

Background
The syndrome of heart failure is characterized by increased levels of circulating inflammatory mediators (pro-inflammatory cytokines, cell adhesion molecules, complement and C-reactive protein), which have been implicated in the pathogenesis of heart failure. Recently a number of studies have suggested that statins (3-hydroxy-3-methylglutaryl-CoA [HMG-CoA] reductase inhibitors) may exert salutary effects in patients with heart failure by virtue of their pleiotropic (non-lipid lowering) actions. The following review will focus on the non-lipid lowering effects of statins with an emphasis on the anti-inflammatory properties of these agents.
Discovery of Statins
In 1971 Drs. Akira Endo and Masao Kuroda began to search for HMG-CoA reductase inhibitors of microbial origin. They initially considered that these microbes would synthesize molecules that would inhibit other microbes that required sterols or other isoprenoids for growth. After testing 6000 microbial strains, their efforts were rewarded with the discovery that the mold Pythium ultimum produced a compound called Citrinin, which was able to irreversibly bind the HMG-CoA reductase. By the end of 1971 Dr. Endo and Kuroda were able to determine the structure of a compound they termed mevastatin.[1] From this discovery and based on the pioneering work of Drs. Brown and Goldstein at the University of Texas Southwestern, the use of statins has become a mainstay of therapy in the primary and secondary prevention of cardiovascular disease.[2] Recently, considerable attention has focused on the “off target” or pleiotropic actions of the statin family. As will be discussed below, many of the pleiotropic actions are thought to be mediated through isoprenoids, which affect multiple biochemical signaling pathways.
Effect of Statins on the Small G-Proteins: Rho, Rac, and Ras
As illustrated in Figure 1, statins reduce cholesterol synthesis by inhibiting HMG-CoA reductase, the rate-limiting enzyme in the mevalonate pathway that is responsible for cholesterol synthesis. The important intermediate products in the mevalonate pathway include the isoprenoids, farnesylpyrophosphate, and geranlygeranyl-pyrophosphate.[3, 4] These intermediate products lead to activation of various downstream intracellular signaling molecules by prenylation of the guanosine triphosphate (GTP)-binding proteins Rho, Ras, and Rac, which facilitates attachment of these proteins to the cell membrane.[5] The Rho signal transduction pathway is involved in the activation of inflammatory cytokines and chemokines. Rho also plays an important role in the formation and maintenance of the actin cytoskeleton and thereby affects intracellular transport, mRNA stability, and gene transcription. The Ras proteins are responsible for cell proliferation and hypertrophy, whereas the Rac proteins are involved in the production of reactive oxygen species through activation of NADPH oxidase. Inhibition of HMG-CoA reductase leads to a decrease in farnesylated and geranygeranylated proteins, and a subsequent dose-dependent reduction in downstream signaling pathways mediated by Rho, Ras, and Rac.
Figure 1.
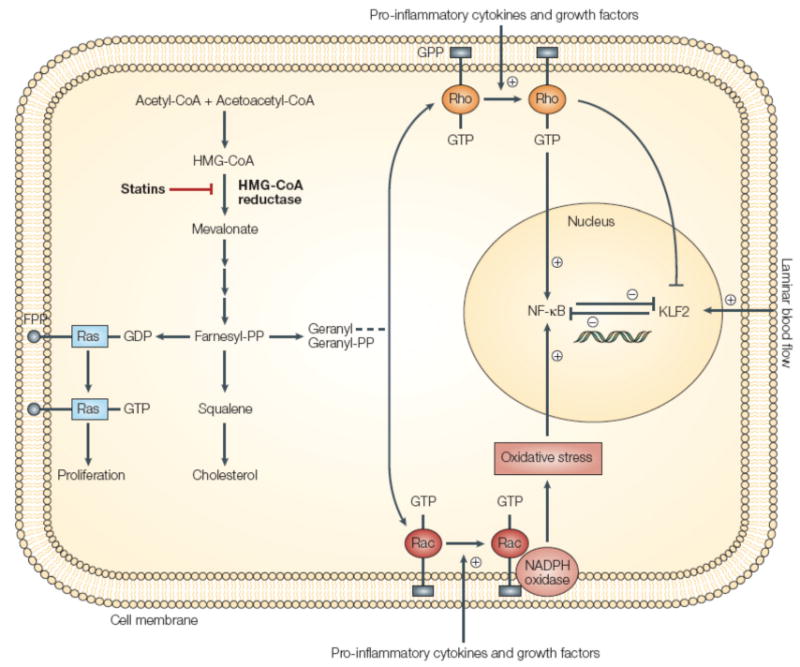
Pleiotropic effects of statins. Inhibition of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase by statins inhibits cholesterol synthesis and isoprenoid production. This results in reduced prenylation of small G-proteins such as Rho and, in turn, reduced nuclear factor-κB (NF-κB) activation and enhanced Kruppel-like factor-2 (KLF2) expression. KLF2 expression can also be enhanced by exposure to blood flow termed laminar flow. FPP, farnesyl pyrophosphate; GPP, geranyl pyrophosphate. (Reprinted by permission from Macmillan Publishers Ltd. Jain MK, Ridker PM. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nature reviews Drug discovery. 2005 Dec 1;4(12):977-87.
