

Abstract
To investigate the polymerase components involved in transcription versus replication of vesicular stomatitis virus (VSV), we sequenced the polymerase gene of a conditionally RNA defective, temperature sensitive VSV: ts(G)114, which has a phenotype upon shift from permissive to non-permissive temperature of shut-down of mRNA transcription and unaffected genome replication. Sequence analysis of the ts(G)114 L gene identified three altered amino acid residues in the L protein. These three changes were specifically engineered individually and in combinations into a functional cDNA clone encoding the VSV genome and tested for association with the temperature sensitive and RNA defective phenotypes in the background of recovered engineered viruses. The data presented in this study show a specific amino acid substitution in domain II of the VSV L protein that significantly affects total RNA synthesis, but when in combination with two additional amino acid substitutions identified in the ts(G)114 L protein, leads to a specific reduction in mRNA transcription, but not replication.
Introduction
Vesicular stomatitis virus (VSV) is the prototypic rhabdovirus belonging to the order Mononegavirales. Viruses classified within this order contain single-stranded, nonsegmented negative sense RNA genomes and display a reasonably conserved gene order and RNA synthetic control elements, which include cis-acting sequences and trans-acting viral proteins. The VSV genomic RNA contains a 3’ terminal leader region and a 5’ terminal 59 nt trailer region and in between these terminal regions are the genes for five major proteins: the nucleocapsid (N) protein, the phosphoprotein (P), which is a cofactor of the viral RNA-dependent RNA polymerase (RdRp) and a solubility factor for the N protein (Davis et al., 1986; Green et al., 2000; Howard and Wertz, 1989), the matrix (M) protein, the glycoprotein (G), and the large (L) protein, the major component of the RdRp (Abraham and Banerjee, 1976; Ball, 1977; Ball and White, 1976). During transcription, the first biosynthetic event in the viral replication process, the virion-associated RdRp complex, composed of the L and P proteins, transcribes each of the five genes into discrete mRNAs in an obligatorily sequential manner (Abraham and Banerjee, 1976; Ball and White, 1976; Villarreal et al., 1976). There is a gradient of viral mRNA synthesis, such that 3’ proximal genes are transcribed more abundantly than 3’ distal genes, and each mRNA is capped and methylated at the 5’ end and polyadenylated at the 3’ end (Iverson and Rose, 1981). Primary transcription by the infecting viral ribonucleoprotein (RNP) complex can occur prior to any viral protein synthesis using the input polymerase complex.
Replication of the VSV RNA genome however, requires in addition to the polymerase complex, de novo synthesis of the viral nucleocapsid protein, N, to encapsidate the nascent viral anti-genomic and genomic RNAs (Patton et al., 1984). Replication initiates at the 3’ end of the viral genome with the RdRp synthesizing a complementary copy of the negative sense genome, which is then used as a template for the asymmetric synthesis of progeny genomes that can be assembled into virus particles. This process requires the RdRp to ignore the conserved gene junctions known to regulate mRNA synthesis, capping, and polyadenylation (Barr and Wertz, 2001; Barr et al., 1997a; Barr et al., 1997b; Hinzman et al., 2002; Wang et al., 2007). The dichotomy between the influences of the cis-acting regulatory sequences located at each gene junction on the RdRp during transcription, which results in the synthesis of discrete mRNAs, versus replication, in which a full-length genome is synthesized, is not understood.
Numerous studies have investigated the differences between mRNA transcription and genome replication. It was initially shown that, unlike transcription, genomic replication required de novo protein synthesis, and N protein synthesis alone fulfilled this requirement in a concentration-dependent manner (Patton et al., 1984; Wertz et al., 1987). While the concentration of N protein is a critical determinant in the ability to replicate, as it is needed in stoichiometric amounts to encapsidate newly synthesized genomes and anti-genomes, it is not thought to be the sole regulator of replication.
It was found that VSV transcription and replication initiate at separate sites on the genome, suggesting that these two synthetic processes are regulated by the choice of initiation site (Whelan and Wertz, 2002). These data suggested that a regulatory event might take place prior to initiation of transcription or replication to determine where the RdRp will enter the genome. It is unclear what factor(s) influence the polymerase to initiate at the 3` end versus the N gene start, but it was suggested that it could be a modification of the RdRp or template (Whelan and Wertz, 2002). The VSV P protein, which is a co-factor of the RdRp, has been shown to require phosphorylation within domain II in order to signal the RdRp to replicate genomic RNA (Hwang et al., 1999). Also, it was shown using immunoaffinity chromatography that two RdRp complexes exist in cells. One complex, which has been proposed as the transcriptase contains VSV L and P proteins, in addition to translation elongation factor-1α, heat shock protein 60, and a sub-molar amount of cellular guanylyltransferase, and the other complex, shown to contain the VSV proteins N, P, and L, has been proposed as the replicase (Qanungo et al., 2004). The factors that control transcription and replication, however, are not understood.
To further investigate factors potentially involved in discriminating transcription and replication, we used a forward genetic approach to identify L protein residues that might be selectively involved in transcription. A temperature sensitive mutant of VSV, ts(G)114, was isolated after exposure to 5-fluorouracil based upon its ability to grow at 31°C but not at 39°C (Pringle, 1970). It was classified as complementation group I, which mapped to a lesion in the L gene as responsible for the temperature sensitive and RNA negative phenotypes (Pringle, 1970). Previous work showed that at the permissive temperature (31°C), the RNA profile of ts(G)114 was indistinguishable from wt. However, if infection was initiated at the permissive temperature and then shifted to the non-permissive temperature (39°C), transcription was shut down while replication was largely unaffected (Perlman and Huang, 1973; Wertz, 1978). In the work described here, we sequenced the L gene of ts(G)114 and identified three predicted amino acid substitutions compared to wt. These mutations were introduced individually or collectively into the L gene of a full-length functional cDNA clone of the VSV genome. The resultant viruses were recovered and assayed for temperature sensitivity. The RNA profiles of each recombinant virus were analyzed at permissive and non-permissive temperatures, as well as after temperature shift in order to identify the mutation(s) responsible for the conditional defect in transcription. The data presented here identify specific amino acids that, together, affect transcription, but not replication.
