

Abstract
Introduction
Familial tumoral calcinosis (FTC) and hyperostosis-hyperphosphatemia syndrome (HHS) are caused by mutations in FGF23, GALNT3, or KLOTHO. They are characterized by hyperphosphatemia, increased phosphate reabsorption, and elevated or inappropriately normal serum 1, 25-dihydroxyvitamin D3 (1,25-D); FTC is associated with calcific masses, and HHS with diaphyseal hyperostosis.
Methods
A 36-year-old woman presented with abnormal dental x-rays at age 12, and was hyperphosphatemic at 22. She underwent radiographic, biochemical and genetic testing, and medical treatment.
Results
Serum phosphorus was 7.3 mg/dl (2.5-4.8), TmP/GFR 6.99 mg/100 ml (2.97-4.45), 1,25-D3 35 pg/ml (22-67). Radiographs revealed tooth anomalies, thyroid cartilage calcification, calcific masses in vertebral spaces, calcification of the interstitial septae of the soft tissue in the lower extremities, and cortical thickening of the long bones. Her total hip Z-Score was 1.9. C-terminus serum FGF23 was 1210 RU/ml (20-108), but intact FGF23 was 7.4 pg/ml (10-50). DNA sequencing determined she was a compound heterozygote for mutations in GALNT3. Treatment with niacinamide and acetazolamide decreased TmP/GFR and serum phosphate, which was paralleled by a decrease in serum C-terminus FGF23.
Conclusions
This case broadens the spectrum of phenotypic and genotypic features of FTC/HHS, and suggests treatments to decrease renal phosphate reabsorption in the setting of a low intact FGF23.
Introduction
Familial tumoral calcinosis (FTC) and hyperostosis-hyperphosphatemia syndrome (HHS) are rare disorders that are characterized by hyperphosphatemia due to increased renal phosphate reabsorption, and elevated or inappropriately normal serum 1,25-dihydroxyvitamin D3 (1, 25-D) levels(1,2). Phenotypically, these two disorders have been distinguished by the fact that FTC is associated with subcutaneous calcific masses, and HHS with painful diaphyseal hyperostosis. Recently it has been shown that FTC can be due to mutations in fibroblast growth factor-23 (FGF23)(1), N-acetylgalactosaminyltransferase 3 (GALNT3)(2), or KLOTHO.(3) Those cases that have been phenotypically characterized as HHS have only been shown to be caused by mutations in GALNT3.(4-7) Common to all the patients with disease caused by these mutations is the fact that they have very low serum levels of intact FGF23, but high levels of C-terminus FGF23. FGF23 is secreted by bone cells(8), and it is believed that the intact form of FGF23 acts at the kidney to regulate phosphate reabsorption and vitamin D production.(9) GALNT3 is a glycosyltransferase that is presumed to be involved in FGF23 glycosylation (10,11), and KLOTHO is a co-receptor for FGF23.(12) Significant phenotypic overlap exists between FTC and HHS, and it is possible that these disorders represent not so much distinct entities, but phenotypic variations of disruption of the FGF23/GALNT3/KLOTHO axis. In this study we present a new case of FTC/HHS. Careful characterization revealed a phenotype with aspects of both FTC and HHS, as well as several novel findings. Genetic testing revealed a novel compound heterozygous genotype of two previously described mutations in GALNT3.
Patient and Methods
The patient was a 36-year-old Caucasian woman of French-Canadian descent who presented at the age of 12 with an asymptomatic abnormal dental radiographs showing hypercalcification of multiple tooth roots. Starting at age 17, she complained of bilateral knee pain and swelling, and several years later developed discomfort in her hands. These symptoms waned with time and were not present on the current examination. Hyperphosphatemia was not noted until the age of 22. Subsequently, she had been treated intermittently with calcium carbonate as a phosphate binder and a low phosphorus diet. Since 2000, her phosphate level had consistently been documented to be in the 6-7 mg/dl range. She had no other signs or symptoms of FTC or HHS. At the age of 34, a 1.5 cm thyroid nodule was detected on routine examination. It proved to be a focus of stage 2 papillary thyroid cancer, for which she was treated with thyroidectomy, radioactive iodine, and TSH suppression. Her family history was negative for disorders of mineral metabolism. Serum phosphorus was checked in her parents and one of the two siblings and was found to be normal.
