

Abstract
Numerous pathological states including cancer, autoimmune diseases, and viral/bacterial infections are often attributed to uncontrollable DNA replication. Inhibiting this essential biological process provides an obvious therapeutic target against these diseases. A logical target is the DNA polymerase, the enzyme responsible for catalyzing the addition of mononucleotides into a growing polymer using a DNA or RNA template as a guide for directing each incorporation event. This review provides a summary of therapeutic agents that target polymerase activity. A discussion of the biological function and mechanism of polymerases is first provided to illustrate the strategy for therapeutic intervention as well as the rational design of various nucleoside analogs that inhibit various polymerases associated with viral infections and cancer. The development of nucleoside and non-nucleoside inhibitors as anti-viral agents is discussed with particular emphasis on their mechanism of action, structure-activity relationships, toxicity, and mechanism of resistance. In addition, commonly used anti-cancer agents are described to illustrate the similarities and differences associated with various nucleoside analogs as therapeutic agents. Finally, new therapeutic approaches are discussed that include the inhibition of selective polymerases involved in DNA repair and/or translesion DNA synthesis as anti-cancer agents.
Keywords: DNA replication, polymerases, viral replication, anti-cancer agents, mutagenesis, fidelity
DNA Replication as a Therapeutic Target
DNA replication is the process of duplicating DNA to generate two copies of an organism's genetic information (Figure 1A). This complex biological process is catalyzed by DNA polymerases that add mononucleotides into a growing primer using nucleic acid templates as a guide for directing each incorporation event (Figure 1A). DNA replication is absolutely essential for the proliferation and survival of all forms of life ranging from simple viruses and bacteria to more complex organisms including humans. The importance of DNA replication is often highlighted by the variety of pathological states that result from dysfunctional and/or unregulated activity. For example, certain genetic diseases such as ataxia telangiectasia (1) are associated with the generation of mutations caused by decreased fidelity during replication. Hyperproliferative diseases such as cancer, autoimmune conditions, and viral/bacterial infections are associated with uncontrollable and, in some cases, pro-mutagenic DNA replication. As such, a major goal in biomedical science is to develop therapeutic agents to combat these pathological states.
Figure 1.
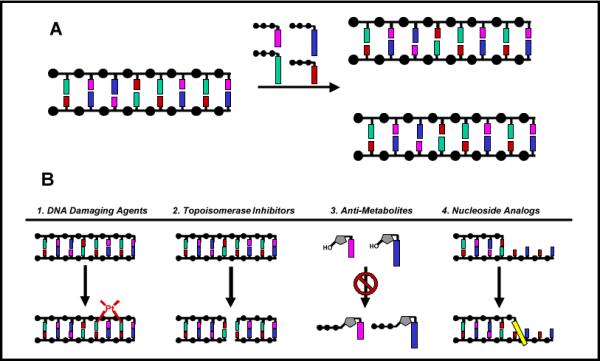
(A) DNA replication is catalyzed by a DNA polymerase. During this process, the polymerase incorporates 2'-deoxynucleoside triphosphates opposite a DNA template which guides each incorporation event. (B) Chemotherapeutic strategies used to inhibit DNA replication include 1.) DNA damaging agents, 2.) inhibition of DNA modifying enzymes, 3.) inhibition of enzymes involved in nucleotide metabolism, and 4.) nucleoside analogs that directly inhibit DNA polymerase activity.
As outlined in Figure 1B, there are several strategies currently used to inhibit DNA replication. One approach is the use of DNA damaging agents (Step 1) that modify nucleic acid so that it no longer acts as an effective substrate for the DNA polymerase. Examples include chemotherapeutic agents such as temozolomide and cisplatin that modify the composition and structure of DNA to inhibit DNA synthesis and prevent cellular proliferation (2). While effective, their utility is typically limited by the severity of side effects caused by the non-selective killing of cancerous and healthy cells in addition to the potential to induce mutagenic events that can actually accelerate disease development (3). Another strategy is to modulate the activity of individual enzymes involved in DNA replication (Step 2). Agents such as etoposide inhibit the activity of topoisomerase, an enzyme that resolves the “knots” in DNA formed during replication (4). Inhibition of this enzyme causes apoptosis by creating single- and double-stranded DNA breaks that interrupt the continuity of DNA synthesis. A third approach includes reducing the availability of dNTPs that are the building blocks for DNA synthesis (Step 3). By inhibiting enzymes involved in nucleotide metabolism, anti-metabolites such as methotrexate and hydroxyurea reduce nucleotide pools to indirectly hinder DNA synthesis (5). Perhaps the most direct approach, however, is the use of nucleoside analogs such as AZT (6) or fludaribine (7) that target the enzymatic activity of the DNA polymerase, the workhorse of the DNA replication apparatus (Step 4). This last strategy represents the focus of this review. In this article, we shall discuss the design, mechanism of action, and pitfalls associated with various nucleoside analogs that are used as therapeutic agents against common hyperproliferative diseases including viral infections and cancer.
Mechanism and Dynamics of the DNA Polymerization Cycle
Before describing how nucleoside analogs act as therapeutic agents, it is first necessary to understand the mechanism of DNA polymerization and how perturbations in various kinetic steps can influence their effectiveness. A general mechanism that applies to both prokaryotic and eukaryotic DNA polymerases is provided in Figure 2. As illustrated, this intricate biological process occurs via a multiplicative mechanism in which the polymerase binds DNA prior to the binding of dNTP. The first point for generating high catalytic efficiency and fidelity occurs through the binding of dNTP to the polymerase:DNA complex (Step 2). The binding affinity (Kd value) for a nucleotide opposite its correct pairing partner is generally ∼10 μM while Kd values for incorrect nucleotides are usually 10-fold higher. After dNTP binding, an enzymatic conformational change (Step 3) is proposed to align the incoming dNTP into a precise geometrical orientation that is necessary for phosphoryl transfer (Step 4). This conformational change step reflects an “induced-fit” mechanism that also imposes discrimination against nucleotide misinsertion as misaligned intermediates perturb the geometry of the polymerase's active site to prevent the chemical step (8). Another point for error discrimination can occur during the phosphoryl transfer step (Step 4) which may be rate-limiting for nucleotide incorporation with certain polymerases (9). Regardless, the maximal rate constant for polymerization (denoted as kpol) for the incorporation of a correct nucleotide opposite its cognate partner generally range from 20 to 200 sec−1. Thus, the relatively low binding affinity for a correct nucleotide can be offset by a fast kpol value such that the overall catalytic efficiency (kpol/Kd) for correct polymerization is very high. Indeed, kpol/Kd values of ∼107 M−1sec−1 are at the limits of diffusion control and place polymerases within the top tier of most proficient enzymes (10). It is also important to emphasize that kpol/Kd values for incorrect DNA synthesis, i.e., the misincorporation of dATP opposite 2-deoxycytidine, are typically several orders of magnitude lower due to decreases in kpol coupled with increases in Kd. This reduction plays a large role in the maintenance of genomic fidelity.
Figure 2.
