

Abstract
Clostridium difficile causes serious and potentially fatal inflammatory diseases of the colon. Two large protein toxins, TcdA and TcdB, have been clearly implicated in pathogenesis. The goal of this study was to determine whether the glucosyltransferase activity of the toxins is critical for the induction of tumor necrosis factor-α (TNF-α), an important cytokine mediating both local and systematic inflammatory response. A dose-dependent TNF-α secretion was demonstrated in murine macrophage cell line RAW 264.7 after exposure to TcdA or TcdB. TNF-α production was blocked by anti-toxin antibodies, indicating that the cytokine-driven response is mediated by the toxins. Both toxins disrupted the cytoskeleton of host cells, while cytoskeleton disruptions using cytochalasin D and latrunculin B did not affect TNF-α production. The TNF-α synthesis was inhibited by reagents that target clathrin-dependent endocytosis or prevent endosomal acidification, suggesting that the endocytosis pathway is necessary for the induction of TNF-α. Furthermore, knockout of the enzymatic activity by mutating two key amino acids in the catalytic domain of TcdA abolished its cytokine-inducing activity. Our studies demonstrated a crucial role of the glucosyltransferase activity of C. difficile toxins in the induction of TNF-α in macrophages.
Keywords: Clostridium difficile, toxin A (TcdA), toxin B (TcdB), glucosyltransferase, macrophage, tumor necrosis factor-alpha (TNF-α)
Introduction
Clostridium difficile is a Gram-positive, spore-forming, strict anaerobic microorganism which is responsible for one-quarter of the cases of antibiotic-associated diarrhea and all pseudomembranous colitis in humans [1, 2]. C. difficile infection is highly prevalent in hospitals and nursing homes where patients frequently receive antibiotics and represents one of the most common hospital infections with rapidly escalating annual health care costs in the United States [3-5]. More recently, a hyper-virulent strain of C. difficile has emerged in Europe, the USA and Canada [6].
Two high-molecular-weight protein toxins, toxin A (TcdA) and toxin B (TcdB) are major virulent factors released by C. difficile [7]. TcdA and TcdB share a high amino acid homology and exhibit a very similar domain structure including the N-terminal glucosyltransferase domain (GT), the newly identified cysteine proteinase domain (CPD), the central translocation domain covering a hydrophobic region (HR), and the N-terminal receptor binding domain (RBD) consisting of clostridial repetitive oligopeptides (CROPs) [8-14]. TcdA and TcdB are able to mono-glucosylate Rho GTPases including Rho (A, B, C), Rac1 and Cdc42 [12]. Toxin-mediated glucosylation inactivates the Rho GTPases, leading to disruption of cytoskeletal integrity (cytopathic effect) and cell death (cytotoxic effect) [15, 16]. TcdA and TcdB may utilize distinct cell surface receptors which mediate distinct cell signaling pathways [17-19]. Previous studies in animals have suggested only TcdA causes inflammation and intense fluid accumulation [20-22], but recent study has described the enterotoxic and proinflammatory activities of TcdB in human intestinal xenografts in severe combined immunodeficient (SCID) mice [23]. Furthermore, the TcdA-B+C. difficile strains are responsible for pseudomembranous colitis in some patients [24].
It is now clear that TcdA and TcdB can elicit the production of immune mediators, leading to subsequent neutrophil infiltration and severe colitis [25, 26]. TcdA induces the production of IL-6, IL-8 by human intestinal epithelial cells [23, 27, 28] and IL-1, IL-6, IL-8, TNF-α by human monocytes [29, 30]. However, it remains unclear whether or not the glucosyltransferase activity of the toxins is essential for the induction of the proinflammatory cytokines. The ability of C. difficile toxins to induce the release of immune mediators by epithelial cells and immune cells may govern the inflammatory process of the intestine. Macrophages are key sources of inflammatory mediators including prostaglandins, leukotriene B4, IL-1, IL-8, tumor necrosis factor-α (TNF-α) and nitric oxide (NO). Of these mediators, TNF-α is one of the central mediators of inflammation and plays a critical role in host response to infection and cell injury [31]. Only a few reports documented TNF-α production by murine peritoneal macrophages in response to TcdA or TcdB treatments [32-34]. In the present study, we used RAW 264.7 macrophages to dissect the role of TcdA in the induction of TNF-α. By application of a mutant TcdA with deficient enzyme activity, we demonstrated that the glucosyltransferase activity of C. difficile toxins was required for the induction of TNF-α in RAW 264.7 macrophages.
Materials and methods
Cell culture
The murine macrophage cell line RAW 264.7 was obtained from the American Type Culture Collection (ATCC). Monoclonal HEK293 cells expressing human Toll-like receptor 2 (TLR2) was kindly provided by Dr. Douglas Golenbock (UMass Medical Center). The cells were transfected with pNiFty-SEAP (Invivogen, San Diego, CA) that carries a gene for secretory alkaline phosphatase (SEAP) under NF-κB promoter. This new monoclonal cell line was designated as hT2Y. HEK-Blue™ cells were purchased from InvivoGen. All cells were cultured under standard conditions in Dulbecco's Modified Eagle Medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine and 1mM pyruvate. For growing hT2Y cells, antibiotics zeocin (400 μg/ml) and G418 (500 μg/ml) were supplemented in the medium. HEK-Blue™ cells were cultured in the media supplemented with undisclosed antibiotics from InvivoGen.
Preparation of toxins
Recombinant TcdA and TcdB [35], and the glucosyltransferase-deficient mutant TcdA D285/287N (mTcdA) [36] were purified from cell lysate of Bacillus megaterium as described previously [35]. Briefly, after a Ni-affinity purification of crude bacterial extract, TcdA was further purified via a thyroglobulin-affinity chromatography and TcdB through an ion-exchange chromatography. The purity of these recombinant toxins was assessed by gel electrophoresis, and the protein with only a single band on the SDS-PAGE gel [35] was used for this study. Native TcdA was purified from C. difficile strain VPI 10463 (kindly provided by Abraham L. Sonenshein, Tufts University School of Medicine) as described previously [37].
Determination of endotoxin and TLR2 stimuli in toxin preparations
To determine whether the recombinant toxins contain endotoxins and TLR2 ligands, we utilized bioassays. LPS detection was performed using HEK-Blue™ LPS detection kit (InvivoGen) according to the manufacturer's instruction. TLR2 ligand contamination was determined using hT2Y cells. The cells were incubated at 37°C for 24 h with various concentrations of toxins. The TLR2 signaling was monitored by the expression of a reporter SEAP (secretory alkaline phosphatase) under an NF-κB promoter. Heat-killed Listeria monocytogenes was used as a positive control. The amount of SEAP secreted into culture media was determined by SEAP Reporter Assay (Cat#rep-sap, Invivogen) according to the manufacturer's instructions. No detectable LPS or TLR2 ligand activity was observed when up to 200 ng/ml of TcdA or 20 ng/ml of TcdB were used (data not shown). These highly purified recombinant holotoxins were used for subsequent studies.