Her height and weight were 149 cm and 62.6 kg. She was normotensive, and general appearance was that of a normal female without dysmorphic features. On examination, a firm nodule on the right lower gum line and a solid submental calcification of the salivary gland were noted. The remainder of the physical examination was normal. She underwent radiographic, biochemical and genetic testing.
In an effort to determine whether renal phosphate reabsorption could be decreased, she was treated with niacinamide and acetazolamide, both of which have been shown to decrease renal phosphate reabsorption. She received two days of niacinamide, 500 mg twice a day, followed by a 24 hour washout period, and was then treated for three days with acetazolamide, 500 mg twice daily.
Results
Serum phosphorus was 7.3 mg/dl (2.5-4.8), TmP/GFR 6.99 mg/100 ml (2.97-4.45), 1,25-vitamin D3 35 pg/ml (22-67), intact PTH 23 pg/ml (16-87), serum calcium was 2.2 mmol/L, (2.05-2.5), albumin 3.4 g/dl, (3.7-4.7), 25-vitamin D 21ng/ml, creatinine and BUN levels were normal. Alkaline phosphatase was 37u/l (37-116), 24- urine Ca 4.69 mmol/24h (1.25-6.25), 24- urine creatinine 1.29 g/24h(0.8-1.8), 24-urine phosphorus 0.41 g/24h(0.4-1.30), TSH 0.28 mIU/ml(0.4-4), free T4 1.5 ng/dl(0.8-1.9). Serum sodium, chloride, potassium and magnesium were normal.
Panoramic and periapical dental radiographs revealed normal enamel but anomalies of the roots and dentin were evident. Anomalies of root formation included dramatically shortened maxillary premolar roots and mid-root enlargement prominently affecting the premolars and canines (Fig. 1A). Root dilacerations were evident on several molar and premolar teeth. The dental pulps were significantly altered, showing unique thistle shapes in the upper third of the root. The apical extents of the thistle shaped pulps were delineated by radiopacities below which the root canal was again apparent (Fig. 1B). The molars showed an apical extension of the crown body at the expense of the roots.
Figure 1.


Radiographic findings. A. Periapical radiograph of maxillary right sextant showing: shortened premolar (solid white arrow), mid root bulge on canine tooth (dashed arrow), and root dilacerations (arrowheads). B. Periapical radiograph of mandibular right sextant showing thistle-shaped pulps of premolars and canine teeth (small white arrows), mid root bulges (dashed arrows), and apical extension of molar crown (thick white arrow). Molar pulps and root canals appear mineralized (asterisk), and are not evident. C. Normal maxillary sextant for comparison. D. Normal mandibular sextant for comparison. E. Age-inappropriate prominent calcification of the thyroid cartilage (arrow). F. Discrete calcification of the interstitial septae of the subcutaneous soft tissue in the calves (arrows) adjacent to the tibia (thick black arrow). G. Mild femoral cortical thickening, more prominent in the diaphyses (arrows). H. Round, small calcifications in the disk spaces of C2-3, L2-3,L3-4, and L5-S1(arrows).
There was age-inappropriate prominent calcification of the thyroid cartilage (Fig. 1C) and anterior costal cartilage of the lower ribs (not shown). There were multiple foci of vertebral disc calcification; involving the nucleus pulposis and the annulus fibrosis (Fig. 1F). There was discrete calcification of the interstitial septae in the calves (Fig. 1D). The cortices of the long bones, particularly the tibia and fibula, were diffusely and mildly thickened (Fig. 1E).
Bone density showed a total hip Z-Score of 1.9, AP spine Z-Score 0.3, left forearm Z-Score 1.0. There were no calcific masses or evidence of coronary calcifications as determined by CT calcium scoring.
Results of gene testing identified a previously reported benign polymorphism in exon 3 of FGF-23, P195S, (dbSNP # rs13312793), and compound heterozygous p.R438C and p.Q592X mutations in GALNT3 as etiologic for the disease (Fig. 2A). These mutations result in a stop codon at position 592, and a missense mutation known to be causative for HHS, as previously identified in a report by Olauson et. al. (13).
Figure 2.
