

Abstract
Cadmium (Cd2+) is a heavy metal ion known to have a long biological half-life in humans. Accumulating evidence shows that exposure to Cd2+ is associated with neurodegenerative diseases characterized by the retention of ubiquitinated and misfolded proteins in the lesions. Here, we report that Cd2+ directly induces the formation of protein inclusion bodies in cells. The protein inclusion body is an aggresome, a major organelle for collecting ubiquitinated or misfolded proteins. Our results show that aggresomes are enriched in the detergent-insoluble fraction of Cd2+-treated cell lysates. Proteomic analysis identified 145 proteins in the aggresome-enriched fractions. One of the proteins is the highly conserved valosin-containing protein (VCP), which has been shown to colocalize with aggresomes and bind ubiquitinated proteins through its N domain (#1–200). Our subsequent examination of VCP's role in the formation of aggresomes induced by Cd2+ indicate that the C-terminal tail (#780-806) of VCP interacts with histone deacetylase HDAC6, a mediator for aggresome formation, suggesting that VCP participates in transporting ubiquitinated proteins to aggresomes. This function of VCP is impaired by inhibition of the deacetylase activity of HDAC6 or by over-expression of VCP mutants that do not bind ubiquitinated proteins or HDAC6. Our results indicate that Cd2+ induces the formation of protein inclusion bodies by promoting the accumulation of ubiquitinated proteins in aggresomes through VCP and HDAC6. Our delineation of the role of VCP in regulating cell responses to ubiquitinated proteins has important implications for understanding Cd2+ toxicity and associated diseases.
Keywords: Cadmium, Aggresome, Ubiquitin proteasome system, HDAC6
Introduction
Cadmium (Cd2+) poisoning is a serious health threat due to an increased level of industrial pollution and lack of an effective therapy (Shih et al., 2004). Cd2+ has an extremely long half-life in the human body (Elinder et al., 1976), and causes disorders of the renal, skeletal, vascular, and respiratory systems (Nordberg, 2004). Chronic exposure to Cd2+ is associated with neurodegenerative diseases such as amyotrophic lateral sclerosis (Bar-Sela et al., 1992; Bar-Sela et al., 2001) and Alzheimer's disease (Lui et al., 1990), which are characterized by the presence of protein inclusion bodies in the lesions (Li et al., 2004). It is not known, however, whether Cd2+ directly induces the formation of protein inclusion bodies, thus constituting an etiologic factor for neurodegenerative disorders. A common etiology of protein inclusion body formation in related neurodegenerative disorders is the generation of aggresomes, a type of cellular organelle in which ubiquitinated and misfolded proteins accumulate (Kopito, 2000; Kawaguchi et al., 2003). An aggresome is located near the centrosome and is formed through an active transport process involving multiple factors. When cells are treated with proteasome inhibitors, polyubiquitinated protein aggregates are transported to aggresomes via microtubules by the deacetylase HDAC6 and the dynein motor complex (Wojcik et al., 1996; Garcia-Mata et al., 1999; Johnston et al., 2002). Thus, depolarization of microtubules, inhibition of the deacetylase activity of HDAC6, or inactivation of the dynein motor all suppress aggresome formation. During the process of aggresome formation, HDAC6 interacts with the dynein motor complex through a dynein-binding domain and with ubiquitinated proteins through its ZnF-UBP zinc finger, which is a motif of the isopeptidase T class of deubiquitinating enzymes (Kawaguchi et al., 2003). Because the ZnF-UBP motif interacts not only with polyubiquitin chains attached to proteins (Kawaguchi et al., 2003) but also with free ubiquitin molecules (Seigneurin-Berny et al., 2001), the presence of free ubiquitin in the cell may decrease the capacity of HDAC6 to transport ubiquitinated proteins. Therefore, it has been postulated that other cellular proteins may facilitate the interaction between HDAC6 and ubiquitinated protein aggregates (Kopito, 2003).
Valosin-containing protein (VCP), an AAA family ATPase (Peters et al., 1990; and reviewed in Wang et al., 2004), binds to both ubiquitinated proteins and HDAC6. VCP also colocalizes with aggresomes induced by proteasome inhibitors (Kitami et al., 2006), suggesting that VCP may be involved in Cd2+-induced aggresome formation. Structurally, VCP consists of an N domain, two conserved ATPase domains D1 and D2, and a rather flexible C-terminal tail. The N domain is responsible for the binding of VCP to ubiquitin chains and to VCP cofactors, such as p47 and the Ufd1-Npl4 complex. Since these cofactors also bind to polyubiquitinated proteins, VCP is able to associate with large complexes containing ubiquitinated proteins and transport the complexes to aggresomes (Meyer et al., 2000; Dai and Li, 2001a; and reviewed in Wang et al., 2004).