In addition to affecting small GTP-binding proteins, statins affect several important downstream transcription factors, including nuclear factor- kappa B (discussed below), peroxisome proliferators activated receptors (PPARs), as well as Kruppel-Like-Factor-2 (KLF-2). PPARs are nuclear receptors that function as ligand activated transcription factors. Fatty acid derivatives and retinoids serve as the natural ligands for PPARs. After heterodimerization with the 9-cis-retinoic acid receptor (RXR), PPARs bind to specific PPAR response elements in the promoter regions of target genes.[6] Statins activate PPAR α signaling by inhibiting activation of Rho A.[6] Importantly, PPARs may inhibit the activation of pro-inflammatory signaling cascades through Nuclear Factor Kappa B (NF-κB) and activator protein-1 (AP-1) dependent pathways.[7]
KLF-2 is a zinc finger containing transcription factor that is involved in blood vessel development and T lymphocyte activation. KLF-2 is expressed in endothelial cells and is up regulated by laminar sheer stress. KLF-2 is upregulated by statins, and is required for statin dependent endothelial transcriptional changes.[8] KLF-2 is involved in the induction of heme oxygenase-1 (HO), which is the rate-limiting step in the catabolism of heme. HO-1 activates anti-apoptotic, anti-inflammatory, and anti-oxidant signaling pathways. Deficiency of HO-1 results in widespread vascular injury. Thus, by upregulating HO-1 through KLF-2, statins may upregulate an ensemble of protective genes within the vasculature and counteract the endothelial dysfunction (see below) that occurs in heart failure patients.[9]
Effects of Statins on Oxidative Stress
“Oxidative stress” occurs when the production of reactive oxygen species (ROS) exceeds the buffering capacity of antioxidant defense systems, leading to an excess of ROS within the cell. At low levels ROS activate a variety of important intracellular signaling pathways, whereas at higher levels ROS causes damage to cell membranes and can provoke cell death. Not surprisingly, oxidative stress has been implicated in the progression of cardiac hypertrophy and adverse remodeling.[10] In cardiac myocytes NAPDH oxidase is an important source of ROS. As noted above Rac1 is a small GTP binding protein that regulates NAPDPH oxidase activity in myocytes. Insofar as statins inhibit isoprenylation of Rac1, these drugs also decrease the production of ROS.[11] Indeed, in rat models of heart failure treatment with statins was associated with decreased progression to heart failure after aortic banding. Importantly, simvastatin treated rats displayed lower tissue levels of superoxide anion production. In rat myocytes exposed to angiotensin II (which activates NADPH oxidase), simvastatin treatment resulted in decreased superoxide anion production and intracellular oxidation.[12]
Anti-Inflammatory Properties of Statins
Statins have been shown to decrease the expression of pro-inflammatory cytokines in multiple clinical heart failure studies (see article by Kahn in this issue). Herein we will review the molecular mechanisms by which statins may reduce inflammation. Nuclear factor- kappa B is an important transcription factor that is responsible for the activation of a variety of inflammatory pathways. Under normal conditions, NF-κB resides in the cytoplasm, where it is bound by its inhibitor, I kappa B (IκB). In response to inflammatory stimuli, IκB is phosphorylated and targeted for degradation by the ubiquitin proteasome system, thereby releasing NF-κB and allowing it to translocate to the nucleus where it is capable of inducing the expression of inflammatory cytokines and cell adhesion molecules. Statins decrease NF-κB activation by decreasing the levels of oxidative stress within cells (which can directly activate NF-κB), as well as by stabilization of IκB.[13],[14]. Statins decrease the expression of monocyte chemoattractant protein-1 (MCP-1), a cell adhesion molecule that is upregulated by NF-κB. MCP-1 is involved in the chemotaxis of monocytes and smooth muscle cells into tissue beds. In a transgenic mouse model, cardiac restricted overexpression of MCP-1 resulted in myocarditis with the subsequent development of a heart failure phenotype. [15] In a mouse model, atorvastatin and pravastatin decreased the expression of MCP-1 in carotid, femoral, and thoracic aortic vascular beds.[16]
Persistent activation of the renin angiotensin system with increased levels of angiotensin II (Ang II) contributes to disease progression in heart failure. Ang II not only causes peripheral vasoconstriction but can also lead to remodeling in heart failure, by provoking cardiac myocyte hypertrophy, cardiac myocyte cell death, and progressive myocardial fibrosis. [17] Relevant to the current discussion, Ang II stimulates MCP-1 gene expression in cultured rat aortic smooth muscle cells through an AT1 receptor mediated mechanism. Recent studies suggest that statins may interdict Ang II mediated signaling. When double transgenic rats that harbor both the human renin and angiotensinogen genes were treated with cerivastatin, there was a decrease in mortality, cardiac hypertrophy, macrophage infiltration, as well as reduced collagen I, laminin, and fibronectin deposition. Cerivastatin treated rats also had reduced expression of basic fibroblast growth factor mRNA, and IL-6 expression, and less activation of NF-κB and activator protein-1 (AP-1).[18]
Endothelial Dysfunction