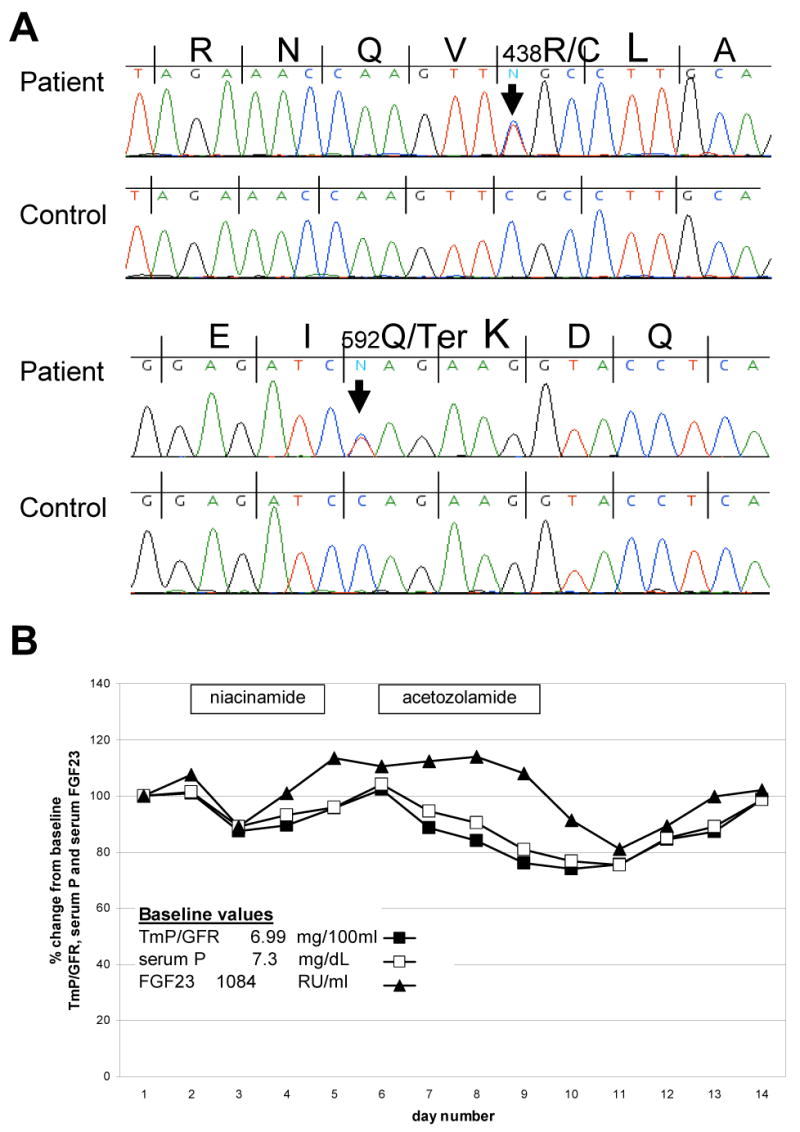
Results of GALNT3 Gene Sequencing and Response to Niacinamide and acetazolamide treatment. A. DNA sequencing of GALNT3 revealed a C→T transversion resulting in the replacement of an arginine with a cysteine at the position 438 (R438C) and a nonsense mutation (C→T) at position 592, resulting in a premature stop codon (Q592X). B. The percent change in TmP/GFR, serum phosphorus and C-terminus FGF23 response to treatment with niacinamide and acetazolamide are shown. The treatment periods are indicated by the boxes. There were responses in TmP/GFR and phosphorus to treatment that were accompanied by changes in serum C-terminus FGF23, which, in general, paralleled the changes in TmP/GFR and serum phosphorus.
Consistent with all other reported cases of FTC/HHS, serum FGF-23 was elevated when measured by a C-terminus assay (Immunotopics, Inc, CA), 1210 RU/ml (20-108), but low on an intact assay (Kainos, Tokyo, Japan), 7.4 pg/ml (10-50). Daily serum phosphorus, calcium, and serum FGF23 (intact and C-terminus) were monitored during a trial of nicotinic acid and acetazolamide (Fig. 2B). There was a minor decrease in TmP/GFR and serum phosphorus in response to niacinamide treatment, and a slightly greater decrease in response to acetazolamide treatment. During niacinamide and acetazolamide treatment, there was a variable change in C-terminus FGF23 levels, but in general the levels paralleled the changes in serum phosphorus (Fig. 2B).