In this study, we report the capacity of Cd2+ to directly induce aggresome formation in human cells. Proteomic analysis of Cd2+-induced aggresomes shows that VCP is present in detergent-insoluble fractions of aggresome-containing cell lysates. In addition, we found that VCP plays a key role in aggresome formation by acting as an adaptor to the deacetylase HDAC6 for transporting ubiquitinated protein aggregates. While the N domain of VCP (#2–207) binds ubiquitinated proteins, its C-terminal tail (#779–806) interacts with HDAC6. Consequently, VCP loads ubiquitinated cargo proteins onto the HDAC6/dynein complexes and transports the complexes to aggresomes via the microtubule network. Thus, VCP meditates Cd2+-induced aggresome formation and serves as a molecular target for the development of therapeutics for Cd2+ poisoning.
Methods
Wild-type and mutant VCP-GFP expression vector construction. The VCP gene was amplified from the pQE60-VCP plasmid (Dai and Li, 2001a) with a sense primer 5′<GGAATTCCATATGGCC TCTGGAGCCGATTC>3′ and an antisense primer 5′>CGGGATCCCATACAGGTCATCGTC ATTGTCTTC>3′, containing a restriction site at the 5′ (Nde I) or the 3′ (BamH 1) end, respectively. The amplified VCP gene was cloned into the pDNR-Dual vector (Clontech, Mountain View, CA), then transferred to pLPS-3′EGFP acceptor vector by a recombination reaction according to the manufacturer's protocol (Clontech, Mountain View, CA). Similarly, #1–200-GFP was constructed using the #1–200 fragment, which was amplified using wild-type VCP as the template and the following primers: a sense primer 5′>GGAATTCCATATGGCCTCTGGAGCCGATTC>3′ and an antisense primer 5′>CGGGATCCCTTCATTCAAGGATTCCTCCTCATC>3′. Deletion and site-specific mutants of VCP were generated using QuickChange® Site-Directed mutagenesis kit (Stratagene, La Jolla, CA) with pDNR-Dual-VCP as the template and the following sense primers: 5′>GGTACCGGACATATGGGTGGTTGCAG GAAG<3′ for #207–806, 5′>GGAACCAGGGTGGAATCCGCTCGAGAAG<3′ for #1–779, 5′> GGTCCCAGCCAGATCCGCTCGAGAAG<3′ for #1–785, 5′>GGCAGTGTGTACACAA TCCG CTCGAGAAGC<3′ for #1–796, 5′>CTGGG ACAGGGGCGACCCTGATTGC<3′ for A1, an ATP-binding site mutant in the VCP D1 domain, 5′>CTGGCTGTGGGGCAACCTTA CTGGC<3′ for A2, an ATP-binding site mutant in the VCP D2 domain. The complementary antisense primers are not shown.
HDAC6-shRNA expression vector construction
Nucleotide sequence #117–136 (Iwata et al., 2005) or #141–159 of the HDAC6 (NM_006044) open reading frame, as the small hairpin RNA interference expression cassette, was inserted into the retroviral expression vector pSIREN-RetroQ (Clontech, Mountain View, CA) according to the procedures described previously (Le et al., 2004).
Cell culture and transfection
Human embryonic kidney 293 (HEK293) cells were grown in DMEM with 10% FBS and 2% glutamine at 37 °C with 5% CO2. Cells growing on the 24-well plate or chambered coverglass (Nalgene Nunc Int. Corp., Rochester, NY) were transiently transfected with various DNA constructs using SuperFect Transfection Reagent (Qiagen, Valencia, CA) according to the manufacturer's instructions. The cell line stably expressing VCP-GFP was established by using flow cytometry sorting to select the green fluorescence-positive cells.
Fluorescent cell imaging
HEK293 cells transfected with wild-type or mutant VCP-GFP expression constructs were fixed with cold methanol for 10 min, then immunostained with primary antisera followed by fluorescent secondary antibodies. Cell images were acquired using a Zeiss LSM510 confocal microscope or a Nikon Diaphot inverted fluorescence microscope equipped with a Sony DKC-5000 digital photo camera.