Endothelial dysfunction is an important mechanism that contributes to disease progression in heart failure. The mechanistic basis for this abnormality is thought to lie, at least in part, to an imbalance between nitric oxide synthesis (vasodilator) and endothelin-1 (vasoconstrictor) production. In endothelial cells nitric oxide is produced by the endothelial nitric oxide synthase (eNOS) gene. Diminished levels of nitric oxide lead to impaired vascular relaxation, platelet aggregation, and increased vascular smooth muscle cell proliferation and migration.[19] Statins enhance endothelial nitric oxide synthase via stabilization of eNOS mRNA through inhibition of Rho, and phosphorylation of eNOS protein by activation of the phosphatidylinositol 3-kinase/Akt pathway.[20] Simvastatin has been shown to stabilize eNOS mRNA, whereas simvastatin and lovastatin have been shown to upregulate eNOS expression.[21, 22] In an ischemic model of heart failure induced by ligation of the left anterior descending artery low dose simvastatin was shown to decrease the severity of heart failure. The salutary effects of simvastatin were not evident in eNOS deficient mice (eNOS -/-), suggesting that eNOS is required for the beneficial effects of simvastatin in this model.[20] Endothelin-1 is a 21 amino acid peptide that acts on ETA receptors on endothelial cells to cause vasoconstriction. Atorvastatin and simvastatin inhibit pre-proET-1 mRNA expression and ET-1 synthesis. Statins have been demonstrated to decrease endothelin-1 levels in animal models as well as in humans. In a hypertensive rat model of cardiac failure treatment with pitavastatin resulted in lower endothelin-1 levels.[23] Similarly, endothelin-1 levels were decreased in heart failure patients after treatment with statins.[24]
The beneficial effects of statins on endothelial function have also been observed in heart failure patients. Tousoulis used forearm blood flow as a surrogate for endothelial function in patients with ischemic cardiomyopathy. Patients were divided into three groups, the first group received 10mg of atorvastatin a day, the second group received 10mg of atorvastatin and 400 IU/day of Vitamin E and the third group received no stain or vitamin E treatment. In the group that received atorvastatin there was an increase in the maximum hyperemic blood flow, which was slightly attenuated, but also present in the atorvastatin plus vitamin E group. No effect on hyperemic blood flow was found in the placebo group.[25] Node et al examined the use of statins in 51 patients with non-ischemic heart failure and an EF < 40% in Japan. Patients were randomized to either simvastatin or placebo and followed for 14 weeks. Endothelial function was assessed using high resolution B mode ultrasonography of the brachial artery before and after cuff induced transient ischemia of the forearm. In the simvastatin treated group, brachial artery vasodilatation improved from 8 to 13%, and was statistically significant. A statistically significant change was not observed in the placebo group.[26] Thus, by altering the regulation of nitric oxide and endothelin, statins may have a beneficial effect on endothelial function and systemic peripheral vascular resistance.
Angiogenesis
Angiogenesis is a multi-step process by which new blood vessels are created by sprouting from existing blood vessels. Angiogenesis has been implicated in both normal physiological processes such as development, growth and, wound healing. However, angiogenesis has also been linked to pathological processes such as tumor formation. Multiple factors have been shown to be important in angiogenesis, including vascular endothelial growth factor (VEGF), matrix metalloproteinases (MMPs), fibroblast growth factor (FGF), and hepatocyte growth factor (HGF).[27] VEGF levels in patients with hyperlipidemia are elevated, and have been shown in a small study to decrease after treatment with fluvastatin and fenofibrate. [28] Unfortunately the effects of fluvastatin vs fenofibrate vs simple lipid lowering could not be separated in this small clinical study because of the small sample size, but are nonetheless intriguing.[28] Treatment with statins has been shown to produce a biphasic effect on angiogenesis. Endothelial cells treated with low dose cerivastatin increased cell proliferation by 10-20%, while high concentrations decreased proliferation by 38%. The formation of vascular-like structures by human adult dermal microvascular endothelial cells (HMEC-1) was enhanced by low dose cerivastatin and atorvastatin, while the formation of vascular structures at high dose was inhibited.[29] Whether this translates into angiogenesis in humans and/or has an effect on heart failure outcomes remains to be determined. For a more detailed discussion on statin therapy and angiogenesis we refer the reader to the review article by Skaletz-Rorowski and Walsh.[30]
Thrombosis
There is now substantial evidence linking inflammation to a prothrombotic state in heart failure. Studies of heart failure including Studies of Left Ventricular Dysfunction (SOLVD) and Veterans Affairs Vasodilator Heart Failure Trials (V-HeFT) showed that heart failure was associated with an annual 1.5-4% risk of stroke. It is likely that the chronic systemic inflammation in heart failure contributes to the prothrombotic state.[31] Statins may attenuate this pro-thrombotic state by either decreasing inflammation or by more direct interactions with platelets and the coagulation system.[32] Several studies have examined the interaction of statins and coagulation. Reduced levels of plasminogen activator 1 (PAI-1) were found after treatment with platelet derived growth factor or transforming growth factor β in smooth muscles cells and endothelial cells that were exposed to simvastatin. Treatment with simvastatin was also shown to increase the level of tissue-type plasminogen activator (tPA) released from endothelial cells.[33] Another possible mechanism by which statins affect thrombosis involves the protease-activated receptor-1 (PAR-1), which was noted in the PAR-1 inhibition by statins (PARIS) study. PAR-1 is a thrombin receptor that is reduced in statin treated patients.[34, 35] PAR-1 repression by all statins has been found to be nearly identical.[36] Simvastatin decreases the expression of tissue factor (TF) in a monocyte model of in vitro clotting. When monocytes were treated with LPS and simvastatin, they displayed a decreased rate of thrombin generation by interfering with monocyte expression of TF.[37]
