed iNOS expression in IL-1β stimulated hepatocytes through modulation of p38 kinase and the proteosome degradation pathway
CO has been reported to modulate cell signaling through MAP kinase and guanylate cyclase pathways (38; 39). We carried out experiments to determine if CO modulation of MAP or Akt accounted for the inhibition of IL-1β-induced iNOS expression. CO enhanced IL-1β induced p38 activation, had no effect on phosphorylation of ERK and JNK, and suppressed the phosphorylation of Akt (Fig. 8A)(40). To determine whether the enhanced activation of p38 MAPK contributed to the inhibitory effect of CO on iNOS expression, cultured hepatocytes were treated for 24 h with IL-1β and CO in the absence or in the presence of SB203580 (10 μM), a selective inhibitor of p38 MAPK. SB203580 reversed the inhibitory effect of CO on IL-1β-induced iNOS protein expression. As expected the ERK inhibitor, PD98059, had no effect on IL-1β induced iNOS protein levels (Fig. 8B). Inhibitors of guanyl cyclase (ODQ) and Akt (wortmannin) likewise had no effect on the CO-inhibition of iNOS expression (data not shown).
Figure 8.
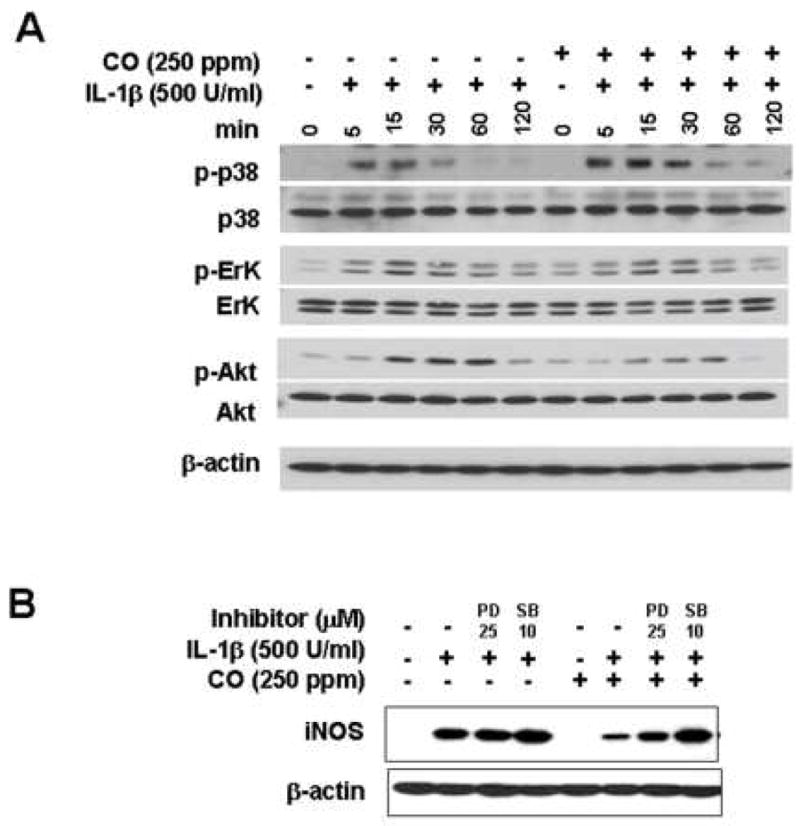
CO enhances phosphorylation of p38 MAPK, while suppressing Akt phosphorylation in IL-1β-treated hepatocytes. (A) Cells were starved for 24h in medium without serum and stimulated with IL-1β (500 U/ml) in the presence or absence of CO (250 ppm). Cell lysed in lysis buffer containing with inhibitor of phosphatase and protease. Western blot was performed from cytosolic fraction using denaturing SDS-PAGE. The SB203580 reversed the inhibitory effect of CO on IL-1β-induced iNOS expression and nitrite production. (B) Cells were incubated with inhibitors of p38 or ERK (SB;10 μM, PD; 25 μM) for 1h and further incubated with IL-1β for 24h in a presence or absence of CO (250 ppm).