Antibodies and reagents
Antibodies used in this study include monoclonal anti-Rac1 (clone 102, BD Biosciences Pharmingen), monoclonal anti-β-actin (clone AC-15, Sigma), anti-TNF-α (Alexa Fluor 647 conjugates, BD Biosciences), monoclonal anti-TcdA (clone A1H3, generated in our laboratory), monoclonal anti-TcdB (clone 5A8-E11, Meridian Life Science, Inc., Memphis, TN), and goat anti-TcdA and TcdB polyclonal antibodies (Techlab Inc., Blacksburg, VA). Brefeldin A, Cytochalasin-D, chlorpromazine hydrochloride, chloroquine diphosphate salt, phenylarsine oxide (PAO) and bafilomycin A1 (BAF) were purchased from Sigma (St. Louis, MO). Latrunculin B was purchased from Invitrogen (Carlsbad, CA).
Enzyme linked immunosorbent sandwich assay (ELISA)
RAW 264.7 cells (3 × 105) were seeded into 24-well plates and incubated at 37°C overnight. Cells were exposed to various concentrations of toxins or LPS (1 μg/ml) for 18 h. The concentration of TNF-α in the supernatants was determined by an ELISA kit (Catlog # CMC3013, Invitrogen) according to the manufacture's instructions.
Immunofluorescence staining
RAW 264.7 cells on coverslips were exposed to TcdA (50 ng/ml) or mTcdA (50 ng/ml) at 37°C for 30 min in case of toxin staining or 2 h for F-actin staining. Cells were fixed with 4% formaldehyde for 15 min at room temperature followed by permeabilization with 0.1% Triton X-100 in PBS for another 15 min. For toxin staining, cells were incubated with anti-TcdA A1H3 followed by a fluorochrome-conjugated secondary antibody staining. The actin cytoskeleton was stained with Alexa 568-phalloidin at 1 μg/ml for 30 min at room temperature. Final counterstaining of the nucleoli with DAPI (0.5 μg/ml) was performed at room temperature for 3 min. The coverslips were mounted and imaged under a confocal microscope (Leica LSM TSC SP2 AOBS).
Intracellular cytokine staining
RAW 264.7 cells were treated with various doses of TcdA or TcdB in the presence or absence of different chemicals. Brefeldin A (20 μM) was added to block cytokine secretion from cells after 3 h treatment. Cells were fixed and permeabilized and then stained with a fluorochrome-conjugated anti-TNF-α antibody. The frequency of TNF-α-producing cells was determined by flow cytometry analysis using FACSCalibur and CellQuest software (BD Biosciences, Mountain View, CA).
Immunoblotting
Protein samples were resolved by SDS polyacrylamide gel, transferred onto nitrocellulose membrane, and blotted with primary (monoclonal anti-Rac1 1:500, monoclonal anti-β-actin 1:10000) and secondary (HRP-conjugated goat-anti-mouse IgG 1:2500) antibodies. The monoclonal anti-Rac1 antibody only recognizes non-glucosylated Rac1 protein [16, 38]. The protein bands were visualized by an enhanced chemiluminescence assay (ECL, Amersham Biochiences).
Results
TcdA and TcdB stimulate TNF-α release by RAW 264.7 macrophages
Previous studies have shown that murine peritoneal macrophages produce TNF-α after exposure to TcdA or TcdB [32]. Here, we have found that a murine macrophage cell line RAW 264.7 produced copious amount of TNF-α after treatments with either TcdA or TcdB. Exposure of RAW 264.7 cells to recombinant TcdA or TcdB induced a dose-dependent release of TNF-α within 18 h incubation as determined by ELISA (Fig. 1A) or intracellular cytokine staining after 6 h of toxin exposure (Fig. 1B and C). TcdB was more potent than TcdA in stimulating TNF-α release since a much lower dose of TcdB was required for the induction of a response similar to that following TcdA treatment (Fig. 1A and C). LPS induced a higher amount of TNF-α production by the macrophages than both toxins (Fig. 1A and B). The purified native TcdA from C. difficile culture supernatant showed a comparable TNF-α production to recombinant TcdA in RAW 264.7 cells (data not shown). Recombinant TcdA was used in all follow-up experiments unless otherwise specified.
Fig. 1. TNF-α release by RAW 264.7 cells stimulated with TcdA or TcdB.
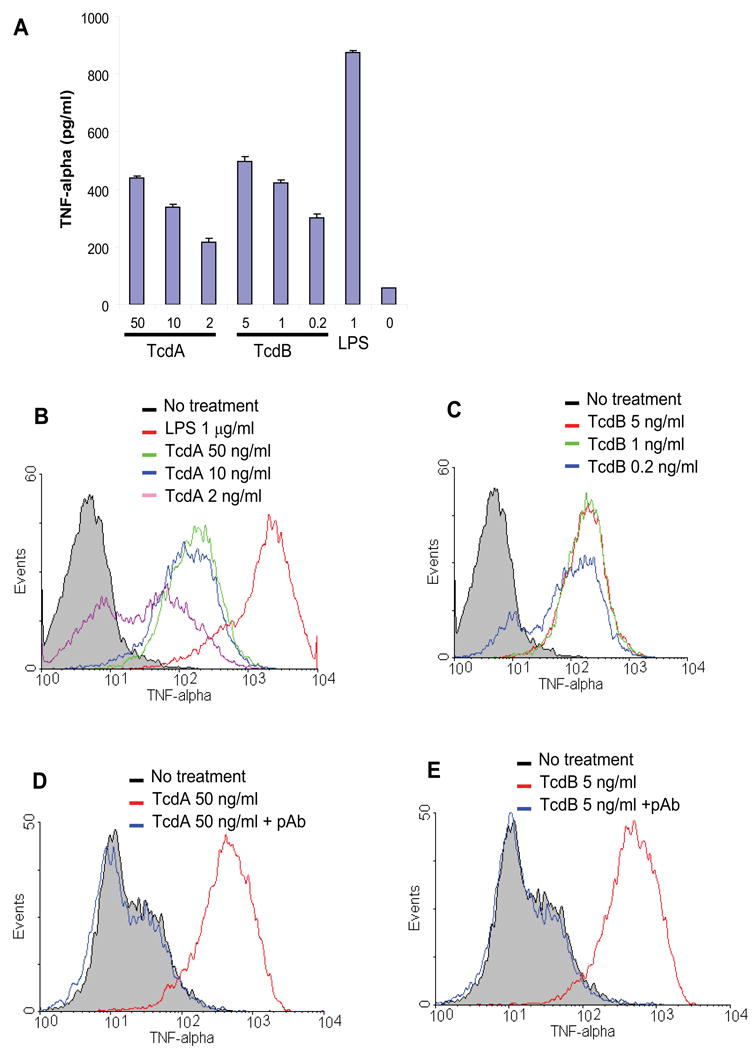
(A) RAW 264.7 cells were cultured for 18 h in the presence of TcdA or TcdB at the indicated doses (ng/ml). Control groups included LPS at 1 μg/ml and medium alone. TNF-α concentration in the medium was determined by ELISA. Data are mean ± SD (n=3). (B) RAW 264.7 cells were treated with TcdA at 50 ng/ml (green line), 10 ng/ml (blue line) or 2 ng/ml (purple line) for 6 h. (C) RAW 264.7 cells were treated with TcdB at 5 ng/ml (red line), 1 ng/ml (green line) or 0.2 ng/ml (blue line) for 6 h. Control groups include no treatment (grey line) or LPS (1 μg/ml, red line in (A)). (D and E) RAW 264.7 cells were treated with TcdA (50 ng/ml) (D) or TcdB (5 ng/ml) (E) for 6 h in the presence (blue line) or absence (red line) of a neutralizing antibody against both TcdA and TcdB. TNF-α was measured by an intracellular cytokine staining followed by flow cytometry analysis.