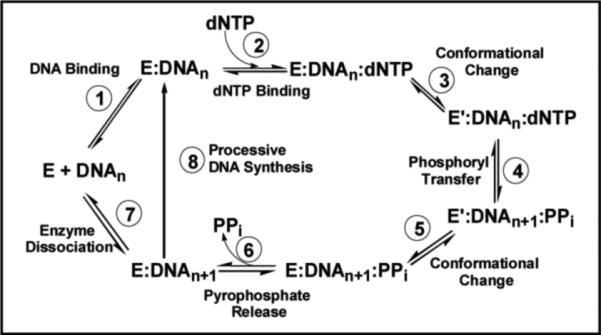
Kinetic mechanism for RNA- and DNA-dependent polymerases. Individual steps along the pathway for DNA polymerization are numbered and identified. Abbreviations: E = polymerase, DNAn = DNA substrate, E’ = conformational change in DNA polymerase, PPi = inorganic pyrophosphate, and DNAn+1 = DNA product (DNA extended by one nucleobase).
Following phosphoryl transfer, there is a second conformational change (step 5) that is required for pyrophosphate release (step 6). After pyrophosphate (PPi) is released, the polymerase can either remain bound to the nucleic acid substrate to continue primer elongation (step 8) or dissociate from the elongated primer (step 7) to initiate DNA synthesis on another usable primer template. The ability to incorporate multiple nucleotides without dissociating from DNA defines the processivity of the polymerase.
Alterations in various kinetic steps can occur to influence the behavior of the polymerase and alter the effectiveness of certain chemotherapeutic agents. One example is the ability of a polymerase to reverse the polymerization step (Figure 2, Step 4) via a process known as pyrophosphorolysis to remove inappropriately incorporated nucleotides (11). Other kinetic steps include the dNTP binding step (Figure 2, Step 2) and the conformational change preceding chemistry (Figure 2, Step 3).
Strategic Interventions
This mechanistic framework highlights the importance of kinetic steps that can be targeted to inhibit replication and prevent proliferation. One potential stage for therapeutic intervention is the use of competitive inhibitors to block dNTP binding to the polymerase's active site (Figure 2, Step 2). Unfortunately, developing a reversible inhibitor that binds tightly and selectively to a DNA polymerase is an extraordinarily difficult task. Much of this challenge arises from the promiscuous and relatively weak binding affinity (Kd ∼ 10 μM) of natural nucleotides to the polymerase. Furthermore, “correct” polymerization occurs with an incredibly high commitment to catalysis (Keq ∼106) since forward kinetic steps such as phosphoryl transfer (kpol ∼100 sec−1) are generally much faster than the corresponding reverse step of pyrophosphorolysis (kpyro ∼0.0001 sec−1) (12). These two features coupled with additional pharmacokinetic problems such as drug absorption (13) and metabolism (14) of nucleoside triphosphates have hindered this conventional approach.
An alternative strategy is to take advantage of the polymerase's high commitment to catalysis and trick the enzyme into using a potential suicide inhibitor. This Trojan Horse strategy outlined in Figure 3 represents the current paradigm in the design and implementation of many nucleoside analogs targeting prokaryotic and eukaryotic DNA polymerases. The polymerase is provided with a modified nucleotide such as AZT-TP that is incorporated into DNA as efficiently as its natural counterpart, dTTP. Once incorporated, however, the modified nucleotide creates a nucleic acid substrate that can not be elongated and thus terminates DNA synthesis. In general, the development of an effective chain-terminating nucleotide depends upon several features including: 1) high catalytic efficiency for incorporation, 2) no potential for subsequent elongation, 3) poor excision after incorporation, 4) selective utilization by polymerases responsible for the pathogenic state, and 5) effective metabolism of the parental nucleoside to form the corresponding nucleoside triphosphate. The following sections describe successes and pitfalls associated with several therapeutically-relevant nucleoside analogs. Our discussion begins by analyzing the development and application of several viral DNA polymerase inhibitors as this represents a major effort in modern pharmaceutical science.
Figure 3.
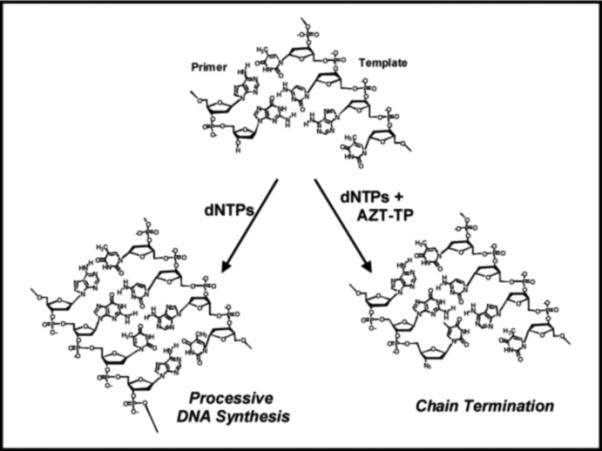
Mechanism by which chain-terminating nucleotides inhibit DNA synthesis. AZT-TP is an effective chain-terminator due to the simple replacement of the hydroxyl group with an azide (-N3) which prevents subsequent primer elongation.
Agents That Target Retroviral Polymerases
Viruses are responsible for a wide variety of human diseases ranging from simple ailments including the common cold, chickenpox, and cold sores to more serious health threats such as Aquired Immune Deficiency Syndrome (AIDS), Severe Acute Respiratory Syndrome (SARS), and cervical cancer (15). In addition, an alarming number of other diseases such as progressive multifocal leukoencephalopathy (PML) and multiple sclerosis may also be linked with viral infections (16). A universal feature for the virulence and propagation of any virus is the requirement for high levels of DNA synthesis. For example, reverse transcription of the viral single-stranded (+) RNA genome of the human immunodeficiency virus (HIV) into double-stranded DNA is catalyzed by the retroviral polymerase, reverse transcriptase (RT). This RNA- and DNA-dependent polymerase is a prime target for anti-viral drug development since it plays an essential role in the HIV life cycle. To date, two distinct types of RT inhibitors with unique pharmacodynamic behavior have been developed for clinical use. These include nucleoside RT inhibitors and non-nucleoside RT inhibitors (NNRTIs).
Nucleoside Reverse Transcriptase Inhibitors
Figure 4 compares the structures of natural nucleosides with several nucleoside analogs approved as anti-viral agents against HIV or herpes simplex virus (HSV), another important viral target. All of these nucleoside analogs share a common feature that distinguishes them from their natural counterparts - the absence of the 3'-OH group that is required for subsequent primer elongation. As previously illustrated in Figure 3, AZT-TP is an effective chain-terminator due to the simple replacement of the hydroxyl group with an azide (-N3). Since the coding potential of the nucleobase remains identical to that of the natural nucleoside, the binding affinities for chain-terminators such as AZT as well as d4T (Stavudine) and ddC (Zalcitabine) are nearly identical to their natural counterparts (17-19). However, the maximal rate constant for the incorporation of AZT-TP is significantly slower than dTTP incorporation (compare kpol values of 0.7 vs ∼20 sec−1 for AZT-TP and dTTP, respectively) (17). Thus, alterations to the ribose moiety appear to influence the phosphoryl transfer step and/or the ability of the polymerase to mediate the conformational change preceding this kinetic step. Regardless, the kpol/Kd values for these modified nucleotides are comparable to their natural counterparts and the inhibition of viral replication via chain-termination corroborates clinical data showing viral load reductions upon treatment with these analogs.