Results
Analysis of ts(G)114 RNA and protein synthesis
We confirmed the RNA synthetic phenotype of ts(G)114 in comparison with WT VSV by direct metabolic labeling with [3H]-uridine in infected BHK cells using a temperature shift assay. Briefly, two sets of BHK cells were infected with either ts(G)114 or WT VSV at a MOI of 3. One set of cultures for each virus was infected at 31°C and the other set at 39°C. At 4 hours post infection (hpi) the set of cells initially infected at 31°C was either maintained at 31°C or shifted up to 39°C. Similarly, the set of cells initially infected at 39°C was either maintained at 39°C or shifted down to 31°C. After a 30-minute incubation at the adjusted temperature, RNAs were labeled with [3H]-uridine for 1 hr at the same temperature as they were adjusted to at 4 hpi. RNAs were isolated from cytoplasmic extracts and resolved on 1.5% acid agarose-urea gels (Figure 1A). Quantitative analysis of three independent experiments showed that ts(G)114 had slightly reduced transcriptional activity compared to WT at 31°C, but when ts(G)114 infections were initiated at 31°C and shifted to 39°C at 4 hpi, transcription was significantly reduced compared to WT VSV(Figure 1A, compare lanes 1 and 5 and lanes 2 and 6; Figure 1B, right-hand graph). No labeled RNA was detected when ts(G)114 infections were initiated at 39°C even if shifted down to 31°C at 4 hpi (Figure 1A, lanes 7 and 8; Figure 1B, right-hand graph). Quantitative analysis of RNA replication products showed that ts(G)114 had increased replication activity compared to WT when assayed at 31°C and under temperature shift-up conditions (Figure 1A, compare lanes 1 and 5 and lanes 2 and 6; Figure 1B, left-hand graph).
Figure 1. Analysis of ts(G)114 RNA synthesis at 31°C and 39°C.
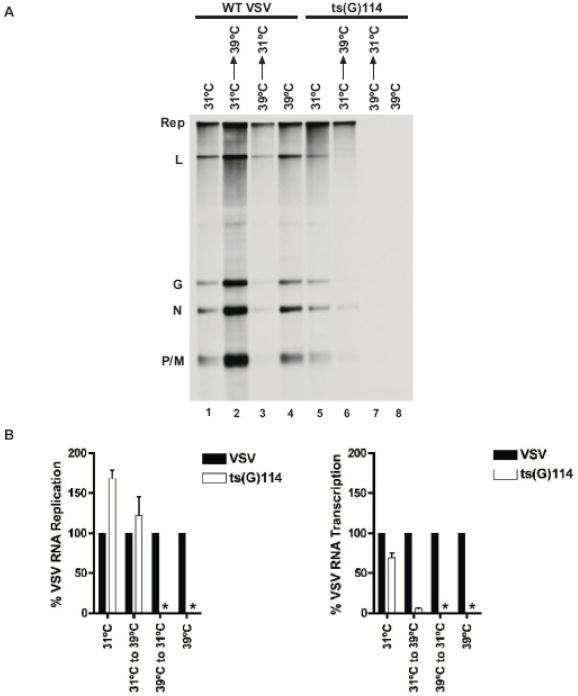
(A) BHK cells were infected at a MOI of 3 at 31°C or 39°C. At 4 hpi, infected cells were pre-treated with actinomycin-D and either maintained at 31°C, shifted from 31°C to 39°C, shifted from 39°C to 31°C, or maintained at 39°C for 30 minutes, at which time RNAs were labeled metabolically with [3H]-uridine at the same temperature as during the pre-treatment period. Radiolabeled RNAs were visualized by acid agarose-urea gel electrophoresis and fluorography. The virus infection from whence the RNAs were generated is noted above the gel along with the temperature conditions. The identities of viral RNAs are noted to the left of the gel. (B) Quantitative analysis of three independent experiments is shown in the graph. The graph on the left represents total viral RNA replication expressed as a percentage of WT at the same temperature. The graph on the right represents total viral transcription expressed as a percentage of WT at the same temperature. (*) indicates no RNA was detected.
Congruent with our analysis of viral RNA synthesis, viral protein synthesis was not observed when infections were initiated at 39°C, which is not surprising given that no mRNA synthesis was detected under these conditions (Figure 2A). Despite the dramatic decrease in transcription when cells infected with ts(G)114 were shifted up to 39°C, protein synthesis under these conditions was indistinguishable from that of ts(G)114 infections conducted entirely at the permissive temperature (Figure 2A and B), suggestive of a long half-life for the viral mRNA.
Figure 2. Analysis of protein synthesis in ts(G)114 infected cells at 31°C and 39°C.

(A) BHK cells were infected at a MOI of 3 at 31°C or 39°C. At 4hpi, infected cells were starved for methionine and either maintained at 31°C, shifted from 31°C to 39°C, shifted from 39°C to 31°C, or maintained at 39°C for 30 minutes, at which time proteins were labeled metabolically with [35S]-methionine. Radiolabeled proteins were visualized by SDS-PAGE and autoradiography. The virus infection from whence the proteins were synthesized is noted above the gel along with the temperature conditions. The identities of viral proteins are noted to the left of the gel. Notably, the M protein in ts(G)114 samples migrates slightly slower than those in WT VSV samples, which is likely due to the size difference between Mudd-Summers (229) and Glasgow (237) M proteins. (B) Quantitative analysis of three independent experiments is shown in the graph. The graph represents total viral protein synthesis expressed as a percentage of WT at the same temperature. (*) indicates no viral protein was detected.
Sequence analysis of the ts(G)114 L gene
We sequenced the L gene of ts(G)114 to identify changes associated with the defect in transcription at the non-permissive temperature. The L gene of ts(G)114 was reverse transcribed from genomic RNA and amplified by PCR as 7 overlapping fragments. Bulk PCR products were sequenced and assembled to yield the full-length ts(G)114 L gene sequence. In addition to the 23 known nt differences, 7 of which result in amino acid changes, between the L genes of the Glasgow and Mudd-Summers strains (Hunt and Hutchinson, 1993), there were four additional nucleotide substitutions: A6256G, A6456G, A8082G, and U10542C (Table 1). Three of the nucleotide substitutions, A6456G, A8082G, and U10542C, resulted, respectively, in changes in the encoded L protein amino acids as follows: D575 to G (DG), E1117 to G (EG), and I1937 to T (IT) (Table 1). One nucleotide substitution was a silent mutation (Table 1).
Table 1.