To determine whether the cytokine induction was indeed caused by toxins, we utilized antibody neutralization assays. Goat polyclonal antibodies against both TcdA and TcdB completely blocked the TNF-α production by RAW 264.7 cells treated with TcdA (Fig. 1D) or TcdB (Fig. 1E), whereas goat normal serum had no blocking effects (data not shown). These data suggest that the stimulation of TNF-α release is not caused by possible contaminants, but rather toxin-specific.
TNF-α production is not induced by the cytoskeleton disruption
Both TcdA and TcdB are glucosyltransferases that mono-glucosylate Rho GTPases including RhoA, Rac1 and Cdc42, leading to their inactivation [7]. The predominant cytopathic effect of TcdA and TcdB is the breakdown F-actin accompanied by a complete loss of cell morphology leading to cell rounding. As shown in Figure 2B, treatment of RAW 264.7 cells with TcdA (50 ng/ml) for 2 h resulted in disintegration of the cytoskeleton and the diminishing of actin stress fibers, as compared to medium control (Fig. 2A). In contrast, the same dose of a mutant form of TcdA (mTcdA), which has point mutations in the enzymatic domain (TcdA D285/287N) and is consequently deficient in glucosyltransferase activity [16], had no effects on the cytoskeleton of RAW 264.7 cells (Fig. 2C).
Fig. 2. TcdA treatment induced the disruption of cytoskeleton in RAW 264.7 cells.
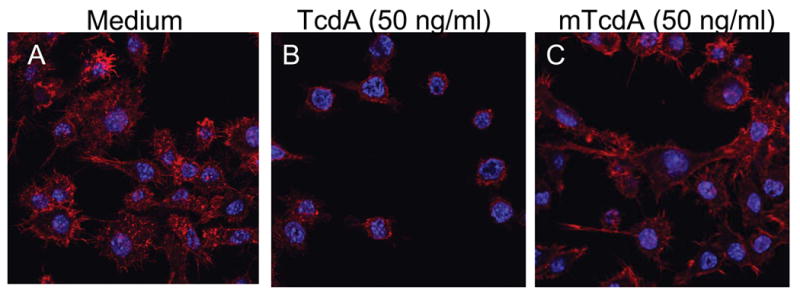
RAW 264.7 cells remained untreated (A) or were exposed to TcdA (50 ng/ml) (B) or mTcdA (50 ng/ml) (C) for 2 h at 37°C. The F-actin of treated cells was stained with Alexa 568-phalloidin. Nucleoli were stained with DAPI. The cells were visualized under a confocal microscope.
We wished to know whether cytoskeleton breakdown might contribute to the activation of TNF-α production. To this end, RAW 264.7 cells were exposed to the pharmacologic agent Cytochalasin-D, known to block actin polymerization by binding to the barbed (plus) end of the actin filament [39]. As expected, treatment of RAW 264.7 cells with Cytochalasin-D at 3 μg/ml resulted in cell rounding in 2 h (data not shown). However, it failed to induce TNF-α production as measured by flow cytometry analysis (Fig. 3A). Another chemical, latrunculin B, which inhibits actin polymerization by a mechanism different from Cytochalasin-D, did not induce TNF-α secretion either (Fig. 3B). The exposure of RAW 264.7 cells to either Cytochalasin-D or Latrunculin B up to 24 h failed to induce TNF-α (data not shown). These data indicate that the remodeling of the cytoskeleton does not necessarily lead to TNF-α production.
Fig. 3. TNF-α production is not induced by the disruption of cytoskeleton.
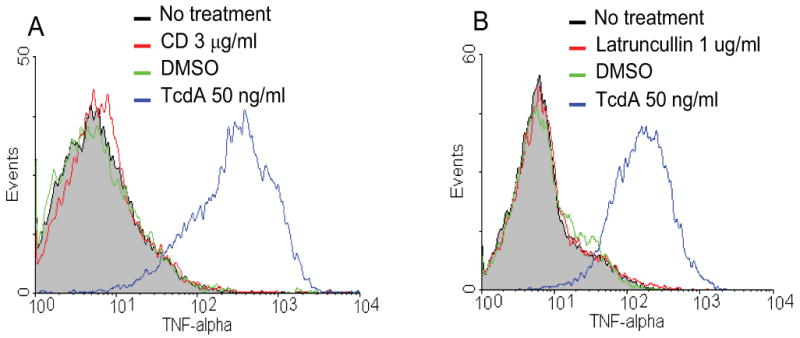
RAW 264.7 cells were incubated with cytochalasin-D (CD) at 3 μg/ml (A, red line) or latrunculin B at 1 μg/ml (B, red line) at 37°C for 6 h. Control groups include no treatment (grey line), DMSO (vehicle, green line) or TcdA (50 ng/ml, blue line). Cells were harvested and TNF-α was determined by flow cytometry analysis.