Figure 4.
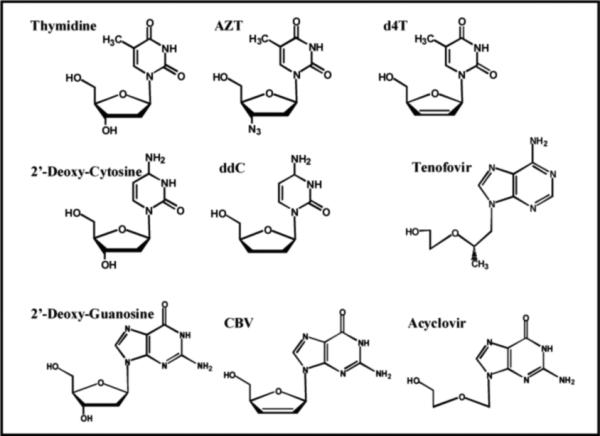
Structures of several nucleoside analogs that are commonly used as anti-viral agents. See text for details.
Acyclovir (Figure 4) is a unique guanine analog that is widely used in the treatment of herpes simplex and herpes zoster (shingles) infections (reviewed in 20). Acyclovir differs significantly from the other aforementioned anti-viral agents as the 2'-deoxyribose sugar is replaced by an open-chain structure which accounts for its chain-termination capabilities. However, this mofication confers two additional pharmacodynamic properties that make it a highly effective anti-viral agent. The first is an unusually high selectively for conversion into acyclo-guanosine monophosphate (acylo-GMP) by the virally-encoded thymidine kinase as opposed to the host kinase. The 3,000-fold higher activity of the viral enzyme leads to higher concentrations of acylo-GMP in infected cells as opposed to healthy, unfected cells. Secondly, the triphosphate form of acyclovir (acyclo-GTP) is incorporated 100-fold more efficiently by the viral polymerase compared to the host enzyme. These two features make acyclovir one of the safest anti-viral agents currently on the market. The success of acyclovir has spawned the development of a number of acyclonucleosides as anti-viral agents. Page limitations prohibit a thorough discussion of these analogs. However, an excellent overview of these unique anti-viral agents is provided by Hewlett et al. (21).
Pitfalls Associated with Chain-Terminating Nucleoside Analogs
Despite their widespread use, the therapeutic utility of most nucleoside analogs is often restricted by major complications including the development of resistance and the induction of adverse side effects. Resistance to all HIV-related nucleoside analogs invariable develops in clinical settings through one of several mechanisms. First, conversion of the nucleoside into the required triphosphate form is limited by the action of the host thymidine kinase and results in lower plasma concentrations that may limit their effectiveness (22). A second mechanism occurs via the generation of selective point mutations in RT that hinders incorporation of the chain-terminator but that have no effect on the utilization of natural nucleotides. One example is the M184V mutant that causes resistance to 3TC by simple steric hindrance between the β-branched amino acid side chain (valine) with the β-L-oxathialone ring of the nucleoside triphosphate (23). This simple mutation decreases the binding affinity of the chain-terminator but has no effect on the binding of the natural substrate, dTTP. The generation of point mutations also accounts for the third most common mechanism of resistance - enhanced enzymatic removal of the chain-terminator from the viral genome via pyrophosphorolysis, the reversal of polymerization (11).
Adverse side effects arise from the inherently low selectivity of these nucleoside analogs as they can be effectively utilized by either viral or host DNA polymerases to inhibit DNA synthesis. In the case of HIV, however, it was hypothesized that viral replication would be selectively inhibited since HIV RT does not possess exonuclease activity to remove the chain terminators from the viral genome. In contrast, most eukaryotic polymerases have robust exonuclease activities that can rapidly excise the analog and allow for replication of the host's genome. In hindsight, these assumptions proved inaccurate as many anti-viral agents are slowly excised by host enzymes and/or are rapidly removed from the viral genome. In the former case, chronic administration of certain nucleoside analogs can cause side effects resembling heritable mitochondrial diseases (24). Patients treated with one or more nucleoside analogs generally display phenotypes ranging from peripheral neuropathy, cardiac and skeletal muscle myopathy, pancreatitis, and bone marrow suppression (24). The molecular basis for most of these side effects appears linked with the inhibition of mitochondrial DNA replication catalyzed by pol γ (25). In fact, a recent review by Lee et al. (26) provides an excellent description of a predictive index correlating the clinical toxicity of a nucleoside analog with the probability of the chain terminating nucleotide to be incorporated and subsequently excised from DNA by pol γ.
It is clear that a diminution in exonuclease proofreading by human polymerases can cause an increased risk in toxicity. However, a different complication arises if the viral polymerase efficiently excises the chain-terminator from the viral genome via pyrophosphorolysis (11). As described earlier, pyrophosphorolysis plays an important role in drug resistance as the efficient removal of these chain terminators allows RT to re-initiate viral DNA synthesis. During long-term treatment with AZT, a set of mutations including M41L, D67N, K70R, T215F/Y, and K219Q develop within the active site of HIV RT that confer resistance (27). The structures of these mutants complexed with chain-terminated DNA (reviewed in 28) has been instrumental in understanding the mechanism of this resistance. In this model, RT can position the 3’-OH of the primer into two distinct locations denoted as the priming site (P-site) or the nucleotide-binding site (N-site). During normal replication, RT binds the 3’-OH in the P site while dNTP binding occurs in the N-site. After phosphoryl transfer, the newly extended primer is transferred from the N-site to the P-site and the catalytic cycle continues. However, the dynamics of this cycle are altered if a chain-terminator is incorporated as the absence of the 3'-OH on a chain-terminated primer prevents elongation. Thus, the binding of the next correct dNTP in the N-site typically causes the formation of a dead-end ternary complex to prevent DNA synthesis. However, the aforementioned mutations allows RT to partition the chain-terminated primer from the P-site back into the N-site. This movement allows for PPi binding and subsequent pyrophosphorolysis to excise the chain-terminator so that DNA synthesis can resume. Although PPi is the natural substrate for this excision reaction, ATP can also act as the acceptor substrate (29). ATP appears to interact directly within the active site through stacking interactions between the purine moiety and the aromatic ring of Y215 (30).
Since pyrophosphorolysis plays an important role in drug resistance, significant effort has been placed in developing molecules to inhibit this process. One example is foscarnet, a structural mimic of PPi, that selectively inhibits the binding of pyrophosphate to various viral DNA polymerases (31). Foscarnet is widely used as an anti-viral agent against cytomegalovirus retinitis and HSV type 1 and 2 infections. However, its use in HIV therapy is limited as chronic administration causes electrolyte imbalance and nephrotoxicity due to alterations in calcium, magnesium, potassium, and phosphate levels.
Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs)
In addition to nucleoside analogs, another important class of RT inhibitors widely used in the treatment against HIV infection are non-nucleoside reverse transcriptase inhibitors (NNRTIs). It is clear from the structures provided in Figure 5 that these analogs do not bear any resemblance to nucleoside analogs such as AZT and ddC. In fact, this class of agents was initially identified as potent inhibitors of HIV RT through high throughput screening efforts rather than through rational drug design targeting the active site of the polymerase (32). Steady-state inhibition studies determined that NNRTIs such as Efavirenz act as uncompetitive inhibitors at low drug concentrations (<50 nM) (33). This result is consistent with a mechanism in which Efavirenz binds to the ternary RT:DNA:dNTP complex with higher affinity than to either free RT or the RT:DNA complex (33). Transient kinetic studies monitoring polymerization revealed that NNRTIs block the phosphoryl transfer reaction without interfering with nucleotide binding (Figure 2, Step 2) or the nucleotide-induced conformational change (Figure 2, Step 3) (33). Instead, NNRTI binding allows the incoming nucleoside triphosphate to bind tightly (Kd ∼100 nM) but in a nonproductive manner (34). This represents one of the first examples of a small molecule that binds to an allosteric site to cause potent inhibition. Indeed, structural analysis reveals that the NNRTI binds to hydrophobic pocket on the polymerase lined with large and hydrophobic amino acids including Y181, Y188, F227, and L234 which is located ∼ 10 Å from the enzyme's polymerase active site (35).
Figure 5.
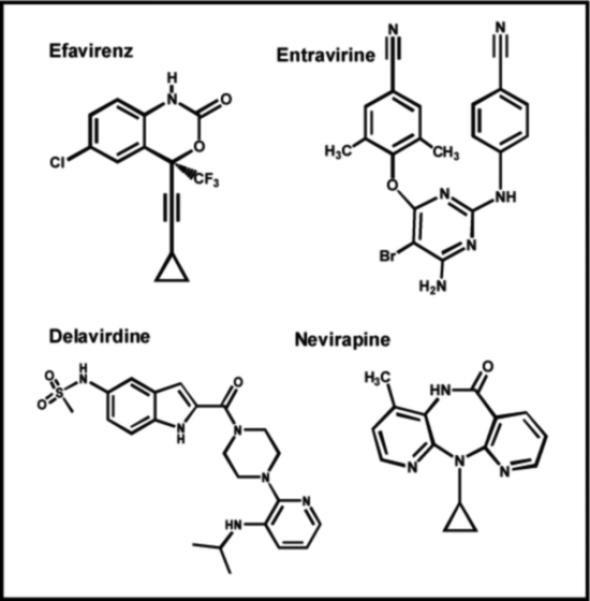
Structures of various non-nucleoside reverse transcriptase inhibitors (NNRTIs) that target HIV reverse transcriptase. See text for details.
Not surprisingly, resistance to NNRTIs develops rapidly through the generation of point mutations within this hydrophobic binding site. For example, a recent retrospective cohort study of HIV-infected patients treated with Nevirapine revealed that resistant isolates developed in 92% of treated patients within 24 weeks (36). Furthermore, cross-resistance to other NNRTIs such as Efavirenz and Delavirdine were detected in ∼70% of these patients, suggesting that a common set of mutations lead to resistance (36). In general, drug resistance arises due to the replacement of a large amino acid with a smaller one that weakens the molecular interactions of the inhibitor with RT. For example, Nevirapine binds 500-fold more weakly to the Y181C mutant than to wild-type RT, and the reduced affinity reflects an increase in the off-rate of the inhibitor from the RT:DNA complex rather than simple steric hindrance (37). Surprisingly, Entravirine is a new NNRTI that remains active as an allosteric inhibitor against mutant RTs that are resistant to other currently used NNRTIs such as Nevirapine (38).
Combination therapy, the co-administration of multiple drugs targeting various pathological pathways, is a strategy that is widely used in the treatment of hyper-proliferative diseases such as HIV. In fact, nucleoside inhibitors are often combined with NNRTIs such as Nevirapine to synergistically reduce viral loads in vivo. This particular drug combination is beneficial as it delays the onset of resistance to nucleoside analogs that occurs through pyrophosphorolysis (39). This occurs since the binding of an NNRTI to the allosteric site inhibits the pyrophosphorylytic activity of RT by reducing the binding affinities for the phosphoryl acceptor (PPi or ATP) as well as reducing the rate of the chemical step. While this combination of drugs reduces the rate of drug resistance, it does not completely eliminate it as the intrinsically low fidelity of RT eventually generates drug-resistant mutants.
Nucleotide Analogs that Induce Lethal Mutagenesis
Viral DNA synthesis is viewed as an error-prone process in which at least one mutation is introduced per round of replication. The lack of fidelity causes low mutation rates that are considered beneficial for certain viruses as it generates selective point mutations in critical proteins that confer resistance. However, higher rates of mutagenesis should reduce viral fitness and viability. Thus, a provocative therapeutic strategy is to use pro-mutagenic nucleoside analogs to increase the mutation frequency of a virus beyond a critical threshold capable of supporting its viability (40). These nucleoside analogs would have little effect on the integrity of the host's genome since eukaryotic polymerases exhibit higher intrinsic fidelity and would prevent these analogs from being stably incorporated into DNA. Proof-of-concept for this strategy was first demonstrated by the Loeb laboratory using 5-hydroxy-2'-deoxycytidine (Figure 6) as a pro-mutagenic nucleoside that pairs with either guanine or adenine (41). Treatment of HIV-infected cells with this pro-mutagenic nucleoside showed an 80% reduction in viral replication that correlated with a disproportionate increase in G to A substitutions in isolated viral DNA. Another example is Ribavirin, the ribose form of 1,2,4-triazole-3-carboxamide (Figure 6), that is used to treat viral infections such as SARS and hepatitis C. This pro-mutagenic nucleoside reduced infective poliovirus production to as little as 0.00001% that coincides with the induction of mutations in the viral genome caused by the misincorporation of Ribavirin opposite C or U (42). These studies provide an exciting and novel therapeutic strategy to convert a perceived strength of a virus (low polymerization fidelity) into a target for therapeutic intervention.
Figure 6.
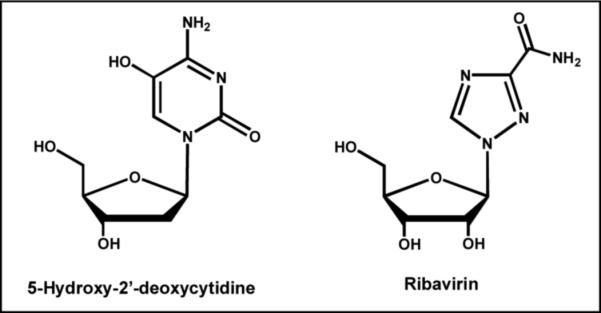
Structure of pro-mutagenic nucleoside analogs that are proposed to induce lethal mutagenesis in various viral systems. See text for details.