Nucleotide and amino acid differences between Wild-type (Glasgow) and ts(G)114
| VSV nucleotide # | WT (Glasgow) | Ts(G)114 | Amino acid |
|---|---|---|---|
| 6256 | A | G | Silent (E508E) |
| 6456 | A | G | D575G |
| 8082 | A | G | E1117G |
| 10542 | U | C | I1937T |
Recovery of recombinant viruses containing ts(G)114 L gene mutations
To analyze the effects of these mutations on viral RNA synthesis and to determine if the changes correlated with the RNA synthesis phenotype associated with ts(G)114, we introduced each L gene mutation individually, in pairs, or collectively into a full-length cDNA (VSV1+) of the well-characterized Mudd-Summers VSV genome, as a cDNA encoding the Glasgow VSV complete genome, which is the parent strain of ts(G)114, is not available. However, as noted above there are only seven amino acid differences between the L proteins of the Glasgow and Mudd-Summers VSV-Indiana strains, all of which are conservative substitutions (Hunt and Hutchinson, 1993).
We recovered infectious viruses having the individual or paired L protein mutations identified by sequence analysis of the ts(G)114 L gene by transfection of engineered viral cDNAs into a T7-expressing cell line, AlphaV-T7, which was described previously and is briefly discussed in Material and Methods (Galloway et al., 2008b). AlphaV-T7-transfected cells were maintained at 31°C for 24-80 hr, supernatants were harvested and passed onto BHK cells. Supernatants from BHK cells that showed cytopathology were harvested 24-98 hpi. Viruses were recovered that contained each mutation individually or together. Recombinant viruses containing the DG mutation took significantly longer to propagate following transfection of cDNA than viruses carrying the other mutations. To compare, following passage of supernatants harvested from primary transfected cells onto BHK cells, DG-containing viruses took an average of 98 hr for extensive CPE compared to an average of 38 hr for all other recombinant viruses.
Recombinant viruses were examined first for their ability to plaque on Vero-76 cells at 31°C and 39°C. Plaque assays were incubated for 45 hr and the plaque size of each virus at both temperatures was determined and is shown in table 2. The original stock of ts(G)114 was unable to plaque at 39°C, but was able to form plaques at 31°C, although plaques were much smaller (Table 2). All of the recombinant viruses containing the ts(G)114 L protein mutations were able to form plaques, although some with greatly reduced plaque sizes. Recombinant viruses DG, EG, EG/IT, and DG/EG/IT formed plaques at 31°C and 39°C; however, the sizes of the plaques at 39°C and those yielded by the DG recombinant virus at 31°C were smaller than 1mm in diameter (Table 2). Of the three mutations in ts(G)114, the IT mutation did not appear to cause temperature sensitivity of the L protein, as the plaques formed at 31°C and 39°C were similar in size (Table 2).
Table 2.
Summary of growth characteristics
| Virus | Plaque diameter (mm) | 24 hr titer Log10 (pfu/ml) | Percent inhibition at 39°Cb | ||
|---|---|---|---|---|---|
| 31°C | 39°C | 31°C | 39°C | ||
| ts(G)114 | 1.5 ± 0.2 | NDa | 8.8 | 2.0 | >99.9% |
| rVSV | 2.0 ± 0.1 | 2.2 ± 0.2 | 8.5 | 8.0 | 66.7% |
| D575G | < 1mm | < 1mm | 7.3 | 5.0 | 99.5% |
| E1117G | 1.5 ± 0.3 | < 1mm | 8.3 | 8.2 | 27.3% |
| I1937T | 1.8 ± 0.2 | 1.9 ± 0.2 | 8.3 | 8.0 | 50.0% |
| EG/IT | 1.6 ± 0.3 | < 1mm | 8.5 | 8.6 | NIc |
| DG/EG/IT | 1.1 ± 0.2 | < 1mm | 8.6 | 4.0 | >99.9% |
ND = None detected
Percent inhibition was determined by expressing the virus titer at 39°C as a percentage of the virus titer at 31°C, which was then subtracted from 100% using the equation 100%-[(virus titer at 39°C/virus titer at 31°C)×100%].
NI = No Inhibition
To determine whether the reduced plaque size displayed by some of the recombinant viruses was indicative of replication restriction, we determined viral titers at 24 hr in a single-step growth experiment for each recombinant virus at 31°C and 39°C (Table 2). BHK cells were infected at a MOI of 3 and incubated at 31°C or 39°C for 24 hr. Supernatants were harvested and titrated on Vero-76 cells at 31°C. The results in table 2 show that virus replication for the original ts(G)114 virus was similar to WT rVSV at 31°C. However, at 39°C replication of ts(G)114 was reduced by six logs, representing a greater than 99.9% reduction, confirming the temperature sensitivity of ts(G)114. The recombinant viruses containing the EG or IT L protein mutations individually or together, replicated to titers very similar to WT rVSV at 31°C and 39°C, indicating that neither of these two mutations was responsible for the temperature sensitivity associated with ts(G)114. Recombinant virus containing the DG mutation alone replicated 10-fold less well at 31°C compared to WT and was reduced further by 2.3 logs at 39°C compared to 31°C. DG in combination with EG and IT, the triple mutant DG/EG/IT, showed similar virus replication to WT rVSV at 31°C, however, the DG/EG/IT recombinant virus showed growth restriction at 39°C of 4.6 logs, which is a >99.9% reduction in viral titer compared to WT rVSV (Table 2). These data indicated that the DG mutation was a major cause of temperature sensitivity associated with ts(G)114.