Cellular internalization of TcdA is required for TNF-α secretion
Both TcdA and TcdB are thought to bind to specific cellular receptor(s), which mediate their cellular uptake through endocytosis [17, 19, 40]. Lysosomotropic agents such as chloroquine, NH4Cl can inhibit toxin-mediated cytotoxicity, suggesting that the endosomal acidification is involved in toxin internalization [41, 42]. To determine whether or not the interaction of toxin with host cell receptor is sufficient for the induction of TNF-α secretion, RAW 264.7 cells were pretreated with lysosomotropic agents for 30 min prior to the addition of TcdA. All three lysosomotropic agents (i.e., chloroquine, NH4Cl and bafilomycin A1 (BAF)) effectively blocked TcdA-induced TNF-α production (Fig. 4A, 4B and 4D). Moreover, chlorpromazine [43] and phenylarsine oxide (PAO) [44, 45], respective, inhibitors of clathrin-coated pit formation and clathrin-coated vesicle formation/budding, dramatically inhibited TNF-α secretion (Fig. 4B and 4C). Neither lysosomotropic agents nor chlorpromazine affected the binding of TcdA to the surface of the macrophage assessed by immunofluorescent staining (data not shown). Taken together, these data strongly suggest that cellular uptake and internalization through a receptor mediated endocytosis are required for TcdA to effectively induce TNF-α secretion by RAW 264.7 cells.
Fig. 4. TNF-α production induced by TcdA in macrophages is dependent upon the endocytosis pathway.
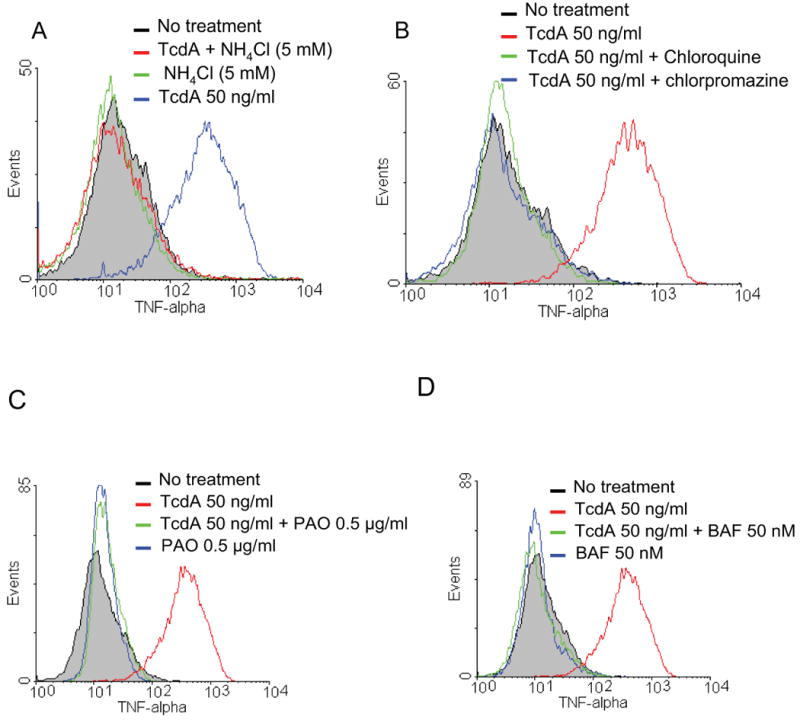
(A) RAW 264.7 cells were pretreated with NH4Cl at 5 mM for 30 min at 37 °C, TcdA (50 ng/ml, red line) or nothing (green line) was then added. (B) RAW 264.7 cells were pretreated with chloroquine (0.1 mM, green line) or chlorpromazine (5 μg/ml, blue line) for 30 min at 37 °C, TcdA (50 ng/ml) was then added. (C) RAW cells were pretreated with phenylarsine oxide (PAO) at 0.5 μg/ml (green line), TcdA (50 ng/ml) or nothing (blue line) was then added. (D) RAW cells were pretreated with bafilomycin A1 (BAF) at 50 nM (green line), TcdA (50 ng/ml) or nothing (blue) was then added. After addition of reagents cells were incubated for an additional 6 h at 37 °C. TNF-α was determined by flow cytometry analysis. TcdA (50 ng/ml) treatments alone were shown in blue in (A) and red in (B), (C) and (D), respectively. Grey lines represent no treatment.
The induction of TNF-α by TcdA is dependent on the enzymatic activity of toxin
After receptor binding and internalization, TcdA releases its enzymatic domain into cytosol where the toxin executes its glucosyltransferase activity [8]. To determine whether the glucosyltransferase activity of TcdA is ultimately responsible for the induction of TNF-α, we took advantage of a mutant form of holotoxin (mTcdA) which has two point mutations in the DXD motif of the catalytic domain [16]. TcdA and mTcdA were compared for their ability in inducing TNF-α from RAW 264.7 cells. As shown in Figure 5A, a dramatic increase of the fluorescence intensity was seen in TcdA (10 ng/ml)-treated macrophages as compared with untreated cells. In contrast, mTcdA at both doses (10 and 50 ng/ml) did not induce a measurable TNF-α synthesis. TNF-α level from RAW 264.7 cells treated with TcdA or mTcdA was also measured by ELISA (Fig. 5B) and the results were consistent to those obtained by intracellular cytokine staining.
Fig. 5. mTcdA treatment of macrophages failed to induce TNF-α production.
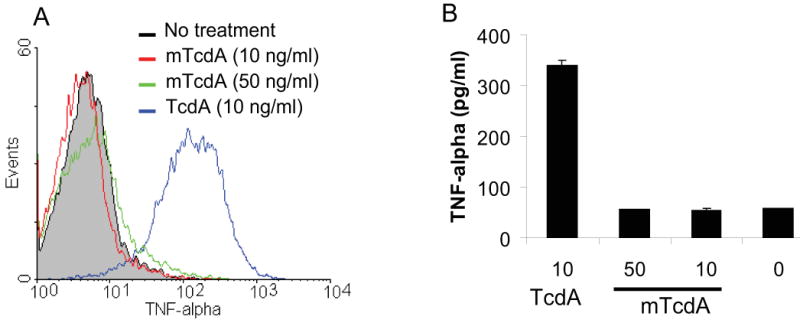
(A) RAW 264.7 cells were incubated with TcdA (10 ng/ml, blue line) or its enzymatic mutant mTcdA at 50 ng/ml (green line) or 10 ng/ml (red line) for 6 h. TNF-α was measured by flow cytometry analysis. Grey line represents no treatment. (B) RAW 264.7 cells were cultured for 18 h in the presence of TcdA or mTcdA at the indicated doses (ng/ml). TNF-α concentration in the medium was determined by ELISA. Data are mean ± SD (n=3).