Nucleoside Analogs as Anti-Cancer Agents
Despite advances in the detection and prevention of cancer, this disease still remains the second-leading cause of death in industrialized nations. Cancer is best defined as a hyperproliferative disease since the hallmark of this pathological malignancy is uncontrolled DNA synthesis. Treatment modalities vary depending upon whether the cancer is a solid tumor (brain, lung, pancreatic, colon, etc.) or a hematological malignancy (leukemia and lymphomas). However, combination therapy is typically employed either case in which various chemotherapeutic agents are used to target the different biological pathways that are responsible for sustaining the uncontrollable growth of the cancer. One important class of chemotherapeutic agents are nucleoside analogs such as fludarabine2, cladribine, gemcitabine, and cytarabine (Figure 7). Their mechanism of action is similar to previously discussed anti-viral agents such as AZT as the ribose moiety that is essential for primer elongation is modified compared to the natural nucleoside. However, a major difference is that the deoxyribose sugar is replaced with arabinose or another modified sugar moiety. This simple replacement causes a number of pharmacodynamic effects that make these analogs effective anti-cancer agents by inhibiting DNA synthesis via multiple mechanisms (refer to Figure 1B). For example, fludarabine behaves as a classic chain-terminator of DNA synthesis (43) since the alteration in sugar composition makes it refractory to elongation after incorporation into DNA. However, fludarabine exhibits additional chemotherapeutic properties by acting as an anti-metabolite by inhibiting the activity of ribonucleotide reductase and adenosine deaminase (44), two key enzymes involved in purine metabolism. Since this agent hits multiple targets, it is highly effective as a single agent in the treatment of chronic lymphocytic leukemia. However, it is also used in combinations with other chemotherapeutic agents such as DNA damaging agents (cyclophosphamide and mitoxantrone) and glucocortcoid steroids (dexamethasone).
Figure 7.
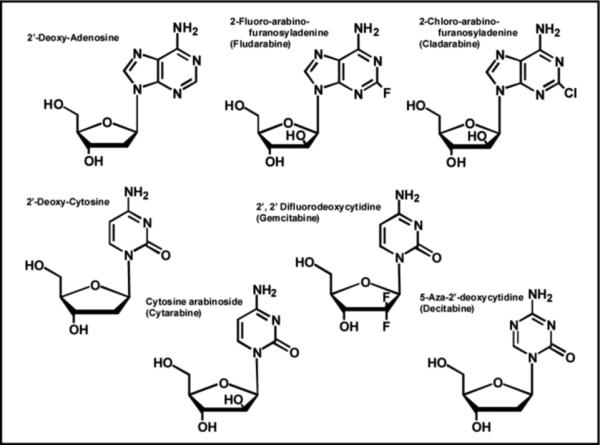
Structures of several nucleoside analogs that are widely used as anti-cancer agents. See text for details.
Cytarabine (Figure 7) is a pyrimidine analog that acts as a substrate for both eukarytoic DNA and RNA polymerases and exerts its cytotoxic effects by terminating replication and transcription (45). This pyrimidine analog is similar to fludarine as it also indirectly inhibits DNA synthesis by disrupting the proper balance of nucleotide pools by inhibiting ribonucleotide reductase activity. 5-aza-2'-deoxycytidine (Decitabine) is another pyrimidine analog that is a near perfect mimetic of 2-deoxy-cytidine as only the C5 carbon is replaced with nitrogen (Figure 7). This simple substitution does not influence the ability to 5-aza-2'-deoxycytidine to properly base-pair with a guanine so there is no overt inhibitory effect on DNA synthesis. Instead, this subtle modification prevents the ability of nucleobase to be modified by a class of DNA-modfiying enzymes known as DNA methyltransferases which influence transcription rather than replication (46). DNA methylation is an epigenetic modification that reversibly regulates gene expression without changing the original DNA sequence of the gene promoter. In eukaryotes, the expression of many genes is inversely correlated with the methylation status of its promoter region. In certain forms of cancer, the promoter region of critical genes that regulate important cellular functions can become hypermethylated (47). These inappropriate epigenetic modifications lower gene expression to reduce the intacellular level of critical proteins. For example, many sporadic colon cancers show hypermethylation of the hMLH1 gene promoter which correlates which lower expression of gene product, hMLH1, an essential DNA repair protein (48). Reduced levels of this repair enzyme plays important roles in the development of carcinogenesis in addition to drug-resistance to various chemotherapeutic agents such as temozolomide that damage DNA. However, cells treated with 5-aza-2'-deoxycytidine as a “demethylating agent” regain expression of the hMLH1 protein which then sensitizes cells to the cytotoxic effects of certain chemotherapeutic agents. Although 5-aza-2'-deoxycytidine is used as a chemotherapeutic agent, it possesses a relatively narrow therapeutic window as it affects the methylation patterns of normal and cancer cells (49).
Pitfalls Associated with Nucleoside Analogs in Cancer Chemotherapy
While nucleoside analogs are important in treating cancer, their utility is often limited by dose-limiting toxicity that arises due to their non-selective nature. As expected, a number of the body's normal cells also divide rapidly and are therefore killed by this class of chemotherapeutic agent. This non-selective killing contributes to the development of debilitating side effects that include alopecia (temporary hair loss), mucositis (mouth sores), anemia (reduced red blood cell number), leukopenia (reduced white blood cell number), thrombocytopenia (decreased platelet numbers), and gastrointestinal symptoms such as nausea, vomiting, and diarrhea. The intensity of these side effects depends heavily upon the dose and duration of exposure to the chemotherapeutic agent. For example, patients treated with high doses of fludarabine often develop leukopenia and thrombocytopenia which leads to opportunistic infections and bleeding disorders. As a result, patients receiving fludarabine are sometimes co-adminsitered antibiotics as prophylactic measures against opportunistic infections.
New Frontiers in the Development of Nucleoside Analogs
A major disadvantage with the current arsenal of nucleoside analogs is dose-limiting toxicity that arises from the lack of selectivity to inhibit one DNA polymerase versus another. This is not unexpected since all current analogs bear structural identity to natural nucleosides and are thus incorporated opposite templating nucleobases with high catalytic efficiencies. An additional complication is that all DNA polymerases characterized to date share common structural features that prohibit the use of rational drug design to selectively target the active site. The overall structure of DNA polymerases resembles a “right hand” containing fingers, palm, and thumb subdomains (50). The palm domain is the most closely conserved structural feature as it contains the amino acids required for phosphoryl transfer. The fingers domain interacts with the incoming dNTP as well as the templating base and thus plays an important role in nucleotide selection. The thumb domain plays dual roles by positioning duplex DNA for the incoming dNTP as well as in the processivity and translocation of the polymerase.
Rather than directly target the active site of the polymerase, an alternative approach is to develop nucleoside analogs that can be selectively incorporated opposite modified or damaged DNA. This approach would inhibit pro-mutagenic DNA synthesis and could be advantageous for several reasons. First, this approach represents a chemopreventive strategy as the generation of disease-causing mutations caused by the misreplication of damaged DNA would be inhibited. Secondly, this approach could be used as an adjunctive therapy to sensitize cancer cells to the effects of other chemotherapeutic agents that damage DNA (refer to Figure 1B, Step 1). The in vitro applications of this strategy have been demonstrated in our lab using the abasic site as a model form of DNA damage. Abasic sites are a commonly formed DNA lesion that result from the hydrolysis of the C-N bond between the 2'-deoxyribose and a purine/pyrimidine. Hydrolysis occurs spontaneously but is significantly enhanced by DNA damaging agents such as temozolomide (51). The net effect of this hydrolysis reaction is that templating information originally present in the DNA is lost which causes the abasic site to be highly pro-mutagenic.