Statins may alter platelet function by altering the cholesterol content of platelet membranes and thus membrane fluidity, or they may inhibit platelet aggregation indirectly by increasing NO bioavailability.[38] β–thromboglobulin and platelet factor 4 (PF4) are platelet-specific proteins that are secreted from the α–granules during the release reaction induced by ADP, epinephrine, arachidonic acid, collagen, and thrombin. In mice treated with atorvastatin, PF4 and β-thromboglobulin levels were decreased. [39] In eNOS deficient mice, atorvastatin treatment did not alter serum levels of PF4 or beta thromboglobulin, which suggests that the major effect of atorvastatin on platelet activity is mediated by eNOS.[40] In a human study, fluvastatin was shown to decrease platelet aggregation in a mechanism thought to be mediated by increased platelet derived nitric oxide release.[41] In a rat model of streptozotocin-induced diabetes, rosuvastatin has been shown to reduce fibrinogen binding to activated GPIIb/IIIa and P-selectin surface expression on platelets.[42] In a small clinical study, 17 patients received atorvastatin 10mg/day, whereas 18 patients received no statin therapy. Treatment with atorvastatin decreased plasma levels of antithrombin III, protein C, factor V, tPA, and PAI-1.[43] Thus, statins may alter the prothrombotic state of chronic heart failure through several different pathways, including reduced inflammation, decreased platelet activity, and/or improved endothelial function.
Mobilization of Bone-Marrow Cells
The exact mechanisms by which mobilization of bone marrow cells may benefit patients in heart failure remains to be determined. Interestingly, statins have been shown to increase the number of circulating bone-marrow derived endothelial progenitor cells. In mice fed 20mg/kg of simvastatin for three weeks the number of circulating endothelial progenitor cells (EPCs) was increased. The number of c-kit/Sca-1 double positive cells increased from 0.78% to 8.2%. It is suggested that statins increase the levels of EPCs by acting through the PI3-kinase/AKT pathway.[44] Statins have been shown previously to activate PI-3 Kinase and AKT.[45, 46] When inhibitors of PI-3 kinase were added the increase in EPCs was abolished.[44] In a murine infarct model, atorvastatin was found to augment the number of circulating EPCs after infarction. Interestingly, increased levels of circulating EPCs were not seen after treatment with atorvastatin in eNOS -/- mice, suggesting that eNOS has a role in mobilization of EPCs.[47, 48] Although speculative, it is possible that statin-induced enhancement of EPCs may contribute to improved cardiac regeneration and/or repair.
Ventricular Remodeling
Treatment with statins has also shown to have important effects on cardiac remodeling. As noted above, activation of Ras signaling has been shown to initiate a cascade of intracellular signaling events that contribute to myocardial hypertrophy.[49] Germane to the present discussion, statins have been shown to prevent or reverse cardiac hypertrophy in humans.[50] In animal infarct models, suppression of Rho kinase has been associated with attenuated LV remodeling.[51, 52] Atorvastatin has been shown to reduce collagen synthesis, α-1-procollagen mRNA expression, and expression of the profibrotic peptide connective tissue growth factor in cell cultures of rat and human cardiac fibroblasts.[53] In a rat model of cardiac hypertrophy, treatment with pitavastatin led to a decrease in the expression of hypertrophic and profibrotic genes that was accompanied by significant decrease in interstitial fibrosis and collage deposition.[53] Cerivastatin has been shown to decrease the levels of matrix metalloproteinase-1 (MMP-1), MMP-3, and MMP-9 in macrophages of Watanabe heritable hyperlipidemic rabbits.[54] In canine model of heart failure induced by microembolization dogs were treated with three months of rosuvastatin. Treatment with rosuvastatin resulted in the prevention of progressive LV dysfunction and remodeling and improvement in remodeling was similar to that seen after angiotensin converting enzyme inhibitor treatment. Additionally, treatment with rosuvastatin resulted in decreased levels of TNF and MMP2, and increased circulating levels of bone marrow derived stems cells.[55] A rabbit model of hypertrophic cardiomyopathy has been used to examine the effects of simvastatin on cardiac hypertrophy and fibrosis. Simvastatin was able to reduce LV mass, wall thickness, and filling pressures in this model. It was postulated that simvastatin was able to provide protection by down-regulating the levels of ERK1/2 kinases, which have been implicated in hypertrophic growth.[56] Dhal salt sensitive rats develop cardiac hypertrophy and congestive heart failure after a being fed a high salt diet. When these rats were fed a high salt diet and given cerivastatin they displayed decreased left ventricular wall thickness and myocardial fibrosis, as well as decreased heart weight, and LV diameter, and improved LV function at 17 weeks (Figure 2).[57] Finally, treatment with 20mg/kg of simvastatin for 6 weeks reduced caspase-3 activation in a hamster model with a recessive mutation in delta sarcoglycan, which has been shown to lead to a heart failure phenotype.[58] Taken together, these observations suggest that statins may have important effects on cardiac remodeling, which has been implicated in the pathogenesis of heart failure.