Although knowledge about toxin uptake process is still fragmentary, it is generally accepted that the first step of toxin action is receptor binding and endocytosis. To rule out the possibility that the dramatic discrepancy between TcdA and mTcdA in the induction of TNF-α resulted from a defective binding by mTcdA, we compared binding and internalization of TcdA and mTcdA by confocal microscopy. As shown in Figure 6, both TcdA and mTcdA bound to and entered into cells similarly well. However, only wild type TcdA exhibited glucosyltransferase activity (Fig. 7). While TcdA showed a time-(Fig. 7A) and dose-dependent (Fig. 7C) glucosylation of Rac1, mTcdA at 50 ng/ml failed to induce any detectable Rac1 glucosylation in RAW 264.7 cells within 8 h of treatment (Fig. 7B). Taken together, these results demonstrate that the glucosyltransferase activity of TcdA is essential for the induction of TNF-α in macrophages.
Fig. 6. Binding and endocytosis of TcdA and mTcdA.
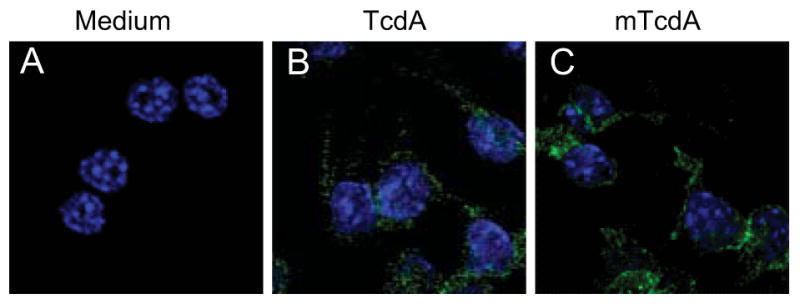
RAW 264.7 cells were exposed to medium (A), or to TcdA (50 ng/ml) (B) or mTcdA (50 ng/ml) (C) for 30 minutes at 37°C. Cells were then permeabilized and incubated with anti-TcdA monoclonal antibody A1H3 followed by staining with Alexa Fluor 488-conjugated goat anti-mouse IgG and DAPI. The cells were visualized under a confocal microscope.
Fig. 7. mTcdA has a deficient glucosyltransferase activity.
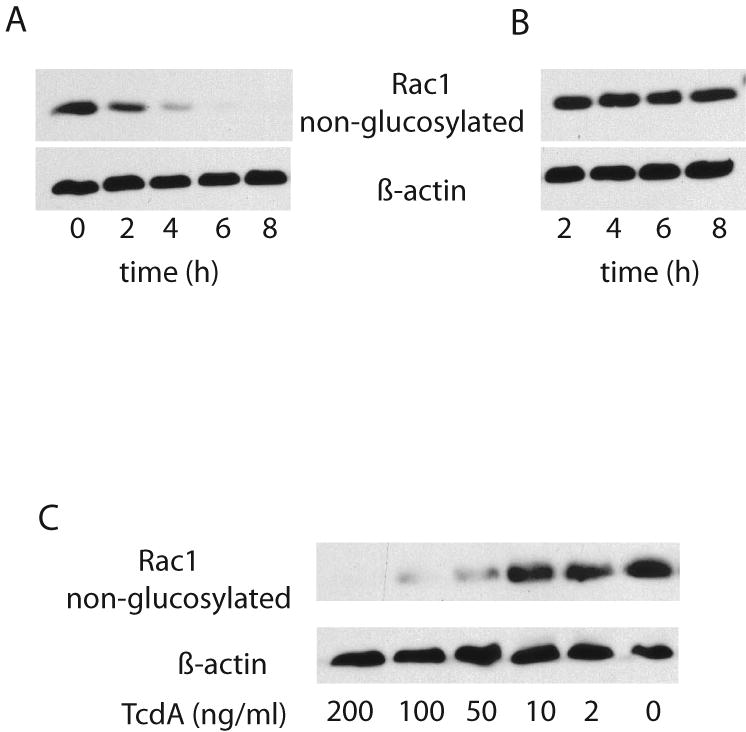
RAW 264.7 cells were treated with TcdA (A) or mTcdA (B) at 50 ng/ml for the indicated time, or treated with TcdA at the indicated doses (C) for 4 h. Cells were harvested and western blot was performed as described in the Materials and Methods. Monoclonal antibody clone 102 only recognizes non-glucosylated Rac1 and has a reduced affinity to glucosylated Rac1. Anti-actin antibody was used to monitor an equal loading of samples.
Discussion
Macrophages play an important role in the pathogenesis of C. difficile toxin-induced colitis [5, 29]. In C. difficile colitis, tissue macrophages may well become exposed to TcdA and TcdB especially when colonic microulceration has developed [46, 47]. One of the most important cytokine mediators released by macrophages is TNF-α, which is a central mediator of inflammation and plays a critical role in host response to infection and tissue injury [48]. However, only a few reports documented C. difficile toxin-induced TNF-α production in murine peritoneal macrophages [32-34]. The underlying mechanism of the induction of TNF-α by C. difficile toxins however has not been investigated. This study showed that both TcdA and TcdB induced TNF-α production in murine macrophages in a dose-dependent manner. TcdB was more potent than TcdA in stimulation of TNF-α, which seems to correlate to the cytotoxic activity of the toxins. We further demonstrated that the glucosyltransferase activity of TcdA is required for the induction of TNF-α in macrophages.
To study the host immune response to the toxins, it is critically important to obtain highly purified TcdA and TcdB free of molecules that potentially modulate immune response. Native toxins can be purified from toxigenic C. difficile culture supernatant. The purification involves multiple steps such as ultrafiltration, ammonium sulfate precipitation, and ion-exchange chromatography [49, 50]. In the case of TcdA, affinity purification through thyroglobulin is available and pure TcdA can be obtained [51]. TcdB, however, is often contaminated with some unknown substance even after extensive purification steps [50]. We therefore generated His(6)-tagged recombinant TcdA and TcdB in B. megaterium system and both toxins were fully active [35]. One of the advantages of using B. megaterium for expressing heterologous proteins is that the bacterium is free of the endotoxins LPS [52, 53], which is important because LPS is a potent TNF-α inducer in RAW 264.7 cells (Fig. 1A and B). To obtain highly purified recombinant toxins, several purification steps were performed, which resulted in recombinant toxins free of detectable LPS or TLR2 ligand contaminants under sensitive bioassays (data not shown).