Despite the non-templating nature of an abasic site, most DNA polymerases preferentially incorporate dATP opposite the lesion (52). We applied this information to rationally design a series of 5-substituted indolyl-2’-deoxyriboside triphosphates that mimic the core structure of dATP (Figure 8) (reviewed in 53 and 54). In vitro kinetic analyses reveal that analogs containing significant π-electron density such as 5-NITP, 5-Ph-ITP, and 5-CE-ITP are incorporated opposite the abasic site with high catalytic efficiencies of 107 M−1sec−1. These values are ∼1,000 fold higher than that for the preferred natural substrate, dATP. In contrast, non-natural nucleotides that lack extensive π-electron density such as 5-FITP, 5-AITP, and 5-CH-ITP are poorly inserted opposite the abasic site. These kinetic studies demonstrate that simple alterations to the π-electron surface area of a non-natural nucleotide can modulate its efficiency for incorporation opposite an abasic site. In addition, there is evidence that certain non-natural nucleotides are utilized by various DNA polymerases more effectively than other analogs in the same class (55). One possibility is that while the functions of the fingers and thumb subdomains are conserved amongst DNA polymerases, there are subtle differences with respect to sequence and structural characteristics such that selective inhibitors could be developed that exploit these nuances. The development and application of this class of non-natural nucleoside opens new avenues toward understanding the mechanism and biological consequences of translesion DNA synthesis which can then be exploited as adjunctive chemotherapeutic agents.
Figure 8.
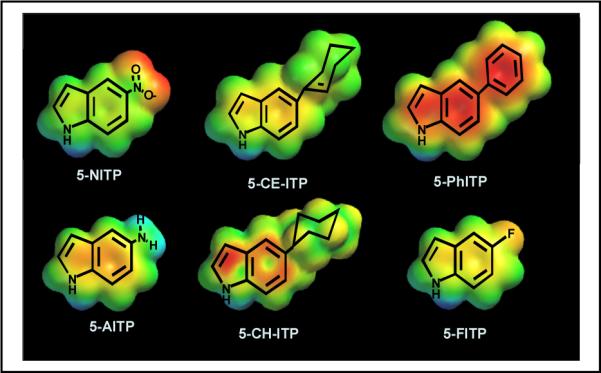
Structures of a new series of 5-substituted-indolyl-2’-deoxyribosides that show selectivity for incorporation opposite damaged versus un-damaged DNA.
Conclusions and Prospectus
The inhibition of DNA replication remains an important therapeutic strategy against numerous diseases including viral infections and cancer. The current arsenal of agents targeting polymerase activity are generally based upon modifications to the 2'-deoxyribose moiety of a nucleotide which are designed to prevent elongation of nucleic acid. While effective, the vast majority of these nucleoside analogs display pharmacodynamic complications that include an intrinsic lack of selectivity and the development of drug resistance. In addition, pharmacokinetic problems such as conversion to the triphosphate form and degradation through salvage pathways can limit their effectiveness. However, new therapeutic approaches are currently being developed to circumvent these complications that include developing selective inhibitors for polymerases involved in DNA repair and/or translesion DNA synthesis.
Acknowledgments
This article was supported through funding from the National Institutes of Health CA118408.
Abbreviations
- dNTP
deoxynucleoside triphosphate
- RT
reverse transcriptase
- AZT
3-azidothymidine
- d4T
2'-3'-didehydro-2'-3'-dideoxythymidine
- ddC
2'-3'-dideoxycytidine
- (−) 3TC
2',3'-dideoxy-3'-thiacytidine
- CBV
(9-[4-alpha-(hydroxymethyl)cyclopent-2-ene-1-alpha-yl)guanine
- acyclovir
2-amino-1,9-dihydro-9-[(2-hydroxyethoxy)methyl]-6H-purin-6-one
- NNRTI
non-nucleoside reverse transcriptase inhibitor
- efavirenz
8-chloro-5-(2-cyclopropylethynyl)- 5-(trifluoromethyl)- 4-oxa-2-azabicyclo [4.4.0]deca-7,9,11-trien-3-one
- nevirapine
1-cyclopropyl-5,11-dihydro-4-methyl-6H-dipyrido [3,2-b:2',3'-e][1,4] diazepin-6-one
- delavirdine
N-[2-[4-[3-(1-methylethylamino) pyridin-2-yl] piperazin-1-yl] carbonyl-1H-indol-5-yl] methanesulfonamide
- entravine
4-[6-Amino-5-bromo-2- [(4-cyanophenyl)amino] pyrimidin-4-yl]oxy- 3,5-dimethylbenzonitrile
- fludaribine
2-fluoro-arabinofuranosyladenine
- cladarabine
2-chloro-arabinofuranosyladenine
- cytarabine
1-β-D-arabinofuranosylcytosine
- gemcitabine
2',2'-difluoro-2'-deoxycytidine
- decitabine
5-aza-2-deoxy-2'-cytidine
- 5-NITP
5-nitro-indolyl-2’-deoxyriboside triphosphate
- 5-CE-ITP
5-cyclohexene-indolyl-2’-deoxyriboside triphosphate
- 5-PhITP
5-phenyl-indolyl-2’-deoxyriboside triphosphate
- 5-FITP
5-fluoro-indolyl-2’deoxyriboside triphosphate
- 5-AITP
5-amino-indolyl-2’-deoxyriboside triphosphate
- 5-CH-ITP
5-cyclohexyl-indolyl-2’deoxyriboside triphosphate
Footnotes
The monophosphate form of this analog is used clinically as the addition of the monophosphate increases its solubility. The monophosphate is rapidly hydrolyzed in plasma so that the free nucleoside enter cells via passive diffusion.