Analysis of RNA synthesis by ts(G)114 recombinant viruses
To determine whether the identified mutation(s) were responsible for the ts(G)114 conditional RNA phenotype, RNA synthesis profiles were examined under the various temperature conditions as described for figure 1. In comparison to rVSV, mRNA and genome-length RNA synthesis in DG-infected cells were significantly reduced at 31°C and undetectable at 39°C, or after shift from 31°C to 39°C (Figure 3A, lanes 2, 8 (4X exposure), 10, 18, and 25 and Figure 3B). The viruses carrying the EG and IT mutations individually, and the virus containing both mutations, the EG/IT virus, showed significantly greater levels of transcription and replication at 31°C, after shift from 31°C to 39°C, and at 39°C compared to WT rVSV (Figure 3A, lanes 3-5, 11-13, 19-21, 26-28 and Figure 3B). Notably, there were two additional RNA species synthesized in EG/IT-infected cells that were not synthesized in cells infected with rVSV: one migrated slightly slower than the L mRNA and the other migrated slightly slower than the G mRNA. We determined that these RNAs were polycistronic mRNAs representing the G-L and N-P mRNAs, respectively (data not shown). However, when the DG mutation was examined with the EG and IT L protein mutations at 31°C and under the temperature shift-up condition, viral transcription was preferentially inhibited over replication, recapitulating the ts(G)114 phenotype (Figure 3A, lanes 6, 14, and 16 (4X exposure) and Figure 3B). When viral RNA synthesis was examined for the DG/EG/IT recombinant virus at 39°C or under the temperature shift-down condition, no RNA was detected (Figure 3A, lanes 22 and 29 and Figure 3B). The data show the DG mutation is the mutation responsible for the temperature sensitive phenotype of ts(G)114, but that it is DG in combination with EG and IT that recapitulate the ts(G)114 phenotype, in that together they allow viral genome replication at the non-permissive temperature, whereas transcription is still severely inhibited. Notably, however, the ts(G)114 phenotype is not fully reconstituted with the DG/EG/IT rVSV, in that the selective inhibition of transcription over genome replication is observed at 31°C, as well as under temperature shift-up conditions with the recombinant virus, but only under temperature shift-up conditions with ts(G)114. We favor the explanation that this disparity is likely due to the seven amino acid differences in the L proteins of the Glasgow and Mudd-Summers strains (Hunt and Hutchinson, 1993).
Figure 3. Analysis of viral RNA synthesis for recombinant ts(G)114 VSVs.

(A) BHK cells were infected at a MOI of 3 at 31°C or 39°C. At 4 hpi, infected cells were pre-treated with actinomycin-D and either maintained at 31°C, shifted from 31°C to 39°C, shifted from 39°C to 31°C, or maintained at 39°C for 30 min, at which time RNAs were labeled metabolically with [3H]-uridine. Radiolabeled RNAs were visualized by acid agarose-urea gel electrophoresis and fluorography. A representative set of gels is shown; all panels shown are from the same experiment. The virus infection from whence the RNAs were synthesized is noted above the gel, as are the temperature conditions. The identities of viral RNAs are denoted to the left of the gel. Lanes 8 and 16 represent 4 times longer exposures of film of RNAs from D575G-infected cells at 31°C and DG/EG/IT-infected cells under temperature shift-up conditions, respectively. Notably, two additional RNAs were synthesized in EG/IT-infected cells that were not synthesized in WT rVSV infected-cells, and which migrated slightly slower than the G or L mRNA. The additional RNA that migrates slightly slower than the G mRNA represents a polycistronic mRNA corresponding to the N and P genes, and the RNA that migrates slightly slower than the L mRNA represents a polycistronic mRNA corresponding to the G and L genes, both of which arose due to selection of variations in the N or G gene end sequence during passage of the EG/IT recombinant virus. (B) Quantitative analysis of three independent experiments is shown in the graphs. The graph on the left represents total viral RNA transcription expressed as a percentage of WT at the same temperature. The graph on the right represents total viral replication expressed as a percentage of WT at the same temperature. (*) indicates no RNA was detected.
Discussion
The NNS L proteins are large multifunctional proteins that provide a significant portion of the enzymatic activity required for viral genome replication and gene expression. These activities include transcription, replication, polyribonucleotidyltransferase (PRNTase), methyltransferase, and poly(A) polymerase activities (Li et al., 2005; Li et al., 2008; Li et al., 2006; Ogino and Banerjee, 2007; Rhodes et al., 1974; Testa and Banerjee, 1977). Extensive sequence and phylogenetic analyses have subdivided the NNS L proteins into six domains of conservation linked by variable regions (Poch et al., 1990; Poch et al., 1989; Svenda et al., 1997). There have been numerous investigations aimed at addressing the significance of the various domains using both forward and reverse genetic approaches. We used a forward genetic approach to identify L protein residues that were responsible for the conditional RNA synthesis phenotype of ts(G)114, which results in abundant genomic replication and extremely low levels of transcription. The mutations that we identified in the L protein were amino acids D575 to G, E1117 to G, and I1937 to T.
The D575 residue is located in domain II of the VSV L protein, E1117 is in domain V, and I1937 is in a variable region beyond domain VI. Domain II has been hypothesized to function in template recognition due to the abundance of positively charged amino acids (Poch et al., 1990). Domain V contains a concentration of invariant cysteine and histidine residues (Poch et al., 1990), the significance of which is not known, and recently, a motif was identified in domain V that is important for the PRNTase activity associated with capping of VSV mRNAs (Li et al., 2008).
Mutations within domain II of the Sendai virus L protein were previously shown to significantly diminish viral RNA synthesis (Smallwood et al., 1999). The D575, or its homologous residue in other NNS L proteins, has not been examined previously. It is unclear how the D575G mutation might cause the observed defect in transcription, but given the polarity of aspartic acid, it is likely positioned on the surface of the L protein and might be involved in an intra-molecular interaction with a separate domain of the L protein or with another trans-acting factor, viral or cellular, that is necessary for transcription, such as the VSV P protein or an as yet unidentified protein. Considering the RNA synthetic activity of the L protein containing the D575G mutation alone, versus in combination with the E1117G and I1937T mutations, the D575G mutation alone affected both mRNA transcription and RNA replication, but when in combination with the other two mutations, the ability to synthesize genome-length RNA was restored. The reason for this is unknown. However, in considering the causes of temperature sensitivity, it possible that the D575G mutation results in an improperly folded L protein that is defective for transcription and replication. The presence of the E1117G and I1937T mutations might provide enough structural flexibility to the L protein, either directly or by virtue of the L proteins ability to interact with another trans-acting factor, to support genome-length RNA synthesis. However, due to the paucity of structural data for the NNS L proteins, more extensive investigation is required to examine this.
Mutations within domain V were shown previously to disparately affect transcription and replication, and, as noted above have been shown to affect capping of VSV mRNAs (Cortese et al., 2000; Li et al., 2008). The homologous residue in the Sendai virus L protein (S1174), based on a published sequence alignment (Svenda et al., 1997), was previously mutated as a set of three residues and these combined mutations resulted in decreased viral RNA replication (Cortese et al., 2000). However, the E1117G mutation identified in ts(G)114 did not have a significant effect on transcription or replication by itself (Figure 3). Further, given the lack of similarity between serine and glutamic acid, as well as the general lack of conservation of the E1117 residue among NNS L proteins, it is not surprising that mutation of this residue in the Sendai virus and VSV L proteins did not yield similar results. Notably, because mutation of S1174 in the Sendai virus L protein was in the context of mutations to two adjacent amino acids (E1172 and G1173) it is impossible based on the available data to determine which of these residues, or if all three were responsible for the negative affect on replication.