In addition to the use of highly purified toxins, several lines of evidence also indicated that the TNF-α induction in macrophage was mediated via toxin. First, chemicals such as lysosomotropic agents or chlorpromazine that do not affect TLR signaling completely abolished TNF-α production (Fig. 4). Second, the mutant form of TcdA that was purified in the same way as the wild type recombinant one failed to stimulate TNF-α production (Fig. 5). Third, neutralizing antibodies against TcdA or TcdB completely blocked toxin-induced TNF-α production (Fig 1D and E). Therefore, we believed that TNF-α induction in RAW 264.7 cells was not due to contaminants, but rather mediated by the toxins.
The exposure of RAW 264.7 cells to TcdA inactivated Rho GTPases and disrupted the actin cytoskeleton (Fig. 2). Disruption of the actin cytoskeleton was said to be involved in IL-8 secretion in human intestinal epithelial HT-29 and Caco-2 cells [54]. The integrity of the cytoskeleton also plays a role in LPS-induced TNF-α release in human monocytes [55]. We therefore hypothesized that the alteration of the cytoskeleton can trigger TNF-α production. This hypothesis was disproved by using Cytochalasin D and latrunculin B, both of which disrupt the cytoskeleton through different mechanisms. Cytochalasin-D is known to block actin polymerization by binding to the barbed end of the actin [56], and latrunculin B forms 1:1 complexes with actin monomers and thereby inhibit actin polymerization [57]. Both cytoskeletal disrupting agents did not induce TNF-α secretion (Fig. 3), while they did result in a complete cell rounding at the experimental doses (data not shown). This result is not surprising because cytoskeleton breakdown is only one of the outcomes of signaling pathways affected by toxin-induced Rho/Rac glucosylation. It has been reported that the mere destruction of the actin cytoskeleton by either latrunculin B or the ADP-ribosylating C2 toxin from C. botulinum does not reflect the functional outcome of Rho inhibition by TcdA/TcdB [16, 39]. One recent study has shown that both TcdB and latrunculin B induced the activation of p38 MAPK and ERK1/2 in human mast cells, and TcdB, but not latrunculin B, stimulated a strong IL-8 production [58]. The authors further hypothesized that the strong IL-8 release required a simultaneous activation of two parallel signaling pathways, e.g., NF-κB and p38 MAPK, which may be only driven by TcdB but not latrunculin B.
The C- terminal receptor binding domain of TcdA has an immune modulating activity [59, 60]. Whether or not C-terminus alone is sufficient to induce cytokine production appears to depend upon the specific cell types. The recombinant TcdA fragment containing only the receptor-binding domain was reported to stimulate human epithelial cells [19] or umbilical vein endothelial cells [61] to express cytokines and chemokines. However, in human monocytes the C-terminal binding domain alone is not sufficient for inducing IL-8 secretion, and cellular uptake of the holotoxin is required for such an activity [42]. In this study we have demonstrated that the N-terminal catalytic domain of TcdA with glucosylating activity is essential for TcdA-induced TNF-α production in RAW 264.7 macrophages. This conclusion was drawn based on several lines of evidence. First, TcdA and a glucosyltransferase-deficient mutant TcdA D285/287N (mTcdA) [16] bound to and entered into RAW 264.7 cells equally well (Fig. 6), but mTcdA failed to induce the release of TNF-α. Second, cellular endocytosis and endosomal acidification, necessary steps for the N-terminus of the holotoxin entering into cytosol, are required for TcdA to stimulate TNF-α production in macrophages. The chemicals chlorpromazine and PAO that block endocytosis completely inhibited TcdA-induced cytokine production (Fig. 4B). NH4Cl, chloroquine and bafilomycin A1, all of which block endosomal acidification, also abolished TcdA-induced TNF-α production. Finally, mTcdA that contains an intact C-terminal receptor binding domain could neither glucosylate Rac1 nor induce TNF-α production in RAW 264.7 cells. These data clearly demonstrate that TcdA-induced TNF-α production depends on its glucosyltransferase activity. In agreement of this, the enzymatic activity of TcdA was indicated to be responsible for TcdA-induced IL-8 production in human monocytes [42]. A recent report showed that the enzymatic activity was also involved in IL-8 production in human mast cells stimulated by TcdB [58].
In conclusion, the present study has demonstrated that both TcdA and TcdB can induce TNF-α production in RAW 264.7 macrophages. A receptor-mediated endocytosis pathway is required for the induction of TNF-α, and cell cytoskeleton breakdown is not sufficient to induce TNF-α. TcdA-induced TNF-α secretion is not mediated by cell surface binding, but rather by glucosyltransferase of the N-terminal domain of TcdA.
Acknowledgments
This work was supported by NIH N01AI30050, and in part by NIH K01DK076549 to HF and by the German Research Foundation, SFB621, B5 to RG. The authors thank Weijia Nie for technical supports.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bartlett JG. Historical perspectives on studies of Clostridium difficile and C. difficile infection. Clin Infect Dis. 2008;46 1:S4–11. doi: 10.1086/521865. [DOI] [PubMed] [Google Scholar]
- 2.Johnson S, Gerding DN. Clostridium difficile--associated diarrhea. Clin Infect Dis. 1998;26(5):1027–34. doi: 10.1086/520276. quiz 1035-6. [DOI] [PubMed] [Google Scholar]
- 3.Owens RC, Jr, et al. Antimicrobial-associated risk factors for Clostridium difficile infection. Clin Infect Dis. 2008;46 1:S19–31. doi: 10.1086/521859. [DOI] [PubMed] [Google Scholar]
- 4.Kelly CP, Pothoulakis C, LaMont JT. Clostridium difficile colitis. N Engl J Med. 1994;330(4):257–62. doi: 10.1056/NEJM199401273300406. [DOI] [PubMed] [Google Scholar]
- 5.Pothoulakis C, LaMont JT. Clostridium difficile colitis and diarrhea. Gastroenterol Clin North Am. 1993;22(3):623–37. [PubMed] [Google Scholar]
- 6.Blossom DB, McDonald LC. The challenges posed by reemerging Clostridium difficile infection. Clin Infect Dis. 2007;45(2):222–7. doi: 10.1086/518874. [DOI] [PubMed] [Google Scholar]
- 7.Just I, Gerhard R. Large clostridial cytotoxins. Rev Physiol Biochem Pharmacol. 2004;152:23–47. doi: 10.1007/s10254-004-0033-5. [DOI] [PubMed] [Google Scholar]