Figure 2.
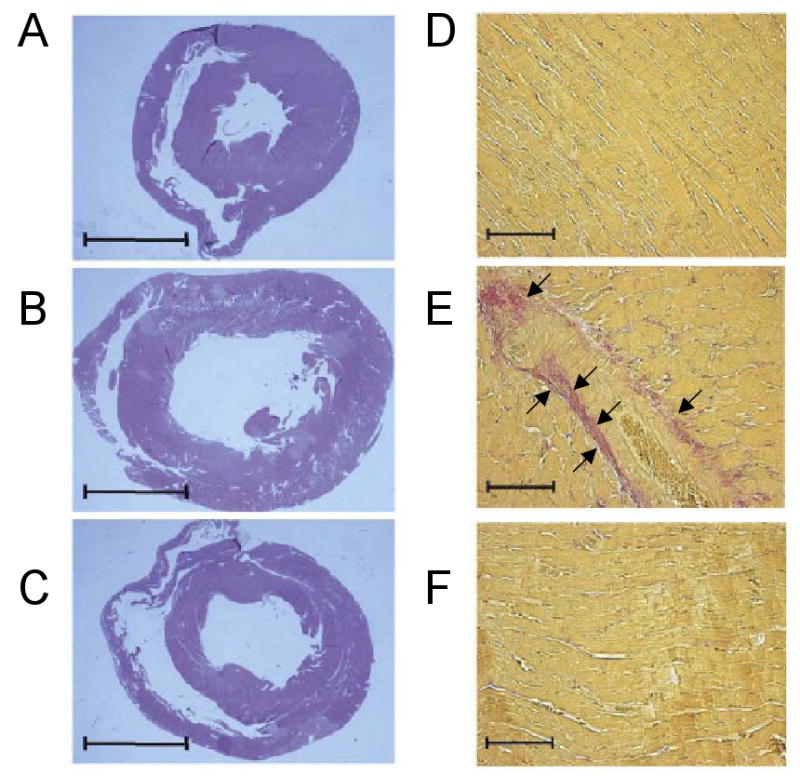
Effects of statins on LV remodeling. Dahl salt sensitive rats (17 weeks) were treated with (A) low salt diet (B) high salt diet (C) high salt + cerivastatin. Panels D – F show the resultant cardiac fibrosis (arrow heads) at 17 weeks in (D) low salt diet (E) high salt diet and (F) high salt diet + cerivastatin treated rats (Modified with permission from Elsevier. Hasegawa H, Yamamoto R, Takano H, et al: 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors prevent the development of cardiac hypertrophy and heart failure in rats. J Mol Cell Cardiol 35:953, 2003)
Conclusion
In the foregoing review, we have discussed the spectrum of salutary non-lipid lowering effects of statins in patients with heart failure, including decreased oxidative stress, improved endothelial function, decreased systemic inflammation, and decreased cardiac remodeling. There is also evidence that statins play an important role in preventing atrial and cardiac arrhythmias, which will be discussed in the accompanying review by Olshansky in this issue.[59] The question that remains is whether the aggregate of these beneficial effects observed in the laboratory will translate into beneficial effects in patients with heart failure, and if so, which of the potential spectrum of mechanisms will prove to be most important. Hopefully, these issues may potentially be gleaned from ongoing large scale clinical trials.
Acknowledgments
This research was supported by research funds from the N.I.H. (RO1 HL58081, RO1 HL61543, HL-42250). Dr. Mathur is supported by an NIH T32 training grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Nitin Mathur, Email: mathur@bcm.tmc.edu.
Kumudha Ramasubbu, Email: kumudhar@bcm.tmc.edu.