iNOS degradation is known to be protesome dependent upon stable expression of human iNOS in human epithelial kidney (HEK293) cells (40). To determine if the effects of CO required protesome activity, IL-1β stimulated hepatocytes were exposed to CO in the presence or absence of the protesome inhibitor MG132 or Lactacystin. The presence of proteosome inhibitors (MG132; 1 and 5 μM or Lactacystin; 0.25 and 2.5 μM) prevented the reduction of iNOS expression in IL-1β treated hepatocytes resulting from CO exposure (Fig. 9).
Figure 9.
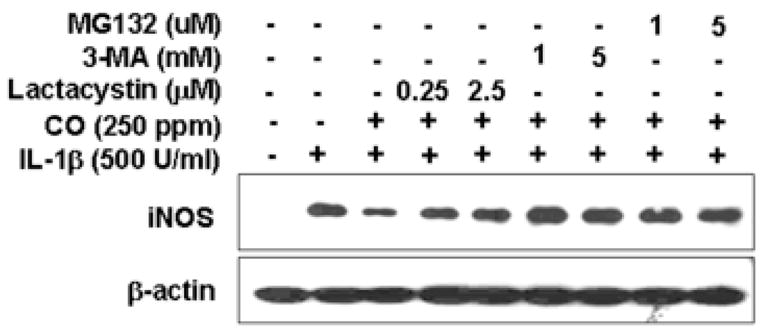
Proteosome inhibitors increased the level of iNOS protein suppressed by CO. Cells were treated with IL-1β for 24h in a presence or absence of CO (250 ppm) following administration of proteosome inhibitor (3-Methyladenine; 1 and 3 mM, MG132;1 and 5 μM or Lactacystin;0.25 and 2.5 μM), western blot was performed from cytosolic fraction.
Discussion
This study was undertaken to determine how CO modulates iNOS protein expression and NO production induced by IL-1β in hepatocytes. CO (250 ppm) down regulated iNOS protein and suppressed the level of active iNOS dimer formation in IL-1β-stimulated hepatocytes. Overexpression of HO-1 using CoPP or adenovirus also effectively decreased the level of iNOS protein, and enzymatically active iNOS. Our results implicate p38 in the CO mediated effects on iNOS expression. The failure to see the reduction in iNOS in response to CO in the presence of proteasome inhibitors suggests that targeting NOS for degradation may be one mechanism by which CO lowers iNOS expression. The suppression of dimer formation raises the possibility that CO might also directly impact on iNOS activity.
CO has been shown to be cytoprotective by modulating NF-κB and iNOS in several types of cells including endothelial cells, hepatocytes and macrophages. We reported that CO increased level of iNOS protein following TNFα/D-galactosamine treatment in vivo, resulting in liver protection (29). Our study presented here shows that CO decreased iNOS protein levels in IL-1β stimulated cells. The interplay between CO/HO-1 and NO/iNOS has been the subject of much debate, not only because of the possibilities of exploiting these pathways as therapeutic targets, but also the possible ramifications of interfering with either CO or NO production. The CO/HO-1 and NO/iNOS pathways are linked in that NO can up regulate HO-1 expression leading to the formation of endogenous CO (41), and CO can bind heme within iNOS and influence the enzyme activity of iNOS and the production of NO (42). Both pathways are expressed in a number of stress conditions, including ischemia/reperfusion and endotoxemia (43). Thus, it is likely that the interplay between CO and NO production takes place. For example, NO can induce HO-1 expression at moderate levels by iNOS. CO produced by HO-1 could then suppress iNOS activity limiting NO production to reduce the risk of iNOS-mediated toxicity.