REFERENCES
- 1.Kulkarni A, Wilson DM., 3rd The involvement of DNA-damage and -repair defects in neurological dysfunction. Am. J. Hum. Genet. 2008;82:539–566. doi: 10.1016/j.ajhg.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Havelka AM, Berndtsson M, Olofsson MH, Shoshan MC, Linder S. Mechanisms of action of DNA-damaging anticancer drugs in treatment of carcinomas: is acute apoptosis an “off-target” effect? Mini Rev. Med. Chem. 2007;7:1035–1039. doi: 10.2174/138955707782110196. [DOI] [PubMed] [Google Scholar]
- 3.Guillem V, Tormo M. Influence of DNA damage and repair upon the risk of treatment related leukemia. Leuk. Lymphoma. 2008;49:204–217. doi: 10.1080/10428190701769657. [DOI] [PubMed] [Google Scholar]
- 4.Baldwin EL, Osheroff N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anticancer Agents. 2005;5:363–372. doi: 10.2174/1568011054222364. [DOI] [PubMed] [Google Scholar]
- 5.McGuire JJ. Anticancer antifolates: current status and future directions. Curr. Pharm. Des. 2003;9:2593–2613. doi: 10.2174/1381612033453712. [DOI] [PubMed] [Google Scholar]
- 6.Yarchoan R, Pluda JM, Perno CF, Mitsuya H, Thomas RV, Wyvill KM, Broder S. Initial clinical experience with dideoxynucleosides as single agents and in combination therapy. Ann. NY Acad. Sci. 1990;616:328–343. doi: 10.1111/j.1749-6632.1990.tb17853.x. [DOI] [PubMed] [Google Scholar]
- 7.Anderson VR, Perry CM. Fludarabine: a review of its use in non-Hodgkin's lymphoma. Drugs. 2007;67:1633–1655. doi: 10.2165/00003495-200767110-00008. [DOI] [PubMed] [Google Scholar]
- 8.Johnson KA. Conformational coupling in DNA polymerase fidelity. Annu. Rev. Biochem. 1993;62:685–713. doi: 10.1146/annurev.bi.62.070193.003345. [DOI] [PubMed] [Google Scholar]
- 9.Showalter AK, Tsai MD. A reexamination of the nucleotide incorporation fidelity of DNA polymerases. Biochemistry. 2002;41:10571–10576. doi: 10.1021/bi026021i. [DOI] [PubMed] [Google Scholar]
- 10.Wolfenden R, Snider MJ. The depth of chemical time and the power of enzymes as catalysts. Acc. Chem. Res. 2001;34:938–945. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- 11.Meyer PR, Matsuura SE, Zonarich D, Chopra RR, Pendarvis E, Bazmi HZ, Mellors JW, Scott WA. Relationship between 3'-azido-3'-deoxythymidine resistance and primer unblocking activity in foscarnet-resistant mutants of human immunodeficiency virus type 1 reverse transcriptase. J. Virol. 2003;77:6127–6137. doi: 10.1128/JVI.77.11.6127-6137.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Capson TL, Peliska JA, Kaboord BF, Frey MW, Lively C, Dahlberg M, Benkovic SJ. Kinetic characterization of the polymerase and exonuclease activities of the gene 43 protein of bacteriophage T4. Biochemistry. 1992;31:10984–10994. doi: 10.1021/bi00160a007. [DOI] [PubMed] [Google Scholar]
- 13.Diab R, Degobert G, Hamoudeh M, Dumontet C, Fessi H. Nucleoside analogue delivery systems in cancer therapy. Expert Opin. Drug Deliv. 2007;4:513–531. doi: 10.1517/17425247.4.5.513. [DOI] [PubMed] [Google Scholar]
- 14.Piliero PJ. Pharmacokinetic properties of nucleoside/nucleotide reverse transcriptase inhibitors. J. Acquir. Immune Defic. Syndr. 2004;37:S2–S12. doi: 10.1097/01.qai.0000137001.40505.56. [DOI] [PubMed] [Google Scholar]
- 15.Bermejo JF, Muñoz-Fernandez MA. Severe acute respiratory syndrome, a pathological immune response to the new coronavirus--implications for understanding of pathogenesis, therapy, design of vaccines, and epidemiology. Viral Immunol. 2004;17:535–544. doi: 10.1089/vim.2004.17.535. [DOI] [PubMed] [Google Scholar]
- 16.Becker LE. Slow infections of the central nervous system. Can. J. Neurol. Sci. 1977;4:81–88. [PubMed] [Google Scholar]
- 17.Kerr SG, Anderson KS. Pre-steady-state kinetic characterization of wild type and 3'-azido-3'-deoxythymidine (AZT) resistant human immunodeficiency virus type 1 reverse transcriptase: implication of RNA directed DNA polymerization in the mechanism of AZT resistance. Biochemistry. 1997;36:14064–14070. doi: 10.1021/bi9713862. [DOI] [PubMed] [Google Scholar]
- 18.Yang G, Wang J, Cheng Y, Dutschman GE, Tanaka H, Baba M, Cheng YC. Mechanism of inhibition of HIV-1 reverse transcriptase by a stavudine analogue 4'-ethynyl-D4TTP. Antimicrob. Agents Chemother. 2008;52:2035–2042. doi: 10.1128/AAC.00083-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu Z, Arts EJ, Parniak MA, Wainberg MA. Mutated K65R recombinant reverse transcriptase of human immunodeficiency virus type 1 shows diminished chain termination in the presence of 2',3'-dideoxycytidine 5'-triphosphate and other drugs. Proc. Natl. Acad. Sci. USA. 1995;92:2760–2764. doi: 10.1073/pnas.92.7.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reardon JE, Spector T. Herpes simplex virus type 1 DNA polymerase. Mechanism of inhibition by acyclovir triphosphate. J. Biol. Chem. 1989;264:7405–7411. [PubMed] [Google Scholar]
- 21.Hewlett G, Hallenberger S, Rubsamen-Waigmann H. Antiviral against DNA viruses (hepatitis B and the herpes viruses). Curr. Opin. Pharm. 2004;4:453–464. doi: 10.1016/j.coph.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 22.Gao WY, Shirasaka T, Johns DG, Broder S, Mitsuya H. Differential phosphorylation of azidothymidine, dideoxycytidine, and dideoxyinosine in resting and activated peripheral blood mononuclear cells. J. Clin. Invest. 1993;91:2326–2333. doi: 10.1172/JCI116463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao HQ, Boyer PL, Sarafianos SG, Arnold E, Hughes SH. The role of steric hindrance in 3TC resistance of human immunodeficiency virus type-1 reverse transcriptase. J. Mol. Biol. 2000;300:403–418. doi: 10.1006/jmbi.2000.3823. [DOI] [PubMed] [Google Scholar]
- 24.Scatena R, Bottoni P, Botta G, Martorana GE, Giardina B. The role of mitochondria in pharmacotoxicology: a reevaluation of an old, newly emerging topic. Am. J. Physiol. Cell Physiol. 2007;293:C12–21. doi: 10.1152/ajpcell.00314.2006. [DOI] [PubMed] [Google Scholar]
- 25.Lim SE, Copeland WC. Differential incorporation and removal of antiviral deoxynucleotides by human DNA polymerase gamma. J. Biol. Chem. 2001;276:23616–23623. doi: 10.1074/jbc.M101114200. [DOI] [PubMed] [Google Scholar]
- 26.Lee H, Hanes J, Johnson KA. Toxicity of nucleoside analogues used to treat AIDS and the selectivity of the mitochondrial DNA polymerase. Biochemistry. 2003;42:14711–14719. doi: 10.1021/bi035596s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naeger LK, Margot NA, Miller MD. Increased drug susceptibility of HIV-1 reverse transcriptase mutants containing M184V and zidovudine-associated mutations: analysis of enzyme processivity, chain-terminator removal and viral replication. Antivir. Ther. 2001;6:115–126. [PubMed] [Google Scholar]