References
- 1.Endo A. The discovery and development of HMG-CoA reductase inhibitors. Journal of Lipid Research. 1992;33:1569–82. [PubMed] [Google Scholar]
- 2.Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat. 1992;1:445–66. doi: 10.1002/humu.1380010602. [DOI] [PubMed] [Google Scholar]
- 3.Liao JK. Isoprenoids as mediators of the biological effects of statins. J Clin Invest. 2002;110:285–8. doi: 10.1172/JCI16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laufs U, Liao JK. Isoprenoid metabolism and the pleiotropic effects of statins. Current atherosclerosis reports. 2003;5:372–8. doi: 10.1007/s11883-003-0008-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edwards PA, Ericsson J. Sterols and isoprenoids: signaling molecules derived from the cholesterol biosynthetic pathway. Annu Rev Biochem. 1999;68:157–85. doi: 10.1146/annurev.biochem.68.1.157. [DOI] [PubMed] [Google Scholar]
- 6.Martin G, Duez H, Blanquart C, et al. Statin-induced inhibition of the Rho-signaling pathway activates PPARalpha and induces HDL apoA-I. J Clin Invest. 2001;107:1423–32. doi: 10.1172/JCI10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li AC, Binder CJ, Gutierrez A, et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J Clin Invest. 2004;114:1564–76. doi: 10.1172/JCI18730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parmar KM, Nambudiri V, Dai G, et al. Statins exert endothelial atheroprotective effects via the KLF2 transcription factor. J Biol Chem. 2005;280:26714–9. doi: 10.1074/jbc.C500144200. [DOI] [PubMed] [Google Scholar]
- 9.Ali F, Hamdulay SS, Kinderlerer AR, et al. Statin-mediated cytoprotection of human vascular endothelial cells: a role for Kruppel-like factor 2-dependent induction of heme oxygenase-1. J Thromb Haemost. 2007 doi: 10.1111/j.1538-7836.2007.02787.x. [DOI] [PubMed] [Google Scholar]
- 10.Seddon M, Looi YH, Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart. 2007;93:903–7. doi: 10.1136/hrt.2005.068270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maack C, Kartes T, Kilter H, et al. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circulation. 2003;108:1567–74. doi: 10.1161/01.CIR.0000091084.46500.BB. [DOI] [PubMed] [Google Scholar]
- 12.Chen MS, Xu FP, Wang YZ, et al. Statins initiated after hypertrophy inhibit oxidative stress and prevent heart failure in rats with aortic stenosis. J Mol Cell Cardiol. 2004;37:889–96. doi: 10.1016/j.yjmcc.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 13.Plutzky J. Medicine. PPARs as therapeutic targets: reverse cardiology? Science. 2003;302:406–7. doi: 10.1126/science.1091172. [DOI] [PubMed] [Google Scholar]
- 14.Plutzky J. The potential role of peroxisome proliferator-activated receptors on inflammation in type 2 diabetes mellitus and atherosclerosis. Am J Cardiol. 2003;92:34J–41J. doi: 10.1016/s0002-9149(03)00614-3. [DOI] [PubMed] [Google Scholar]
- 15.Kolattukudy PE, Quach T, Bergese S, et al. Myocarditis induced by targeted expression of the MCP-1 gene in murine cardiac muscle. Am J Pathol. 1998;152:101–11. [PMC free article] [PubMed] [Google Scholar]
- 16.Martínez-González J, Alfón J, Berrozpe M, et al. HMG-CoA reductase inhibitors reduce vascular monocyte chemotactic protein-1 expression in early lesions from hypercholesterolemic swine independently of their effect on plasma cholesterol levels. Atherosclerosis. 2001;159:27–33. doi: 10.1016/s0021-9150(01)00469-5. [DOI] [PubMed] [Google Scholar]
- 17.Domenighetti AA, Wang Q, Egger M, et al. Angiotensin II-mediated phenotypic cardiomyocyte remodeling leads to age-dependent cardiac dysfunction and failure. Hypertension. 2005;46:426–32. doi: 10.1161/01.HYP.0000173069.53699.d9. [DOI] [PubMed] [Google Scholar]
- 18.Dechend R, Fiebeler A, Park JK, et al. Amelioration of angiotensin II-induced cardiac injury by a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor. Circulation. 2001;104:576–81. doi: 10.1161/hc3001.092039. [DOI] [PubMed] [Google Scholar]
- 19.Diodati JG, Dakak N, Gilligan DM, et al. Effect of atherosclerosis on endothelium-dependent inhibition of platelet activation in humans. Circulation. 1998;98:17–24. doi: 10.1161/01.cir.98.1.17. [DOI] [PubMed] [Google Scholar]
- 20.Greer JJ, Kakkar AK, Elrod JW, et al. Low-dose simvastatin improves survival and ventricular function via eNOS in congestive heart failure. Am J Physiol Heart Circ Physiol. 2006;291:H2743–51. doi: 10.1152/ajpheart.00347.2006. [DOI] [PubMed] [Google Scholar]
- 21.Laufs U, Fata VL, Liao JK. Inhibition of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase blocks hypoxia-mediated down-regulation of endothelial nitric oxide synthase. J Biol Chem. 1997;272:31725–9. doi: 10.1074/jbc.272.50.31725. [DOI] [PubMed] [Google Scholar]