Our results suggest that the effects of CO on iNOS protein levels are mediated through p38 (13) modulation of protein turnover(16; 17). Interplay between p38 and CO to modulate inflammatory cytokines and induction of stress proteins has been established in clinically relevant in vivo and in vitro models (38). For example, CO induced cell death is prevented by blocking p38 activity. Our results suggest that CO promotes iNOS protein turnover through the activation of p38 as seen in other models (44–46). Post-translational modification of iNOS protein by ubiquitination and phosphorylation are mechanisms reported to modulate iNOS protein level (47; 48). These investigators performed co-transfection in HEK293T cells with plasmids for both human iNOS and ubiquitin or an ubiquitin mutant (K48R) to demonstrate that iNOS ubiquination precedes protein degradation. iNOS monomers and dimers had similar half-lives but both increased in the presence of proteasome inhibitors (48). The relatively short half-life of iNOS was a rather surprising finding because iNOS is known for its prolonged production of large amounts of NO. Interestingly in our studies, the effect of protein turnover inhibitors on iNOS protein levels, proteasome inhibitors (MG132 and Lactacystin) and an autophagy inhibitor (3-Methyladenine), superceded the effect of CO on iNOS levels. Our results imply that CO exposure decreases NO production by suppressing dimer formation and increasing iNOS degradation through a process involving p38. Specifically, CO could act as a feedback inhibitor of iNOS when the concentrations of NO gaseous molecule exceed a critical threshold and the products of heme degradation might play a role in this process.
Dimerization is necessary for catalytic activity for each of the three NOS isoforms. Dimerization of iNOS is distinct from the constitutive isoforms (nNOS and eNOS) in that it is independent of increase in intracellular Ca+2 levels and subsequent calmodulin binding for NO synthesis (49). iNOS is synthesized initially as a monomer, which is enzymatically inactive and dependent on the concomitant cytokine stimulated synthesis of BH4 and heme in order to dimerize and generate NO (50). In aqueous solutions, NO reacts to form NO2−, which, in the presence of heme-containing proteins will be further oxidized to form NO3− (51). These properties of NO allow for an indirect measure of the redox environment in which iNOS is found. The generation of NO contributes to the regulation of iNOS enzymatic activity: a high level of NO down regulates heme and BH4 synthesis, thereby creating a mixed population of iNOS monomer and dimer. Panda et al (2002) found in IL-1β treated hepatocytes exposed to exogenous CO had diminished iNOS total protein and active dimer due to the above mentioned feedback inhibition of iNOS/NO generation system. These authors concluded that the differences in the three NOS isoform dimer strength results from the varied cellular environments in which each isozyme is expressed, specifically iNOS is BH4 dependent to form stable dimers (52).
While it is not clear how CO differently regulates iNOS expression in different organs or different stimuli, CO exerts anti-inflammatory and anti-apoptotic effects by modulating iNOS. Considering that pathophysiological conditions such as hemorrhagic shock and ischemic reperfusion injury result in increased overproduction of NO (53; 54), our findings reveal a potential interaction between CO/HO-1 and NO/iNOS in mediating protection against NO/iNOS-mediated liver damage. Depending on the circumstances, sustained NO production up-regulates HO-1. CO generated by HO-1 would then act to suppress initially iNOS activity and then promote iNOS degradation in an important feedback mechanism.