- 8.Jank T, Aktories K. Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol. 2008;16(5):222–9. doi: 10.1016/j.tim.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 9.Hofmann F, et al. Localization of the glucosyltransferase activity of Clostridium difficile toxin B to the N-terminal part of the holotoxin. J Biol Chem. 1997;272(17):11074–8. doi: 10.1074/jbc.272.17.11074. [DOI] [PubMed] [Google Scholar]
- 10.Hofmann F, Busch C, Aktories K. Chimeric clostridial cytotoxins: identification of the N-terminal region involved in protein substrate recognition. Infect Immun. 1998;66(3):1076–81. doi: 10.1128/iai.66.3.1076-1081.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Just I, et al. The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J Biol Chem. 1995;270(23):13932–6. doi: 10.1074/jbc.270.23.13932. [DOI] [PubMed] [Google Scholar]
- 12.Just I, et al. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature. 1995;375(6531):500–3. doi: 10.1038/375500a0. [DOI] [PubMed] [Google Scholar]
- 13.von Eichel-Streiber C, Sauerborn M. Clostridium difficile toxin A carries a C-terminal repetitive structure homologous to the carbohydrate binding region of streptococcal glycosyltransferases. Gene. 1990;96(1):107–13. doi: 10.1016/0378-1119(90)90348-u. [DOI] [PubMed] [Google Scholar]
- 14.Jank T, Giesemann T, Aktories K. Rho-glucosylating Clostridium difficile toxins A and B: new insights into structure and function. Glycobiology. 2007;17(4):15R–22R. doi: 10.1093/glycob/cwm004. [DOI] [PubMed] [Google Scholar]
- 15.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279(5350):509–14. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 16.Nottrott S, et al. Clostridium difficile toxin A-induced apoptosis is p53-independent but depends on glucosylation of Rho GTPases. Apoptosis. 2007;12(8):1443–53. doi: 10.1007/s10495-007-0074-8. [DOI] [PubMed] [Google Scholar]
- 17.Na X, et al. gp96 is a human colonocyte plasma membrane binding protein for Clostridium difficile toxin A. Infect Immun. 2008;76(7):2862–71. doi: 10.1128/IAI.00326-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Na X, et al. Clostridium difficile toxin B activates the EGF receptor and the ERK/MAP kinase pathway in human colonocytes. Gastroenterology. 2005;128(4):1002–11. doi: 10.1053/j.gastro.2005.01.053. [DOI] [PubMed] [Google Scholar]
- 19.Pothoulakis C, et al. Rabbit sucrase-isomaltase contains a functional intestinal receptor for Clostridium difficile toxin A. J Clin Invest. 1996;98(3):641–9. doi: 10.1172/JCI118835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lyerly DM, et al. Biological activities of toxins A and B of Clostridium difficile. Infect Immun. 1982;35(3):1147–50. doi: 10.1128/iai.35.3.1147-1150.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyerly DM, et al. Effects of Clostridium difficile toxins given intragastrically to animals. Infect Immun. 1985;47(2):349–52. doi: 10.1128/iai.47.2.349-352.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Triadafilopoulos G, et al. Comparative study of Clostridium difficile toxin A and cholera toxin in rabbit ileum. Gastroenterology. 1989;97(5):1186–92. doi: 10.1016/0016-5085(89)91689-2. [DOI] [PubMed] [Google Scholar]
- 23.Savidge TC, et al. Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine. Gastroenterology. 2003;125(2):413–20. doi: 10.1016/s0016-5085(03)00902-8. [DOI] [PubMed] [Google Scholar]
- 24.Shin BM, et al. Emerging toxin A-B+ variant strain of Clostridium difficile responsible for pseudomembranous colitis at a tertiary care hospital in Korea. Diagn Microbiol Infect Dis. 2007 doi: 10.1016/j.diagmicrobio.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 25.Kelly CP, et al. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J Clin Invest. 1994;93(3):1257–65. doi: 10.1172/JCI117080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelly CP, LaMont JT. Clostridium difficile infection. Annu Rev Med. 1998;49:375–90. doi: 10.1146/annurev.med.49.1.375. [DOI] [PubMed] [Google Scholar]
- 27.Kim JM, et al. Differential expression and polarized secretion of CXC and CC chemokines by human intestinal epithelial cancer cell lines in response to Clostridium difficile toxin A. Microbiol Immunol. 2002;46(5):333–42. doi: 10.1111/j.1348-0421.2002.tb02704.x. [DOI] [PubMed] [Google Scholar]
- 28.Ng EK, et al. Human intestinal epithelial and smooth muscle cells are potent producers of IL-6. Mediators Inflamm. 2003;12(1):3–8. doi: 10.1080/0962935031000096917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linevsky JK, et al. IL-8 release and neutrophil activation by Clostridium difficile toxin-exposed human monocytes. Am J Physiol. 1997;273(6 Pt 1):G1333–40. doi: 10.1152/ajpgi.1997.273.6.G1333. [DOI] [PubMed] [Google Scholar]
- 30.Flegel WA, et al. Cytokine response by human monocytes to Clostridium difficile toxin A and toxin B. Infect Immun. 1991;59(10):3659–66. doi: 10.1128/iai.59.10.3659-3666.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deleault KM, Skinner SJ, Brooks SA. Tristetraprolin regulates TNF TNF-alpha mRNA stability via a proteasome dependent mechanism involving the combined action of the ERK and p38 pathways. Mol Immunol. 2008;45(1):13–24. doi: 10.1016/j.molimm.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 32.Melo Filho AA, et al. Role of tumor necrosis factor and nitric oxide in the cytotoxic effects of Clostridium difficile toxin A and toxin B on macrophages. Toxicon. 1997;35(5):743–52. doi: 10.1016/s0041-0101(96)00172-9. [DOI] [PubMed] [Google Scholar]
- 33.Rocha MF, et al. Clostridium difficile toxin A induces the release of neutrophil chemotactic factors from rat peritoneal macrophages: role of interleukin-1beta, tumor necrosis factor alpha, and leukotrienes. Infect Immun. 1997;65(7):2740–6. doi: 10.1128/iai.65.7.2740-2746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Souza MH, et al. The involvement of macrophage-derived tumour necrosis factor and lipoxygenase products on the neutrophil recruitment induced by Clostridium difficile toxin B. Immunology. 1997;91(2):281–8. doi: 10.1046/j.1365-2567.1997.00243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang G, et al. Expression of recombinant Clostridium difficile toxin A and B in Bacillus megaterium. BMC Microbiology. 2008 doi: 10.1186/1471-2180-8-192. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Teichert M, et al. Application of mutated Clostridium difficile toxin A for determination of glucosyltransferase-dependent effects. Infect Immun. 2006;74(10):6006–10. doi: 10.1128/IAI.00545-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerhard R, et al. Comparison of wild type with recombinant Clostridium difficile toxin A. Microb Pathog. 2005;38(23):77–83. doi: 10.1016/j.micpath.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 38.Genth H, et al. Cellular stability of Rho-GTPases glucosylated by Clostridium difficile toxin B. FEBS Lett. 2006;580(14):3565–9. doi: 10.1016/j.febslet.2006.04.100. [DOI] [PubMed] [Google Scholar]