- 22.Laufs U, La Fata V, Plutzky J, et al. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–35. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 23.Saka M, Obata K, Ichihara S, et al. Pitavastatin improves cardiac function and survival in association with suppression of the myocardial endothelin system in a rat model of hypertensive heart failure. J Cardiovasc Pharmacol. 2006;47:770–9. doi: 10.1097/01.fjc.0000211791.22411.0d. [DOI] [PubMed] [Google Scholar]
- 24.Mozaffarian D, Minami E, Letterer RA, et al. The effects of atorvastatin (10 mg) on systemic inflammation in heart failure. Am J Cardiol. 2005;96:1699–704. doi: 10.1016/j.amjcard.2005.07.092. [DOI] [PubMed] [Google Scholar]
- 25.Tousoulis D, Antoniades C, Vassiliadou C, et al. Effects of combined administration of low dose atorvastatin and vitamin E on inflammatory markers and endothelial function in patients with heart failure. Eur J Heart Fail. 2005;7:1126–32. doi: 10.1016/j.ejheart.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 26.Node K, Fujita M, Kitakaze M, et al. Short-term statin therapy improves cardiac function and symptoms in patients with idiopathic dilated cardiomyopathy. Circulation. 2003;108:839–43. doi: 10.1161/01.CIR.0000084539.58092.DE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braunwald E. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. Saunders; 2004. [Google Scholar]
- 28.Blann AD, Belgore FM, Constans J, et al. Plasma vascular endothelial growth factor and its receptor Flt-1 in patients with hyperlipidemia and atherosclerosis and the effects of fluvastatin or fenofibrate. Am J Cardiol. 2001;87:1160–3. doi: 10.1016/s0002-9149(01)01486-2. [DOI] [PubMed] [Google Scholar]
- 29.Weis M, Heeschen C, Glassford AJ, et al. Statins have biphasic effects on angiogenesis. Circulation. 2002;105:739–45. doi: 10.1161/hc0602.103393. [DOI] [PubMed] [Google Scholar]
- 30.Skaletz-Rorowski A, Walsh K. Statin therapy and angiogenesis. Curr Opin Lipidol. 2003;14:599–603. doi: 10.1097/00041433-200312000-00008. [DOI] [PubMed] [Google Scholar]
- 31.Chong AY, Lip GY. Viewpoint: the prothrombotic state in heart failure: a maladaptive inflammatory response? Eur J Heart Fail. 2007;9:124–8. doi: 10.1016/j.ejheart.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 32.Kearney D, Fitzgerald D. The anti-thrombotic effects of statins. J Am Coll Cardiol. 1999;33:1305–7. doi: 10.1016/s0735-1097(99)00019-4. [DOI] [PubMed] [Google Scholar]
- 33.Bourcier T, Libby P. HMG CoA reductase inhibitors reduce plasminogen activator inhibitor-1 expression by human vascular smooth muscle and endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:556–62. doi: 10.1161/01.atv.20.2.556. [DOI] [PubMed] [Google Scholar]
- 34.Serebruany VL, Miller M, Pokov AN, et al. Effect of statins on platelet PAR-1 thrombin receptor in patients with the metabolic syndrome (from the PAR-1 inhibition by statins [PARIS] study) Am J Cardiol. 2006;97:1332–6. doi: 10.1016/j.amjcard.2005.11.058. [DOI] [PubMed] [Google Scholar]
- 35.Serebruany VL, Midei MG, Malinin AI, et al. Absence of Interaction Between Atorvastatin or Other Statins and Clopidogrel: Results From the Interaction Study. Arch Intern Med. 2004;164:2051–7. doi: 10.1001/archinte.164.18.2051. [DOI] [PubMed] [Google Scholar]
- 36.Malinin AI, Ong S, Makarov LM, et al. Platelet inhibition beyond conventional antiplatelet agents: expanding role of angiotensin receptor blockers, statins and selective serotonin reuptake inhibitors. International Journal of Clinical Practice. 2006;60:993–1002. doi: 10.1111/j.1742-1241.2006.01063.x. [DOI] [PubMed] [Google Scholar]
- 37.Ferro D, Basili S, Alessandri C, et al. Inhibition of tissue-factor-mediated thrombin generation by simvastatin. Atherosclerosis. 2000;149:111–6. doi: 10.1016/s0021-9150(99)00291-9. [DOI] [PubMed] [Google Scholar]
- 38.Bonetti PO, Lerman LO, Napoli C, et al. Statin effects beyond lipid lowering--are they clinically relevant? Eur Heart J. 2003;24:225–48. doi: 10.1016/s0195-668x(02)00419-0. [DOI] [PubMed] [Google Scholar]
- 39.Kaplan KL, Owen J. Plasma levels of beta-thromboglobulin and platelet factor 4 as indices of platelet activation in vivo. Blood. 1981;57:199–202. [PubMed] [Google Scholar]
- 40.Laufs U, Gertz K, Huang P, et al. Atorvastatin upregulates type III nitric oxide synthase in thrombocytes, decreases platelet activation, and protects from cerebral ischemia in normocholesterolemic mice. Stroke. 2000;31:2442–9. doi: 10.1161/01.str.31.10.2442. [DOI] [PubMed] [Google Scholar]