Conclusions
The biochemical hallmarks of FTC/HHS are hyperphosphatemia, due to increased reabsorption of phosphate at the kidney, and inappropriately normal or elevated levels of 1,25-D3. FTC and HHS can be viewed as the clinical converse of the FGF23 overproduction syndromes, X-linked hypophosphatemia (14) and autosomal dominant hypophosphatemic rickets.(15) The C-terminal FGF-23 level is significantly increased in affected individuals with tumoral calcinosis. However, in spite of significantly increased concentrations of C-terminus FGF23, renal phosphate reabsorption and 1,25-D generation are low, suggesting that intact FGF23, is the active hormone regulating phosphate and vitamin D metabolism at the kidney.
The disease severity of tumoral calcinosis is quite variable, ranging from isolated eyelid calcifications to massive periarticular calcifications. Even considering the phenotypic variability of this disorder, our patient appeared to be considerably less affected than previously reported cases. The greatest morbidity associated with FTC/HHS may be due to vascular calcification.(16) The absence of any coronary calcification in this patient was reassuring. The presenting sign in our patient at age 12 was findings on dental radiographs. Dental radiographs at age 35 revealed the shortened maxillary premolar roots and root dilacerations. In the most dramatic findings, the root pulps demonstrated a unique thistle shaped configuration in the coronal third-half of the root. The thistle shaped pulp terminated at the coronal extent of the mid root bulge (Fig. 1B). The pulps of the molar teeth appeared mineralized, and were not radiographically evident. Our results are consistent with those of Burkes et. al. who noted that radiographically the teeth have short bulbous roots, pulp stones and partial obliteration of pulp cavity.(17) The dramatically shortened maxillary premolars, the canine and premolar mid root bulging and the thistle shaped pulps that end at the area of mid root enlargement are striking and unique. The presence of such unique and dramatic pulp and root changes indicate the development of the teeth is affected between years 4-14, when most root development occurs. These lesions are different from dentin and root changes seen in dentin dysplasia and are unique enough that they may provide a new phenotypic marker for FTC (17). HHS shares several clinical and metabolic features with FTC. In our case, the cortical hyperostosis, and possibly the repeated episodes of “knee” pain and joint swelling, which subsequently resolved, can be interpreted as a HHS manifestation.
Our patient carried a compound heterozygous p.R438C and p.Q592X mutations in GALNT3. These mutations result in a stop codon at position 592, and a missense mutation. Specktor et al described a patient with FTC that carried a homozygous nonsense mutation p.Q592X. He was a 32-year-old man who presented initially in childhood with recurrent episodes of conjunctival irritation, arthralgia and dental abnormalities. He was found to have “salt-like” deposits on the conjunctiva, periarticular calcifications near the acromio-clavicular joints and both elbow joints(18). Olauson et al recently reported a homozygous missense mutation in exon 6 of GALNT3, leading to an amino acid shift from Arg to His at residue 438. In this case, the patient was a 19-year-old woman with HHS who presented with painful cortical lesions in left tibia at 5 years of age. Radiological examinations indicated diaphyseal hyperostosis, periosteal reactions and edema in the surrounding tissue of the affected area (12). It is interesting to note that in most of the cases from the literature (reviewed in (19)), patients with homozygous missense mutations of GALNT3 tend to meet the diagnostic criteria of HHS, while patients with homozygous nonsense mutations tend to fit the diagnosis of FTC phenotype. Our patient, having a compound heterozygote mutation, had manifestations of both FTC and HHS.