As mentioned above, domain V contains a conserved motif among NNS L proteins that is necessary for the PRNTase activity of the VSV L protein, which is the activity utilized to add the G cap to nascent mRNAs (Li et al., 2008; Ogino and Banerjee, 2007). Though the E1117 residue was not examined in the aforementioned study, the nearest residue examined was C1120. Mutation of the C1120 residue in the L protein decreased mRNA synthesis, but retained near WT levels of capping activity (Li et al., 2008). However, mutation of other residues in this region greatly affected the transcriptional processivity and PRNTase activity of the VSV L protein (Li et al., 2008). It will be interesting to see if the E1117G mutation affects the capping activity of the L protein, though we suggest that it does not, as most of the mutations examined in domain V that affected capping also decreased transcriptional processivity, which the E1117G mutation does not (Li et al., 2008) (Figure 3A).
In regard to the EG/IT recombinant virus, we consistently observed two additional RNA species that were not synthesized in rVSV-infected cells. We determined that these two RNAs were polycistronic mRNAs, the larger and smaller representing the G-L and N-P mRNAs, respectively. We sequenced the N-P and G-L gene junctions from the EG/IT virus and found that the gene end U tract had been extended from U7 to U8 at both gene junctions. There was also a minor population of U6 tracts at the N-P gene junction. We do not know why this occurred, but we, and others, have noted this before, particularly in mutants that up-regulate RNA synthesis (Galloway et al., 2008a; Li et al., 2007). We know that both the ratio of N to P proteins and the level of L protein expression are particularly important for the VSV life cycle. It is possible that, in the presence of a polymerase that significantly up-regulates RNA synthesis, such as the EG/IT L protein, it is more efficient for the virus to select for changes in the gene end U tract that affect termination, which would attenuate the expression of downstream genes, rather than to select for changes that attenuate polymerase activity. It has been shown that a recombinant rabies virus that has been engineered to up-regulate the synthesis of the L mRNA, not only synthesized more L protein, but grew to higher titers, had faster growth kinetics, and showed greater cytopathic effects (Finke et al., 2000). Whether viruses with higher L gene expression are more fit than WT has yet to be determined. Evolutionarily speaking, the Mononegavirales have down-regulated the expression of the L gene, placing it at the most 3’ distal location within the genome, which would suggest that a virus that up-regulated the expression of the L protein would be less fit than WT, however, relative levels of gene expression may be more important than overall levels of viral gene expression (Ball et al., 1999; Novella et al., 2004; Wertz et al., 2002). Extension of the U tract from 7 to 8 UMP nucleotides would lead to only modest increases in polycistronic mRNA synthesis, based on analysis of the U8 mutation in a sub-genomic replicon system (Hinzman et al., 2002), but this may be all that is required to achieve optimal levels of gene expression. Serial passage of viruses, like the EG/IT virus, that have polymerase mutations that up-regulate RNA synthesis might yield interesting information about the importance of the level of L gene expression.
The I1937T mutation had no debilitating effect on RNA synthesis at 31°C or 39°C, which indicated that it was not involved directly in transcription or replication (Figure 3). This residue has not been examined previously. We performed a multiple sequence alignment of this region of several related NNS L proteins and found that, although the identity of this amino acid is not conserved, other NNS L proteins have similar amino acids at this position. Similar residues found at this position include valine, alanine, leucine, and methionine.
Considering the available data regarding the regulation of transcription versus replication, particularly the finding that transcription and replication initiate at separate sites on the genome (Whelan and Wertz, 2002), it is possible that the three L protein mutations identified in this study affect the ability of the polymerase to recognize and initiate at the first gene start. It is also possible that the DG/EG/IT polymerase complex is fully capable of initiating transcription at the first gene start, but is incapable of properly recognizing the cis-acting sequences known to regulate transcription that are located at each gene junction. However, if this were the case, one would expect to observe a distinct RNA species similar in length to the genome/anti-genome RNA, but lacking the leader sequence, assuming that in all cases the polymerase initiates transcription at the first gene start. No such RNA was observed in this study, although we did not specifically test this hypothesis. Alternatively, these mutations might affect the stability of the putative “transcription complex” by interfering with the L protein’s ability to interact with other necessary trans-acting factors, such as the P protein, whose phosphorylation state was shown previously to influence transcription and replication (Hwang et al., 1999; Pattnaik et al., 1997). Notably, the original ts(G)114 phenotype is only recapitulated when the D575G mutation is in combination with the E1117G and I1937T mutations, which we suggest might be due to reasons previously discussed regarding the causes of temperature sensitivity. While the DG/EG/IT mutant largely reconstituted the ts(G)114 phenotype of decreased transcription and unaffected replication, it did not fully resemble ts(G)114, in that the mutations also decreased transcription slightly at the permissive temperature. We suggest that this is likely due to the seven amino acid differences between the L proteins of Glasgow, the parent strain of ts(G)114, and Mudd-Summers, the background of the recombinant viruses. Though the precise mechanism of action of the DG/EG/IT L protein is not understood, we suggest that it may be a useful tool to further investigate the regulation of transcription versus replication.
In summary, we have confirmed the conditional RNA phenotype of ts(G)114 by showing that at the permissive temperature, RNA synthesis is very similar to WT VSV, but when infections were initiated at 31°C and shifted to 39°C 4 hpi, transcription was significantly reduced, whereas replication was unaffected (Figures 1 and 3). We have further shown that the L protein residues that are altered in the ts(G)114 L protein are D575G, E1117G, and I1937T (Table 1). Importantly, the D575G rVSV has a defective RNA synthesis phenotype, but we have shown that the ts(G)114 conditional RNA phenotype is recapitulated only when the D575G mutation is present in combination with E1117G and I1937T. This study has identified, using a forward genetic approach, specific residues that affect VSV transcription, but not replication.