- 41.Haramaki N, Ikeda H, Takenaka K, et al. Fluvastatin alters platelet aggregability in patients with hypercholesterolemia: possible improvement of intraplatelet redox imbalance via HMG-CoA reductase. Arterioscler Thromb Vasc Biol. 2007;27:1471–7. doi: 10.1161/ATVBAHA.106.128793. [DOI] [PubMed] [Google Scholar]
- 42.Schäfer A, Fraccarollo D, Vogt C, et al. Improved endothelial function and reduced platelet activation by chronic HMG-CoA-reductase inhibition with rosuvastatin in rats with streptozotocin-induced diabetes mellitus. Biochem Pharmacol. 2007;73:1367–75. doi: 10.1016/j.bcp.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 43.Tousoulis D, Antoniades C, Bosinakou E, et al. Effects of atorvastatin on reactive hyperaemia and the thrombosis-fibrinolysis system in patients with heart failure. Heart. 2005;91:27–31. doi: 10.1136/hrt.2003.027110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dimmeler S, Aicher A, Vasa M, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt. Journal of Clinical Investigation. 2001 doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kureishi Y, Luo Z, Shiojima I, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–10. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerber HP, McMurtrey A, Kowalski J, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–43. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 47.Aicher A, Heeschen C, Mildner-Rihm C, et al. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nature Medicine. 2003 doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- 48.Landmesser U, Engberding N, Bahlmann FH, et al. Statin-induced improvement of endothelial progenitor cell mobilization, myocardial neovascularization, left ventricular function, and survival after experimental myocardial infarction requires endothelial nitric oxide synthase. Circulation. 2004;110:1933–9. doi: 10.1161/01.CIR.0000143232.67642.7A. [DOI] [PubMed] [Google Scholar]
- 49.Brown JH, Del Re DP, Sussman MA. The Rac and Rho hall of fame: a decade of hypertrophic signaling hits. Circ Res. 2006;98:730–42. doi: 10.1161/01.RES.0000216039.75913.9e. [DOI] [PubMed] [Google Scholar]
- 50.Lee TM, Chou TF, Tsai CH. Association of pravastatin and left ventricular mass in hypercholesterolemic patients: role of 8-iso-prostaglandin f2alpha formation. J Cardiovasc Pharmacol. 2002;40:868–74. doi: 10.1097/00005344-200212000-00007. [DOI] [PubMed] [Google Scholar]
- 51.Hattori T, Shimokawa H, Higashi M, et al. Long-term inhibition of Rho-kinase suppresses left ventricular remodeling after myocardial infarction in mice. Circulation. 2004;109:2234–9. doi: 10.1161/01.CIR.0000127939.16111.58. [DOI] [PubMed] [Google Scholar]
- 52.Kobayashi N, Horinaka S, Mita S, et al. Critical role of Rho-kinase pathway for cardiac performance and remodeling in failing rat hearts. Cardiovasc Res. 2002;55:757–67. doi: 10.1016/s0008-6363(02)00457-1. [DOI] [PubMed] [Google Scholar]
- 53.Martin J, Denver R, Bailey M, et al. In vitro inhibitory effects of atorvastatin on cardiac fibroblasts: implications for ventricular remodelling. Clin Exp Pharmacol Physiol. 2005;32:697–701. doi: 10.1111/j.1440-1681.2005.04256.x. [DOI] [PubMed] [Google Scholar]
- 54.Aikawa M, Rabkin E, Sugiyama S, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation. 2001;103:276–83. doi: 10.1161/01.cir.103.2.276. [DOI] [PubMed] [Google Scholar]
- 55.Zacà V, Rastogi S, Imai M, et al. Chronic monotherapy with rosuvastatin prevents progressive left ventricular dysfunction and remodeling in dogs with heart failure. J Am Coll Cardiol. 2007;50:551–7. doi: 10.1016/j.jacc.2007.04.050. [DOI] [PubMed] [Google Scholar]
- 56.Patel R, Nagueh SF, Tsybouleva N, et al. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2001;104:317–24. doi: 10.1161/hc2801.094031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hasegawa H, Yamamoto R, Takano H, et al. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors prevent the development of cardiac hypertrophy and heart failure in rats. J Mol Cell Cardiol. 2003;35:953–60. doi: 10.1016/s0022-2828(03)00180-9. [DOI] [PubMed] [Google Scholar]
- 58.Abraham SS, Osorio JC, Homma S, et al. Simvastatin preserves cardiac function in genetically determined cardiomyopathy. J Cardiovasc Pharmacol. 2004;43:454–61. doi: 10.1097/00005344-200403000-00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kostapanos MS, Liberopoulos EN, Goudevenos JA, et al. Do statins have an antiarrhythmic activity? Cardiovasc Res. 2007;75:10–20. doi: 10.1016/j.cardiores.2007.02.029. [DOI] [PubMed] [Google Scholar]