To investigate whether or not renal phosphate reabsorption could be modulated in FTC/HHS, we treated the patient with niacinamide, followed by acetazolamide, compounds known to affect renal phosphate reabsorption.(20,21) Niacinamide is a B-complex vitamin that is converted to nicotinamide adenosine dinucleotide (NAD) and nicotinamide adenosine dinucleotide phosphate (NADP), which are coenzymes for many oxidation-reduction reactions. Nicotinamide and its analogues, similar to PTH, increase intracellular cAMP in kidney cells, and it is through this mechanism that it is believed to decrease renal phosphate reabsorption.(20) Acetazolamide is an inhibitor of the enzyme carbonic anhydrase, which, like PTH, increases renal cell cAMP and is believed to result in phosphaturia by via this mechanism. (21) There are reports in the literature wherein long term administration of acetazolamide appeared to be useful for the treatment of tumoral calcinosis resistant to phosphorus deprivation by aluminum hydroxide alone.(22) In this study, there were decreases in serum phosphate in response to short term treatment with both niacinamide and acetazolamide. The response to niacinamide and acetazolamide in this patient supports the hypothesis that inhibition of renal phosphate transport via drugs with action at the kidney may have an effect on serum phosphate, and in turn an effect on the serum FGF23 concentration. Longer treatment with higher doses and/or the addition of renal phosphate binders would likely have shown a greater effect. Interestingly, we noted that serum FGF23 appeared to change in parallel in response to changes in serum phosphate, suggesting the possibility of an intact feedback loop in the FGF23-producing cells to changes in serum phosphate. All reported mutations in both GALNT3 and FGF23 that cause either FTC or HHS result in a similar serum FGF23 profile (low levels of intact FGF23 and elevated C-terminal fragments). These findings imply that there is an interaction between GALNT3 and FGF23 and indicate that GALNT3 activity may be involved in the intracellular processing of intact FGF23 to N- and C-terminus FGF23.(23)
This case broadens the spectrum of phenotypic and genotypic features of FTC/HHS and suggests ways to decrease renal phosphate reabsorption in the setting of a low intact FGF23.
Acknowledgments
This research was supported, in part, by the Intramural Research Program of the NIH, National Institute of Dental and Craniofacial Research. KEW and EGF are supported by NIH DK063934
Footnotes
Conflict of Interest Statement: The authors have no conflicts of interest.
References
- 1.Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2004 doi: 10.1093/hmg/ddi034. [DOI] [PubMed] [Google Scholar]
- 2.Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, Khamaysi Z, Behar D, Petronius D, Friedman V, Zelikovic I, Raimer S, Metzker A, Richard G, Sprecher E. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet. 2004;36(6):579–81. doi: 10.1038/ng1358. [DOI] [PubMed] [Google Scholar]
- 3.Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, Goetz R, Mohammadi M, White KE, Econs MJ. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117(9):2684–91. doi: 10.1172/JCI31330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frishberg Y, Topaz O, Bergman R, Behar D, Fisher D, Gordon D, Richard G, Sprecher E. Identification of a recurrent mutation in GALNT3 demonstrates that hyperostosis-hyperphosphatemia syndrome and familial tumoral calcinosis are allelic disorders. J Mol Med. 2005;83(1):33–8. doi: 10.1007/s00109-004-0610-8. [DOI] [PubMed] [Google Scholar]
- 5.Ichikawa S, Guigonis V, Imel EA, Courouble M, Heissat S, Henley JD, Sorenson AH, Petit B, Lienhardt A, Econs MJ. Novel GALNT3 mutations causing hyperostosis-hyperphosphatemia syndrome result in low intact fibroblast growth factor 23 concentrations. J Clin Endocrinol Metab. 2007;92(5):1943–7. doi: 10.1210/jc.2006-1825. [DOI] [PubMed] [Google Scholar]
- 6.Ichikawa S, Imel EA, Sorenson AH, Severe R, Knudson P, Harris GJ, Shaker JL, Econs MJ. Tumoral Calcinosis Presenting with Eyelid Calcifications due to Novel Missense Mutations in the Glycosyl Transferase Domain of the GALNT3 Gene. J Clin Endocrinol Metab. 2006;91(11):4472–5. doi: 10.1210/jc.2006-1247. [DOI] [PubMed] [Google Scholar]