- 28.Tuske S, Sarafianos SG, Clark AD, Jr, Ding J, Naeger LK, White KL, Miller MD, Gibbs CS, Boyer PL, Clark P, Wang G, Gaffney BL, Jones RA, Jerina DM, Hughes SH, Arnold E. Structures of HIV-1 RT-DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nat. Struct. Mol. Biol. 2004;11:469–474. doi: 10.1038/nsmb760. [DOI] [PubMed] [Google Scholar]
- 29.Meyer PR, Matsuura SE, So AG, Scott WA. Unblocking of chain-terminated primer by HIV-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc. Natl. Acad. Sci. USA. 1998;95:13471–13476. doi: 10.1073/pnas.95.23.13471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyer PL, Sarafianos SG, Arnold E, Hughes SH. Selective excision of AZTMP by drug-resistant human immunodeficiency virus type-1 reverse transcriptase. J. Virol. 2001;75:4832–4842. doi: 10.1128/JVI.75.10.4832-4842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crumpacker CS. Mechanism of action of foscarnet against viral polymerases. Am. J. Med. 1992;92:3S–7S. doi: 10.1016/0002-9343(92)90329-a. [DOI] [PubMed] [Google Scholar]
- 32.Yang SS, Fliakas-Boltz V, Bader JP, Buckheit RW., Jr. Characteristics of a group of nonnucleoside reverse transcriptase inhibitors with structural diversity and potent anti-human immunodeficiency virus activity. Leukemia. 1995;9:S75–85. [PubMed] [Google Scholar]
- 33.Maga G, Ubiali D, Salvetti R, Pregnolato M, Spadari S. Selective interaction of the human immunodeficiency virus type 1 reverse transcriptase nonnucleoside inhibitor efavirenz and its thio-substituted analog with different enzyme-substrate complexes. Antimicrob. Agents Chemother. 2000;44:1186–1194. doi: 10.1128/aac.44.5.1186-1194.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spence RA, Kati WM, Anderson KS, Johnson KA. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science. 1995;267:988–993. doi: 10.1126/science.7532321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren J, Bird LE, Chamberlain PP, Stewart-Jones GB, Stuart DI, Stammers DK. Structure of HIV-2 reverse transcriptase at 2.35-A resolution and the mechanism of resistance to non-nucleoside inhibitors. Proc. Natl. Acad. Sci. USA. 2002;99:14410–14415. doi: 10.1073/pnas.222366699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casado JL, Hertogs K, Ruiz L, Dronda F, Van Cauwenberge A, Arnó A, Garcia-Arata I, Bloor S, Bonjoch A, Blazquez J, Clotet B, Larder B. Non-nucleoside reverse transcriptase inhibitor resistance among patients failing a nevirapine plus protease inhibitor-containing regimen. AIDS. 2000;14:F1–7. doi: 10.1097/00002030-200001280-00001. [DOI] [PubMed] [Google Scholar]
- 37.Basavapathruni A, Vingerhoets J, de Bethune MP, Chung R, Bailey CM, Kim J, Anderson KS. Modulation of human immunodeficiency virus type 1 synergistic inhibition by reverse transcriptase mutations. Biochemistry. 2006;45:7334–7340. doi: 10.1021/bi052362v. [DOI] [PubMed] [Google Scholar]
- 38.Udier-Blagović M, Tirado-Rives J, Jorgensen WL. Validation of a model for the complex of HIV-1 reverse transcriptase with nonnucleoside inhibitor TMC125. J. Am. Chem. Soc. 2003;125:6016–6017. doi: 10.1021/ja034308c. [DOI] [PubMed] [Google Scholar]
- 39.Basavapathruni A, Bailey CM, Anderson KS. Defining a molecular mechanism of synergy between nucleoside and nonnucleoside AIDS drugs. J. Biol. Chem. 2004;279:6221–6224. doi: 10.1074/jbc.C300523200. [DOI] [PubMed] [Google Scholar]
- 40.Clementi M. Can modulation of viral fitness represent a target for anti-HIV-1 strategies? New Microbiol. 2004;27:207–214. [PubMed] [Google Scholar]
- 41.Loeb LA, Essigmann JM, Kazazi F, Zhang J, Rose KD, Mullins JI. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc. Natl. Acad. Sci. USA. 1999;96:1492–1497. doi: 10.1073/pnas.96.4.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crotty S, Maag D, Arnold JJ, Zhong W, Lau JY, Hong Z, Andino R, Cameron CE. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- 43.Huang P, Chubb S, Plunkett W. Termination of DNA synthesis by 9-beta-D-arabinofuranosyl-2-fluoroadenine. A mechanism for cytotoxicity. J. Biol. Chem. 1990;265:16617–16625. [PubMed] [Google Scholar]
- 44.Plunkett W, Huang P, Gandhi V. Metabolism and action of fludarabine phosphate. Semin. Oncol. 1990;17:3–17. [PubMed] [Google Scholar]
- 45.Datta R, Kharbanda S, Kufe DW. Transcriptional and posttranscriptional regulation of H1 histone gene expression by 1-beta-D-arabinofuranosylcytosine. Mol. Pharmacology. 1992;41:64–68. [PubMed] [Google Scholar]
- 46.Brueckner B, Kuck D, Lyko F. DNA methyltransferase inhibitors for cancer therapy. Cancer J. 2007;13:17–22. doi: 10.1097/PPO.0b013e31803c7245. [DOI] [PubMed] [Google Scholar]
- 47.Issa JP. CpG island methylator phenotypes in cancer. Nat. Rev. Drug Discov. 2004;4:988–993. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 48.Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, Li GM, Drummond J, Modrich PL, Sedwick WD, Markowitz SD. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc. Natl. Acad. Sci. USA. 1998;95:8698–8702. doi: 10.1073/pnas.95.15.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oki Y, Aoki E, Issa JP. Decitabine--bedside to bench. Crit. Rev. Oncol. Hematol. 2007;61:140–152. doi: 10.1016/j.critrevonc.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 50.Steitz TA. DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- 51.Gentil A, Cabral-Neto JB, Mariage-Samson R, Margot A, Imbach JL, Rayner B, Sarasin A. Mutagenicity of a unique apurinic/apyrimidinic site in mammalian cells. J. Mol. Biol. 1992;227:981–984. doi: 10.1016/0022-2836(92)90513-j. [DOI] [PubMed] [Google Scholar]
- 52.Strauss BS. The “A” rule revisited: polymerases as determinants of mutational specificity. DNA Repair (Amst) 2002;1:125–135. doi: 10.1016/s1568-7864(01)00014-3. [DOI] [PubMed] [Google Scholar]
- 53.Zhang X, Lee I, Berdis AJ. A potential chemotherapeutic strategy for the selective inhibition of promutagenic DNA synthesis by nonnatural nucleotides. Biochemistry. 2005;44:13111–13321. doi: 10.1021/bi050584n. [DOI] [PubMed] [Google Scholar]
- 54.Devadoss B, Berdis AJ. Non-natural nucleotide analogs as probes of DNA polymerase activity. Current Chemical Biology. 2007;1:241–264. [Google Scholar]
- 55.Sheriff A, Motea E, Lee I, Berdis AJ. The mechanism and dynamics of translesion DNA synthesis catalyzed by the Escherichia coli Klenow fragment. Biochemistry. 2008 doi: 10.1021/bi800324r. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]