Material and Methods
Cells and viruses
BHK-21 cells were used for transfections, growth of virus, and metabolic labeling of RNA and protein synthesis. Vero-76 cells were used for titration of virus stocks. For recovery of virus from cDNA clones, a novel BHK-21 cell line, AlphaV-T7, was used for primary transfections, which constitutively expresses T7 RNA polymerase from a self-replicating alphavirus replicon, as previously described (Galloway et al., 2008b).
The stock of ts(G)114 used to generate the stock used in these experiments came originally from Craig Pringle. The stock of ts(G)114 used in these experiments was obtained by an additional passage of the original stock on BHK cells. Cells were infected at a MOI of 0.001 and incubated at 31°C for 36 hr, at which time viral supernatants were harvested, clarified of cell debris, and titrated on Vero-76 cells. The WT VSV stock used for comparison to ts(G)114 was L1VSV, a large plaque isolate of the Mudd-Summers strain of VSV-Indiana (Wertz and Youngner, 1970).
Sequence analysis of the ts(G)114 L gene
One-100mm dish of BHK cells was infected at an MOI of 0.001 with the original stock of ts(G)114. The supernatant was harvested and clarified of cell debris by low-speed centrifugation at 3,000 × g for 10 minutes. Virions from the clarified supernatant were pelleted by ultracentrifugation at 38,000 × g for 90 minutes at 4°C using a Beckman Type 30 rotor. The viral pellet was resuspended with 400μl NTE plus 0.5% SDS. The pellet was incubated on ice for 30 minutes and the virion RNA was then phenol extracted and ethanol precipitated. Purified virion RNA was reverse transcribed using Superscript III (Invitrogen) according to the manufacturer’s recommendation. To the reverse transcription reaction, 2 units of E.coli RNase H were added to remove the complementary RNA. The resultant cDNA was used as a template to generate six overlapping PCR products. The fragments generated corresponded to VSV nts 4723-6044, 5892-7277, 7195-8965, 7941-9200, 8755-10074, and 9911-11081. The PCR products were purified and bulk products were sequenced. ts(G)114 virus was an induced mutant of the VSV-Glasgow strain. We compared the ts(G)114 L gene sequence to the VSV-Mudd-Summers strain taking into account that there are 23 published differences in the L gene between Mudd-Summers and Glasgow, seven of which result in coding changes (Hunt and Hutchinson, 1993).
Plasmid construction and mutagenesis
To introduce L gene mutations, E1117G and I1937T, into a VSV full-length cDNA, we utilized a sub-clone that consisted of 4147 bp of VSV coding sequence (nt 7070-11217). This sub-clone was described previously (Galloway et al., 2008b). To introduce the D575G L gene mutation into the VSV full-length cDNA, we generated a sub-clone that consisted of 4731 bp of VSV coding sequence (nt 3367-8097). The 4731 bp region was PCR amplified from the cDNA encoding the VSV full-length genome (pVSV1(+)) (Whelan et al., 1995) plasmid and cloned into pCR-BLUNT (Invitrogen). The two VSV sub-clones were used as templates for site-directed mutagenesis using the QuikChange methodology (Stratagene). The presence of the desired mutation(s) was verified by sequence analysis of an approximately 3.5 kb fragment that spanned the Avr II and Ava I restriction sites in the case of the 4731 bp sub-clone (D575-containing sub-clone), and Ava I and Afl II in the case of the 4147 bp sub-clone (E1117 and I1937-containing sub-clone). The sub-clone fragment was introduced into the pVSV1(+) backbone using standard cloning techniques. The presence of the desired mutation(s) was further verified via sequence analysis of the full-length cDNA plasmid.
For recovery of recombinant VSV using the AlphaV-T7 cells, we used plasmids that expressed the VSV N, P, and L genes under the control of the T7 RNA polymerase promoter and an internal ribosomal entry site from encephalomyocarditis virus (ECMV) to allow for cap-independent translation of the VSV messages. The IRES containing plasmids have been described previously (Galloway et al., 2008b).
Recovery of recombinant VSV
Recombinant VSV was recovered from cDNA by transfection of AlphaV-T7 cells, as described previously (Galloway et al., 2008b). Five hours post transfection, the transfection medium was replaced with DMEM supplemented with 2% newborn calf serum (NCS) and incubated at 31°C. Transfected cells supernatants were harvested between 24-80 hr and one-quarter of the harvested supernatant was passaged onto BHK-21 cells. Infected cells were incubated at 31°C. Supernatants from infected BHK-21 cells showing cytopathology were harvested between 24-98 hr post infection and clarified to remove cell debris at 3,000 × g for 10 minutes. Viral titers were determined via plaque assay on Vero-76 cells.
Analysis of viral RNA synthesis in cells
For viral RNA analysis, BHK-21 cells were infected at the indicated MOI in DMEM + 2% NCS at 31°C or 39°C. After a 1 hr adsorption period, 1.5 ml of DMEM + 2% NCS that had been pre-warmed to 31°C or 39°C was added to cells and incubated at the noted temperature. At the indicated times post-infection, infected cells were treated with 10μg/ml Act-D and either maintained at 31°C, shifted from 31°C to 39°C, shifted from 39°C to 31°C, or maintained at 39°C for 30 minutes. At 4.5 hpi, RNAs were labeled metabolically with 33μCi/ml [3H]-uridine in the presence of an additional 10μg/ml actinomycin-D for 1 hr at the same temperature as shifted to during the pre-treatment period. RNAs were harvested in lysis buffer, then phenol chloroform extracted and ethanol precipitated. Radiolabeled RNAs were visualized by electrophoresis through 1.5% acid-agarose-urea gels and fluorography.
Analysis of viral protein synthesis in cells
For viral protein analysis, BHK-21 cells were infected at the indicated MOI in DMEM + 2% NCS at 31°C or 39°C. After a 1 hr adsorption period, 1.5 ml of DMEM + 2% NCS that had been pre-warmed to 31°C or 39°C was added to cells and incubated at the noted temperature. At 4 hpi, infected cells were washed twice with HBSS and starved for methionine by the addition of DMEM lacking methionine. At the onset of the starvation period, infected cells were either maintained at 31°C, shifted from 31°C to 39°C, shifted from 39°C to 31°C, or maintained at 39°C for 30 minutes. At 4.5 hpi, proteins were labeled metabolically with 35μCi [35S]-methionine for 1 hr at the same temperature as shifted to during the starvation period. Cell extracts were prepared by the addition of lysis buffer. The nuclei were pelleted and radiolabeled proteins from cytoplasmic extracts were resolved on 10% SDS-PAGE (77 acrylamide:1 bis-acrylamide). Gels were fixed in 50% methanol/10 % acetic acid, dried, and exposed to film.