Abbreviations
- HO
heme oxygenase
- iNOS
inducible nitric oxide synthase
- IL-1β
interleukin-1beta MAPK, mitogen-activated protein kinase
- NOS
nitric oxide synthase
- NF-κB
nuclear factor κB
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang Y, Vodovotz Y, Kim PKM, Zamora R, Billiar TR. Mechanisms of hepatoprotection by nitric oxide. Ann NY Acad Sci. 2001;962:415–422. doi: 10.1111/j.1749-6632.2002.tb04085.x. [DOI] [PubMed] [Google Scholar]
- 2.Zamora R, Vodovotz Y, Billiar TR. Inducible nitric oxide synthase and inflammatory diseases. Mol Med. 2000:347–373. [PMC free article] [PubMed] [Google Scholar]
- 3.Li J, Billiar TR. The anti-apoptotic actions of nitric oxide in hepatocytes. Cell Death Differ. 1999;6:952–955. doi: 10.1038/sj.cdd.4400579. [DOI] [PubMed] [Google Scholar]
- 4.Hierholzer C, Harbrecht B, Menezes JM, Kane J, MacMicking J, Nathan CF, Peitzman AB, Billiar TR, Tweardy DJ. Essential role of induced nitric oxide in the initiation of the inflammatory response after hemorrhagic shock. J Exp Med. 1998;187:917–928. doi: 10.1084/jem.187.6.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim YM, Kim TH, Chung HT, Talanian RV, Yin XM, Billiar TR. Nitric oxide prevents tumor necrosis factor alpha-induced rat hepatocyte apoptosis by the interruption of mitochondrial apoptotic signaling through S-nitrosylation of caspase-8. Hepatology. 2000;32:770–778. doi: 10.1053/jhep.2000.18291. [DOI] [PubMed] [Google Scholar]
- 6.Tzeng E, Billiar TR, Williams DL, Li J, Lizonova A, Kovesdi I, Kim YM. Adenovirus-mediated inducible nitric oxide synthase gene transfer inhibits hepatocyte apoptosis. Surgery. 1998;124:278–283. [PubMed] [Google Scholar]
- 7.Li J, Billiar TR, Talanian RV, Kim YM. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem Biophys Res Commun. 1997;240:419–424. doi: 10.1006/bbrc.1997.7672. [DOI] [PubMed] [Google Scholar]
- 8.Kim YM, Chung HT, Kim SS, Han JA, Yoo YM, Kim KM, Lee GH, Yun HY, Green A, Li J, Simmons RL, Billiar TR. Nitric oxide protects PC12 cells from serum deprivation-induced apoptosis by cGMP-dependent inhibition of caspase signaling. J Neurosci. 1999;19:6740–6747. doi: 10.1523/JNEUROSCI.19-16-06740.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willis D, Moore AR, Frederick R, Willoughby DA. Heme oxygenase: a novel target for the modulation of the inflammatory response. Nat Med. 1996;2:87–90. doi: 10.1038/nm0196-87. [DOI] [PubMed] [Google Scholar]
- 10.Amersi F, Buelow R, Kato H, Ke B, Coito AJ, Shen XD, Zhao D, Zaky J, Melinek J, Lassman CR, Kolls JK, Alam J, Ritter T, Volk HD, Farmer DG, Ghobrial RM, Busuttil RW, Kupiec-Weglinski JW. Upregulation of heme oxygenase-1 protects genetically fat Zucker rat livers from ischemia/reperfusion injury. J Clin Invest. 1999;104:1631–1639. doi: 10.1172/JCI7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartsfield CL. Cross talk between carbon monoxide and nitric oxide. Antioxid Redox Signal. 2002;4:301–307. doi: 10.1089/152308602753666352. [DOI] [PubMed] [Google Scholar]
- 12.Motterlini R, Green CJ, Foresti R. Regulation of heme oxygenase-1 by redox signals involving nitric oxide. Antioxid Redox Signal. 2002;4:615–624. doi: 10.1089/15230860260220111. [DOI] [PubMed] [Google Scholar]
- 13.Foresti R, Motterlini R. The heme oxygenase pathway and its interaction with nitric oxide in the control of cellular homeostasis. Free Radic Res. 1999;31:459–475. doi: 10.1080/10715769900301031. [DOI] [PubMed] [Google Scholar]