- 39.Hippenstiel S, et al. Reduction of tumor necrosis factor-alpha (TNF-alpha) related nuclear factor-kappaB (NF-kappaB) translocation but not inhibitor kappa-B (Ikappa-B)-degradation by Rho protein inhibition in human endothelial cells. Biochem Pharmacol. 2002;64(56):971–7. doi: 10.1016/s0006-2952(02)01162-0. [DOI] [PubMed] [Google Scholar]
- 40.Krivan HC, et al. Cell surface binding site for Clostridium difficile enterotoxin: evidence for a glycoconjugate containing the sequence Gal alpha 1-3Gal beta 1-4GlcNAc. Infect Immun. 1986;53(3):573–81. doi: 10.1128/iai.53.3.573-581.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henriques B, Florin I, Thelestam M. Cellular internalisation of Clostridium difficile toxin A. Microb Pathog. 1987;2(6):455–63. doi: 10.1016/0882-4010(87)90052-0. [DOI] [PubMed] [Google Scholar]
- 42.Jefferson KK, Smith MF, Jr, Bobak DA. Roles of intracellular calcium and NF-kappa B in the Clostridium difficile toxin A-induced up-regulation and secretion of IL-8 from human monocytes. J Immunol. 1999;163(10):5183–91. [PubMed] [Google Scholar]
- 43.Sapinoro R, et al. Fc receptor-mediated, antibody-dependent enhancement of bacteriophage lambda-mediated gene transfer in mammalian cells. Virology. 2008;373(2):274–86. doi: 10.1016/j.virol.2007.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moulakakis C, Stamme C. Role of clathrin-mediated endocytosis of surfactant protein A by alveolar macrophages in intracellular signaling. Am J Physiol Lung Cell Mol Physiol. 2009;296(3):L430–41. doi: 10.1152/ajplung.90458.2008. [DOI] [PubMed] [Google Scholar]
- 45.Bild AH, Turkson J, Jove R. Cytoplasmic transport of Stat3 by receptor-mediated endocytosis. Embo J. 2002;21(13):3255–63. doi: 10.1093/emboj/cdf351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Riegler M, et al. Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in vitro. J Clin Invest. 1995;95(5):2004–11. doi: 10.1172/JCI117885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Price AB, Davies DR. Pseudomembranous colitis. J Clin Pathol. 1977;30(1):1–12. doi: 10.1136/jcp.30.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Means TK, et al. Activation of TNF-alpha transcription utilizes distinct MAP kinase pathways in different macrophage populations. J Leukoc Biol. 2000;67(6):885–93. doi: 10.1002/jlb.67.6.885. [DOI] [PubMed] [Google Scholar]
- 49.Sullivan NM, Pellett S, Wilkins TD. Purification and characterization of toxins A and B of Clostridium difficile. Infect Immun. 1982;35(3):1032–40. doi: 10.1128/iai.35.3.1032-1040.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keel MK, Songer JG. The distribution and density of Clostridium difficile toxin receptors on the intestinal mucosa of neonatal pigs. Vet Pathol. 2007;44(6):814–22. doi: 10.1354/vp.44-6-814. [DOI] [PubMed] [Google Scholar]
- 51.Krivan HC, Wilkins TD. Purification of Clostridium difficile toxin A by affinity chromatography on immobilized thyroglobulin. Infect Immun. 1987;55(8):1873–7. doi: 10.1128/iai.55.8.1873-1877.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Malten M, et al. A Bacillus megaterium plasmid system for the production, export, and one-step purification of affinity-tagged heterologous levansucrase from growth medium. Appl Environ Microbiol. 2006;72(2):1677–9. doi: 10.1128/AEM.72.2.1677-1679.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vary PS, et al. Bacillus megaterium--from simple soil bacterium to industrial protein production host. Appl Microbiol Biotechnol. 2007;76(5):957–67. doi: 10.1007/s00253-007-1089-3. [DOI] [PubMed] [Google Scholar]
- 54.Nemeth ZH, et al. Disruption of the actin cytoskeleton results in nuclear factor-kappaB activation and inflammatory mediator production in cultured human intestinal epithelial cells. J Cell Physiol. 2004;200(1):71–81. doi: 10.1002/jcp.10477. [DOI] [PubMed] [Google Scholar]
- 55.Rosengart MR, et al. The actin cytoskeleton: an essential component for enhanced TNFalpha production by adherent monocytes. Shock. 2002;17(2):109–13. doi: 10.1097/00024382-200202000-00005. [DOI] [PubMed] [Google Scholar]
- 56.Haberzettl P, et al. Actin plays a crucial role in the phagocytosis and biological response to respirable quartz particles in macrophages. Arch Toxicol. 2007;81(7):459–70. doi: 10.1007/s00204-007-0178-5. [DOI] [PubMed] [Google Scholar]
- 57.Wakatsuki T, et al. Effects of cytochalasin D and latrunculin B on mechanical properties of cells. J Cell Sci. 2001;114(Pt 5):1025–36. doi: 10.1242/jcs.114.5.1025. [DOI] [PubMed] [Google Scholar]
- 58.Meyer GK, et al. Clostridium difficile toxins A and B directly stimulate human mast cells. Infect Immun. 2007;75(8):3868–76. doi: 10.1128/IAI.00195-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Castagliuolo I, et al. Clostridium difficile toxin A carboxyl-terminus peptide lacking ADP-ribosyltransferase activity acts as a mucosal adjuvant. Infect Immun. 2004;72(5):2827–36. doi: 10.1128/IAI.72.5.2827-2836.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pavliakova D, et al. Clostridium difficile recombinant toxin A repeating units as a carrier protein for conjugate vaccines: studies of pneumococcal type 14, Escherichia coli K1, and Shigella flexneri type 2a polysaccharides in mice. Infect Immun. 2000;68(4):2161–6. doi: 10.1128/iai.68.4.2161-2166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yeh CY, et al. C-terminal repeats of Clostridium difficile toxin A induce production of chemokine and adhesion molecules in endothelial cells and promote migration of leukocytes. Infect Immun. 2008;76(3):1170–8. doi: 10.1128/IAI.01340-07. [DOI] [PMC free article] [PubMed] [Google Scholar]