The polyacrylamide gel migration of the M protein in ts(G)114 samples is slightly slower than that observed in WT VSV samples, which is likely due to the size difference between the M proteins of Mudd-Summers and Glasgow VSV strains: Mudd-Summers is 229 amino acids (accession #AAA48369) and Glasgow is 237 amino acids (accession #P04876). Mudd-Summers is the background of the WT VSV used in this study and Glasgow is the parent strain of ts(G)114 (Pringle, 1970; Wertz and Youngner, 1970).
Quantitation of RNA and protein
RNA and protein expression levels were quantified by densitometric analysis of fluorographs using the Quantity One software (Biorad) and a PDI model 320i densitometer. Total protein synthesis was determined by normalizing the density of each viral protein to its methionine content, adding together the molar amounts of viral proteins for each virus, and expressing the total as a percentage of WT under the same temperature conditions. Total RNA transcription or replication was determined using the same method described for protein synthesis, except that viral RNAs were normalized to their respective uridine content. All quantities are an average of three independent experiments.
Acknowledgments
We thank Andy Ball, John Barr, and members of the Wertz laboratory for helpful discussions regarding this work and Djamila Harouaka for technical assistance. We also thank Ilya Frolov for the continued use of the AlphaV-T7 cells. This research was supported by NIH grants R37AI12464 and R01AI12464 to G.W.W. and the Freedom to Discover Award from Bristol Myers Squibb to G.W.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham G, Banerjee AK. Sequential transcription of the genes of vesicular stomatitis virus. Proc Natl Acad Sci U S A. 1976;73(5):1504–1508. doi: 10.1073/pnas.73.5.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball LA. Transcriptional mapping of vesicular stomatitis virus in vivo. J Virol. 1977;21(1):411–414. doi: 10.1128/jvi.21.1.411-414.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball LA, Pringle CR, Flanagan B, Perepelitsa VP, Wertz GW. Phenotypic consequences of rearranging the P, M, and G genes of vesicular stomatitis virus. J Virol. 1999;73(6):4705–4712. doi: 10.1128/jvi.73.6.4705-4712.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball LA, White CN. Order of transcription of genes of vesicular stomatitis virus. Proc Natl Acad Sci U S A. 1976;73(2):442–446. doi: 10.1073/pnas.73.2.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr JN, Wertz GW. Polymerase slippage at vesicular stomatitis virus gene junctions to generate poly(A) is regulated by the upstream 3’-AUAC-5’ tetranucleotide: implications for the mechanism of transcription termination. J Virol. 2001;75(15):6901–6913. doi: 10.1128/JVI.75.15.6901-6913.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr JN, Whelan SP, Wertz GW. cis-Acting signals involved in termination of vesicular stomatitis virus mRNA synthesis include the conserved AUAC and the U7 signal for polyadenylation. J Virol. 1997a;71(11):8718–8725. doi: 10.1128/jvi.71.11.8718-8725.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr JN, Whelan SP, Wertz GW. Role of the intergenic dinucleotide in vesicular stomatitis virus RNA transcription. J Virol. 1997b;71(3):1794–1801. doi: 10.1128/jvi.71.3.1794-1801.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese CK, Feller JA, Moyer SA. Mutations in domain V of the Sendai virus L polymerase protein uncouple transcription and replication and differentially affect replication in vitro and in vivo. Virology. 2000;277(2):387–396. doi: 10.1006/viro.2000.0615. [DOI] [PubMed] [Google Scholar]
- Davis NL, Arnheiter H, Wertz GW. Vesicular stomatitis virus N and NS proteins form multiple complexes. J Virol. 1986;59(3):751–754. doi: 10.1128/jvi.59.3.751-754.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finke S, Cox JH, Conzelmann KK. Differential transcription attenuation of rabies virus genes by intergenic regions: generation of recombinant viruses overexpressing the polymerase gene. J Virol. 2000;74(16):7261–9. doi: 10.1128/jvi.74.16.7261-7269.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway SE, Richardson PE, Wertz GW. Analysis of a structural homology model of the 2’-O-ribose methyltransferase domain within the vesicular stomatitis virus L protein. Virology. 2008a;382(1):69–82. doi: 10.1016/j.virol.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway SE, Richardson PE, Wertz GW. Analysis of a structural homology model of the 2’-O-ribose methyltransferase domain within the vesicular stomatitis virus L protein. Virology. 2008b doi: 10.1016/j.virol.2008.08.041. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green TJ, Macpherson S, Qiu S, Lebowitz J, Wertz GW, Luo M. Study of the assembly of vesicular stomatitis virus N protein: role of the P protein. J Virol. 2000;74(20):9515–24. doi: 10.1128/jvi.74.20.9515-9524.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinzman EE, Barr JN, Wertz GW. Identification of an upstream sequence element required for vesicular stomatitis virus mRNA transcription. J Virol. 2002;76(15):7632–41. doi: 10.1128/JVI.76.15.7632-7641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard M, Wertz G. Vesicular stomatitis virus RNA replication: a role for the NS protein. J Gen Virol. 1989;70(Pt 10):2683–94. doi: 10.1099/0022-1317-70-10-2683. [DOI] [PubMed] [Google Scholar]
- Hunt DM, Hutchinson KL. Amino acid changes in the L polymerase protein of vesicular stomatitis virus which confer aberrant polyadenylation and temperature-sensitive phenotypes. Virology. 1993;193(2):786–93. doi: 10.1006/viro.1993.1187. [DOI] [PubMed] [Google Scholar]
- Hwang LN, Englund N, Das T, Banerjee AK, Pattnaik AK. Optimal replication activity of vesicular stomatitis virus RNA polymerase requires phosphorylation of a residue(s) at carboxy-terminal domain II of its accessory subunit, phosphoprotein P. J Virol. 1999;73(7):5613–20. doi: 10.1128/jvi.73.7.5613-5620.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson LE, Rose JK. Localized attenuation and discontinuous synthesis during vesicular stomatitis virus transcription. Cell. 1981;23(2):477–84. doi: 10.1016/0092-8674(81)90143-4. [DOI] [PubMed] [Google Scholar]