- 14.Lin HY, Juan SH, Shen SC, Hsu FL, Chen YC. Inhibition of lipopolysaccharide-induced nitric oxide production by flavonoids in RAW264.7 macrophages involves heme oxygenase-1. Biochem Pharmacol. 2003;66:1821–1832. doi: 10.1016/s0006-2952(03)00422-2. [DOI] [PubMed] [Google Scholar]
- 15.Datta PK, Gross EJ, Lianos EA. Interactions between inducible nitric oxide synthase and heme oxygenase-1 in glomerulonephritis. Kidney Int. 2002;61:847–850. doi: 10.1046/j.1523-1755.2002.00231.x. [DOI] [PubMed] [Google Scholar]
- 16.Foresti R, Hoque M, Bains S, Green CJ, Motterlini R. Haem and nitric oxide: synergism in the modulation of the endothelial haem oxygenase-1 pathway. Biochem J. 2003;372:381–390. doi: 10.1042/BJ20021516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Motterlini R, Foresti R, Intaglietta M, Winslow RM. NO-mediated activation of heme oxygenase: endogenous cytoprotection against oxidative stress to endothelium. Am J Physiol. 1996;270:H107–H114. doi: 10.1152/ajpheart.1996.270.1.H107. [DOI] [PubMed] [Google Scholar]
- 18.Sawle P, Foresti R, Mann BE, Johnson TR, Green CJ, Motterlini R. Carbon monoxide-releasing molecules (CO-RMs) attenuate the inflammatory response elicited by lipopolysaccharide in RAW264.7 murine macrophages. Br J Pharmacol. 2005;145:800–810. doi: 10.1038/sj.bjp.0706241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turcanu V, Dhouib M, Poindron P. Nitric oxide synthase inhibition by haem oxygenase decreases macrophage nitric-oxide-dependent cytotoxicity: a negative feedback mechanism for the regulation of nitric oxide production. Res Immunol. 1998;149:741–744. doi: 10.1016/s0923-2494(99)80050-9. [DOI] [PubMed] [Google Scholar]
- 20.Geller DA, Freeswick PD, Nguyen D, Nussler AK, Di Silvio M, Shapiro RA, Wang SC, Simmons RL, Billiar TR. Differential induction of nitric oxide synthase in hepatocytes during endotoxemia and the acute-phase response. Arch Surg. 1994;129:165–171. doi: 10.1001/archsurg.1994.01420260061008. [DOI] [PubMed] [Google Scholar]
- 21.Taylor BS, de Vera ME, Ganster RW, Wang Q, Shapiro RA, Morris SM, Jr, Billiar TR, Geller DA. Multiple NF-kappaB enhancer elements regulate cytokine induction of the human inducible nitric oxide synthase gene. J Biol Chem. 1998;273:15148–15156. doi: 10.1074/jbc.273.24.15148. [DOI] [PubMed] [Google Scholar]
- 22.Kim YM, de Vera ME, Watkins SC, Billiar TR. Nitric oxide protects cultured rat hepatocytes from tumor necrosis factor-alpha-induced apoptosis by inducing heat shock protein 70 expression. J Biol Chem. 1997;272:1402–1411. doi: 10.1074/jbc.272.2.1402. [DOI] [PubMed] [Google Scholar]
- 23.Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. 1999;274:10689–10692. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- 24.Lowenstein CJ, Alley EW, Raval P, Snowman AM, Snyder SH, Russell SW, Murphy WJ. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proc Natl Acad Sci U S A. 1993;90:9730–9734. doi: 10.1073/pnas.90.20.9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 26.Feinberg AP, Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- 27.Shears LL, Kawaharada N, Tzeng E, Billiar TR, Watkins SC, Kovesdi I, Lizonova A, Pham SM. Inducible nitric oxide synthase suppresses the development of allograft arteriosclerosis. J Clin Invest. 1997;100:2035–2042. doi: 10.1172/JCI119736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otterbein LE, Lee PJ, Chin BY, Petrache I, Camhi SL, Alam J, Choi AM. Protective effects of heme oxygenase-1 in acute lung injury. Chest. 1999;116:61S–63S. doi: 10.1378/chest.116.suppl_1.61s-a. [DOI] [PubMed] [Google Scholar]