- 7.Ichikawa S, Lyles KW, Econs MJ. A novel GALNT3 mutation in a pseudoautosomal dominant form of tumoral calcinosis: evidence that the disorder is autosomal recessive. J Clin Endocrinol Metab. 2005;90(4):2420–3. doi: 10.1210/jc.2004-2302. [DOI] [PubMed] [Google Scholar]
- 8.Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron Robey P. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112(5):683–92. doi: 10.1172/JCI18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Razzaque MS, Lanske B. The emerging role of the fibroblast growth factor-23-klotho axis in renal regulation of phosphate homeostasis. J Endocrinol. 2007;194(1):1–10. doi: 10.1677/JOE-07-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kato K, Jeanneau C, Tarp MA, Benet-Pages A, Lorenz-Depiereux B, Bennett EP, Mandel U, Strom TM, Clausen H. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem. 2006;281(27):18370–7. doi: 10.1074/jbc.M602469200. [DOI] [PubMed] [Google Scholar]
- 11.Frishberg Y, Ito N, Rinat C, Yamazaki Y, Feinstein S, Urakawa I, Navon-Elkan P, Becker-Cohen R, Yamashita T, Araya K, Igarashi T, Fujita T, Fukumoto S. Hyperostosis-hyperphosphatemia syndrome: a congenital disorder of O-glycosylation associated with augmented processing of fibroblast growth factor 23. J Bone Miner Res. 2007;22(2):235–42. doi: 10.1359/jbmr.061105. [DOI] [PubMed] [Google Scholar]
- 12.Strewler GJ. Untangling klotho's role in calcium homeostasis. Cell Metab. 2007;6(2):93–5. doi: 10.1016/j.cmet.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 13.Olauson H, Krajisnik T, Larsson C, Lindberg B, Larsson T. A Novel Missense Mutation In GALNT3 Causing Hyperostosis-Hyperphosphatemia Syndrome. Eur J Endocrinol. 2008 doi: 10.1530/EJE-08-0011. [DOI] [PubMed] [Google Scholar]
- 14.Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Juppner H. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348(17):1656–63. doi: 10.1056/NEJMoa020881. [DOI] [PubMed] [Google Scholar]
- 15.Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. The ADHR Consortium. Nat Genet. 2000;26(3):345–8. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 16.Chefetz I, Heller R, Galli-Tsinopoulou A, Richard G, Wollnik B, Indelman M, Koerber F, Topaz O, Bergman R, Sprecher E, Schoenau E. A novel homozygous missense mutation in FGF23 causes Familial Tumoral Calcinosis associated with disseminated visceral calcification. Hum Genet. 2005;118(2):261–6. doi: 10.1007/s00439-005-0026-8. [DOI] [PubMed] [Google Scholar]
- 17.Burkes EJ, Jr, Lyles KW, Dolan EA, Giammara B, Hanker J. Dental lesions in tumoral calcinosis. J Oral Pathol Med. 1991;20(5):222–7. doi: 10.1111/j.1600-0714.1991.tb00423.x. [DOI] [PubMed] [Google Scholar]
- 18.Specktor P, Cooper JG, Indelman M, Sprecher E. Hyperphosphatemic familial tumoral calcinosis caused by a mutation in GALNT3 in a European kindred. J Hum Genet. 2006;51(5):487–90. doi: 10.1007/s10038-006-0377-6. [DOI] [PubMed] [Google Scholar]
- 19.Garringer HJ, Mortazavi SM, Esteghamat F, Malekpour M, Boztepe H, Tanakol R, Davis SI, White KE. Two novel GALNT3 mutations in familial tumoral calcinosis. Am J Med Genet A. 2007;143A(20):2390–6. doi: 10.1002/ajmg.a.31947. [DOI] [PubMed] [Google Scholar]
- 20.Campbell PI, Abraham MI, Kempson SA. Increased cAMP in proximal tubules is acute effect of nicotinamide analogues. Am J Physiol. 1989;257(6 Pt 2):F1021–6. doi: 10.1152/ajprenal.1989.257.6.F1021. [DOI] [PubMed] [Google Scholar]
- 21.Beck N, Kim KS, Wolak M, Davis BB. Inhibition of carbonic anhydrase by parathyroid hormone and cyclic AMP in rat renal cortex in vitro. J Clin Invest. 1975;55(1):149–56. doi: 10.1172/JCI107905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamaguchi T, Sugimoto T, Imai Y, Fukase M, Fujita T, Chihara K. Successful treatment of hyperphosphatemic tumoral calcinosis with long-term acetazolamide. Bone. 1995;16(4 Suppl):247S–250S. doi: 10.1016/8756-3282(95)00019-a. [DOI] [PubMed] [Google Scholar]
- 23.Benet-Pages A, Lorenz-Depiereux B, Zischka H, White KE, Econs MJ, Strom TM. FGF23 is processed by proprotein convertases but not by PHEX. Bone. 2004;35(2):455–62. doi: 10.1016/j.bone.2004.04.002. [DOI] [PubMed] [Google Scholar]