- Li J, Chorba JS, Whelan SP. Vesicular stomatitis viruses resistant to the methylase inhibitor sinefungin upregulate RNA synthesis and reveal mutations that affect mRNA cap methylation. J Virol. 2007;81(8):4104–15. doi: 10.1128/JVI.02681-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Fontaine-Rodriguez EC, Whelan SP. Amino acid residues within conserved domain VI of the vesicular stomatitis virus large polymerase protein essential for mRNA cap methyltransferase activity. J Virol. 2005;79(21):13373–84. doi: 10.1128/JVI.79.21.13373-13384.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Rahmeh A, Morelli M, Whelan SP. A conserved motif in region v of the large polymerase proteins of nonsegmented negative-sense RNA viruses that is essential for mRNA capping. J Virol. 2008;82(2):775–84. doi: 10.1128/JVI.02107-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang JT, Whelan SP. A unique strategy for mRNA cap methylation used by vesicular stomatitis virus. Proc Natl Acad Sci U S A. 2006;103(22):8493–8. doi: 10.1073/pnas.0509821103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novella IS, Ball LA, Wertz GW. Fitness analyses of vesicular stomatitis strains with rearranged genomes reveal replicative disadvantages. J Virol. 2004;78(18):9837–41. doi: 10.1128/JVI.78.18.9837-9841.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino T, Banerjee AK. Unconventional mechanism of mRNA capping by the RNA-dependent RNA polymerase of vesicular stomatitis virus. Mol Cell. 2007;25(1):85–97. doi: 10.1016/j.molcel.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Pattnaik AK, Hwang L, Li T, Englund N, Mathur M, Das T, Banerjee AK. Phosphorylation within the amino-terminal acidic domain I of the phosphoprotein of vesicular stomatitis virus is required for transcription but not for replication. J Virol. 1997;71(11):8167–75. doi: 10.1128/jvi.71.11.8167-8175.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton JT, Davis NL, Wertz GW. N protein alone satisfies the requirement for protein synthesis during RNA replication of vesicular stomatitis virus. J Virol. 1984;49(2):303–9. doi: 10.1128/jvi.49.2.303-309.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman SM, Huang AS. RNA Synthesis of Vesicular Stomatitis Virus V. Interactions Between Transcription and Replication. J Virol. 1973;12(6):1395–1400. doi: 10.1128/jvi.12.6.1395-1400.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poch O, Blumberg BM, Bougueleret L, Tordo N. Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses: theoretical assignment of functional domains. J Gen Virol. 1990;71(Pt 5):1153–62. doi: 10.1099/0022-1317-71-5-1153. [DOI] [PubMed] [Google Scholar]
- Poch O, Sauvaget I, Delarue M, Tordo N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. Embo J. 1989;8(12):3867–74. doi: 10.1002/j.1460-2075.1989.tb08565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle CR. Genetic characteristics of conditional lethal mutants of vesicular stomatitis virus induced by 5-fluorouracil, 5-azacytidine, and ethyl methane sulfonate. J Virol. 1970;5(5):559–67. doi: 10.1128/jvi.5.5.559-567.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qanungo KR, Shaji D, Mathur M, Banerjee AK. Two RNA polymerase complexes from vesicular stomatitis virus-infected cells that carry out transcription and replication of genome RNA. Proc Natl Acad Sci U S A. 2004;101(16):5952–7. doi: 10.1073/pnas.0401449101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DP, Moyer SA, Banerjee AK. In vitro synthesis of methylated messenger RNA by the virion-associated RNA polymerase of vesicular stomatitis virus. Cell. 1974;3:327–333. doi: 10.1016/0092-8674(74)90046-4. [DOI] [PubMed] [Google Scholar]
- Smallwood S, Easson CD, Feller JA, Horikami SM, Moyer SA. Mutations in conserved domain II of the large (L) subunit of the Sendai virus RNA polymerase abolish RNA synthesis. Virology. 1999;262(2):375–83. doi: 10.1006/viro.1999.9933. [DOI] [PubMed] [Google Scholar]
- Svenda M, Berg M, Moreno-Lopez J, Linne T. Analysis of the large (L) protein gene of the porcine rubulavirus LPMV: identification of possible functional domains. Virus Res. 1997;48(1):57–70. doi: 10.1016/s0168-1702(96)01426-8. [DOI] [PubMed] [Google Scholar]
- Testa D, Banerjee AK. Two methyltransferase activities in the purified virions of vesicular stomatitis virus. J Virol. 1977;24(3):786–93. doi: 10.1128/jvi.24.3.786-793.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal LP, Breindl M, Holland JJ. Determination of molar ratios of vesicular stomatitis virus induced RNA species in BHK21 cells. Biochemistry. 1976;15(8):1663–7. doi: 10.1021/bi00653a012. [DOI] [PubMed] [Google Scholar]
- Wang JT, McElvain LE, Whelan SP. Vesicular stomatitis virus mRNA capping machinery requires specific cis-acting signals in the RNA. J Virol. 2007;81(20):11499–506. doi: 10.1128/JVI.01057-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz GM, Howard MB, Davis N, Patton J. The switch from transcription to replication of a negative-strand RNA virus. Cold Spring Harb Symp Quant Biol. 1987;52:367–71. doi: 10.1101/sqb.1987.052.01.042. [DOI] [PubMed] [Google Scholar]
- Wertz GW. Isolation of possible replicative intermediate structures from vesicular stomatitis virus-infected cells. Virology. 1978;85(1):271–85. doi: 10.1016/0042-6822(78)90431-2. [DOI] [PubMed] [Google Scholar]
- Wertz GW, Moudy R, Ball LA. Adding genes to the RNA genome of vesicular stomatitis virus: positional effects on stability of expression. J Virol. 2002;76(15):7642–50. doi: 10.1128/JVI.76.15.7642-7650.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz GW, Youngner JS. Interferon production and inhibition of host synthesis in cells infected with vesicular stomatitis virus. J Virol. 1970;6(4):476–84. doi: 10.1128/jvi.6.4.476-484.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan SP, Ball LA, Barr JN, Wertz GT. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc Natl Acad Sci U S A. 1995;92(18):8388–92. doi: 10.1073/pnas.92.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan SP, Wertz GW. Transcription and replication initiate at separate sites on the vesicular stomatitis virus genome. Proc Natl Acad Sci U S A. 2002;99(14):9178–83. doi: 10.1073/pnas.152155599. [DOI] [PMC free article] [PubMed] [Google Scholar]