- 29.Zuckerbraun BS, Billiar TR, Otterbein SL, Kim PK, Liu F, Choi AM, Bach FH, Otterbein LE. Carbon monoxide protects against liver failure through nitric oxide-induced heme oxygenase 1. J Exp Med. 2003;198:1707–1716. doi: 10.1084/jem.20031003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geller DA, de Vera ME, Russell DA, Shapiro RA, Nussler AK, Simmons RL, Billiar TR. A central role for IL-1 beta in the in vitro and in vivo regulation of hepatic inducible nitric oxide synthase. IL-1 beta induces hepatic nitric oxide synthesis. J Immunol. 1995;155:4890–4898. [PubMed] [Google Scholar]
- 31.Ghosh DK, Stuehr DJ. Macrophage NO synthase: characterization of isolated oxygenase and reductase domains reveals a head-to-head subunit interaction. Biochemistry. 1995;34:801–807. doi: 10.1021/bi00003a013. [DOI] [PubMed] [Google Scholar]
- 32.Yang ZX, Qin J. Interaction between endogenous nitric oxide and carbon monoxide in the pathogenesis of recurrent febrile seizures. Biochem Biophys Res Commun. 2004;315:349–355. doi: 10.1016/j.bbrc.2004.01.061. [DOI] [PubMed] [Google Scholar]
- 33.Andre M, Felley-Bosco E. Heme oxygenase-1 induction by endogenous nitric oxide: influence of intracellular glutathione. FEBS Lett. 2003;546:223–227. doi: 10.1016/s0014-5793(03)00576-3. [DOI] [PubMed] [Google Scholar]
- 34.Maestrelli P, Paska C, Saetta M, Turato G, Nowicki Y, Monti S, Formichi B, Miniati M, Fabbri LM. Decreased haem oxygenase-1 and increased inducible nitric oxide synthase in the lung of severe COPD patients. Eur Respir J. 2003;21:971–976. doi: 10.1183/09031936.03.00098203. [DOI] [PubMed] [Google Scholar]
- 35.Jozkowicz A, Dulak J. Effects of protoporphyrins on production of nitric oxide and expression of vascular endothelial growth factor in vascular smooth muscle cells and macrophages. Acta Biochim Pol. 2003;50:69–79. [PubMed] [Google Scholar]
- 36.Johnson FK, Durante W, Peyton KJ, Johnson RA. Heme oxygenase inhibitor restores arteriolar nitric oxide function in dahl rats. Hypertension. 2003;41:149–155. doi: 10.1161/01.hyp.0000046923.52222.58. [DOI] [PubMed] [Google Scholar]
- 37.Andersson JA, Uddman R, Tajti J, Cardell LO. Heme oxygenase and nitric oxide synthase in human middle ear epithelium indicates local carbon monoxide and nitric oxide production. Acta Otolaryngol. 2002;122:634–637. doi: 10.1080/000164802320396312. [DOI] [PubMed] [Google Scholar]
- 38.Otterbein LE, Otterbein SL, Ifedigbo E, Liu F, Morse DE, Fearns C, Ulevitch RJ, Knickelbein R, Flavell RA, Choi AM. MKK3 mitogen-activated protein kinase pathway mediates carbon monoxide-induced protection against oxidant-induced lung injury. Am J Pathol. 2003;163:2555–2563. doi: 10.1016/S0002-9440(10)63610-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morse D, Choi AM. Heme oxygenase-1: the “emerging molecule” has arrived. Am J Respir Cell Mol Biol. 2002;27:8–16. doi: 10.1165/ajrcmb.27.1.4862. [DOI] [PubMed] [Google Scholar]
- 40.Jiang B, Xu S, Hou X, Pimentel DR, Cohen RA. Angiotensin II differentially regulates interleukin-1-beta-inducible NO synthase (iNOS) and vascular cell adhesion molecule-1 (VCAM-1) expression: role of p38 MAPK. J Biol Chem. 2004;279:20363–20368. doi: 10.1074/jbc.M314172200. [DOI] [PubMed] [Google Scholar]
- 41.Naughton P, Foresti R, Bains SK, Hoque M, Green CJ, Motterlini R. Induction of heme oxygenase 1 by nitrosative stress. A role for nitroxyl anion. J Biol Chem. 2002;277:40666–40674. doi: 10.1074/jbc.M203863200. [DOI] [PubMed] [Google Scholar]
- 42.McMillan K, Bredt DS, Hirsch DJ, Snyder SH, Clark JE, Masters BS. Cloned, expressed rat cerebellar nitric oxide synthase contains stoichiometric amounts of heme, which binds carbon monoxide. Proc Natl Acad Sci U S A. 1992;89:11141–11145. doi: 10.1073/pnas.89.23.11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee VG, Johnson ML, Baust J, Laubach VE, Watkins SC, Billiar TR. The roles of iNOS in liver ischemia-reperfusion injury. Shock. 2001;16:355–360. doi: 10.1097/00024382-200116050-00006. [DOI] [PubMed] [Google Scholar]
- 44.Ho AK, McNeil L, Terriff D, Price DM, Chik CL. Role of protein turnover in the activation of p38 mitogen-activated protein kinase in rat pinealocytes. Biochem Pharmacol. 2005;70:1840–1850. doi: 10.1016/j.bcp.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 45.vom DS, Dombrowski F, Schmitt M, Schliess F, Pfeifer U, Haussinger D. Cell hydration controls autophagosome formation in rat liver in a microtubule-dependent way downstream from p38MAPK activation. Biochem J. 2001;354:31–36. doi: 10.1042/0264-6021:3540031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jia G, Cheng G, Gangahar DM, Agrawal DK. Insulin-like growth factor-1 and TNF-alpha regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol Cell Biol. 2006;84:448–454. doi: 10.1111/j.1440-1711.2006.01454.x. [DOI] [PubMed] [Google Scholar]
- 47.Vodovotz Y, Russell D, Xie Q-W, Bogdan C, Nathan C. Vesicle association of nitric oxide synthase from primary mouse macrophages. J Immunol. 1995;154:2914–2925. [PubMed] [Google Scholar]
- 48.Kolodziejski PJ, Musial A, Koo JS, Eissa NT. Ubiquitination of inducible nitric oxide synthase is required for its degradation. Proc Natl Acad Sci U S A. 2002;99:12315–12320. doi: 10.1073/pnas.192345199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci U S A. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Albakri QA, Stuehr DJ. Intracellular assembly of inducible NO synthase is limited by nitric oxide-mediated changes in heme insertion and availability. J Biol Chem. 1996;271:5414–5421. doi: 10.1074/jbc.271.10.5414. [DOI] [PubMed] [Google Scholar]
- 51.Ignarro LJ, Fukuto JM, Griscavage JM, Rogers NE, Byrns RE. Oxidation of nitric oxide in aqueous solution to nitrite but not nitrate: comparison with enzymatically formed nitric oxide from L-arginine. Proc Natl Acad Sci U S A. 1993;90:8103–8107. doi: 10.1073/pnas.90.17.8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Panda K, Rosenfeld RJ, Ghosh S, Meade AL, Getzoff ED, Stuehr DJ. Distinct dimer interaction and regulation in nitric-oxide synthase types I, II, and III. J Biol Chem. 2002;277:31020–31030. doi: 10.1074/jbc.M203749200. [DOI] [PubMed] [Google Scholar]
- 53.Menezes JM, Hierholzer C, Watkins SC, Billiar TR, Peitzman AB, Harbrecht BG. The modulation of hepatic injury and heat shock expression by inhibition of inducible nitric oxide synthase after hemorrhagic shock. Shock. 2002;17:13–18. doi: 10.1097/00024382-200201000-00003. [DOI] [PubMed] [Google Scholar]
- 54.Hierholzer C, Menezes JM, Ungeheuer A, Billiar TR, Tweardy DJ, Harbrecht BG. A nitric oxide scavenger protects against pulmonary inflammation following hemorrhagic shock. Shock. 2002;17:98–103. doi: 10.1097/00024382-200202000-00003. [DOI] [PubMed] [Google Scholar]