Aggresome enrichment
HEK293 cells in DMEM with 10% FBS, with or without CdCl2 treatment (20 μM for 6 h), were lysed using DB (20 mM Tris/HCl, pH 7.6, 2 mM EDTA, 150 mM NaCl, 1.2% deoxycholate, 1.2% Triton X-100, 1% β-mercaptoethanol) containing protease inhibitors (1% aprotinin, 70 μg/ml phenylmethanesulfonyl fluoride, 40 μg/ml Tos-Phe-CH2Cl, 5 μg/ml Tos-Lys-CH2Cl, 5 μg/ml leupeptin). The lysates were sonicated (7 × 7 s) on ice with a 50% pulse using a Branson Sonifier 250 Sonicator, followed by centrifugation at 12000 × g for 15 min at 4 °C. The pellets were resuspended in DB and the procedures were repeated twice. The pellets were further analyzed by Western blotting or mass spectrometry.
Immunoprecipitation and Western blot analysis
Cultured cells were lysed in RIPA buffer (20 mM Tris/HCl, pH 7.6, 2 mM EDTA, 150 mM NaCl, 1% deoxycholate, 1% Triton X-100) containing protease inhibitors. The cell extracts were cleared by centrifugation (12000 × g, 10 min) and immunoprecipitated with an anti-GFP antibody (Sigma-Aldrich, St. Louis, MO). Protein samples were electrophoresed using SDS-PAGE, transferred to nitrocellulose membranes, then analyzed by immunoblotting with anti-VCP or HDAC6 antibodies and HRP-labeled secondary antibodies (Dai et al., 1998; Dai and Li, 2001b).
Electron microscopy
In situ cell fixation and processing were carried out as previously described (Gonda et al., 1976). Briefly, cells were cultured on a plate, fixed by 2% glutaraldehyde followed by 1% osmium tetraoxide buffered in 0.1 M sodium cacodylate (pH 7.4). The cells were en bloc stained in 0.5% uranyl acetate aqueous solution and dehydrated in a series of ethanol, embedded in epoxy resin, and cured in a 55 °C oven for 48 h. Thin sections (thickness of 60 to 70 nm) were mounted on 300 mesh grids and stained with uranyl acetate and lead citrate, then stabilized by carbon evaporation. The sections were examined and photographed with an H7000 electron microscope (Hitachi Ltd., Tokyo, Japan)
Analysis of aggresome proteome
The cell pellet containing aggresomes was resuspended in PBS containing 8 M urea and 1% CHAPS. The pellet was vortexed and sonicated twice in a water bath for 5 min each. An equal volume of 25 mM NH4HCO3, pH 8.4, was added to the lysate. The proteins were quantified using the Bradford assay. Sequencing Grade Modified Trypsin (Promega) was added to the lysate at a trypsin: protein ratio of 1:30. The digestion was carried out at 37°C on a shaker overnight.
The tryptic peptides were desalted using an Empore™ 4 mm/1 mL C18 High Performance Extraction Disk Cartridge from 3M, (St. Paul, MN), following the manufacturer's instructions. Peptides bound to the cartridge were eluted in 60% acetonitrile with 0.1% TFA and lyophilized. The peptides were then resuspended in 25% acetonitrile containing 0.1% formic acid and injected onto a strong cation-exchange liquid chromatography column (2.1 mm × 200 mm, polysulfoethyl A; PolyLC, Columbia, MD). Mobile phase A was 25% acetonitrile, mobile phase B was 25% acetonitrile with 0.5 M ammonium formate (pH 3). The flow rate was maintained at 250 μL/min, while the following 96-minute multi-step gradient was used to elute the peptides: 2% B for 5 min, followed by a linear increase to 10% B over 55 min; a further increase to 45% B in 25 min; then an increase to 100% B in 1 min, then continual elution with 100% B for 10 min. Peptide separation was monitored by fluorescence (280 nm excitation/350 nm emission). The peptides were eluted at the rate of one fraction/min for a total of 96 fractions.
The individual peptide fractions were pooled into 28 fractions, lyophilized, and reconstituted in 20 μL of 0.1% formic acid prior to nanoflow reversed-phase liquid chromatography mass spectrometry analysis. A 75 μm i.d. × 360 μm o.d. × 10 cm long fused silica capillary column (Polymicro Technologies, Phoenix, AZ) was packed with 3 μm, 300 Å pore size C-18 silica-bonded stationary RP particles (Vydac, Hysperia, CA). The column was connected to an Agilent 1100 nanoLC system (Agilent Technologies, Palo Alto, CA) and then coupled with a linear ion-trap (LIT) mass spectrometer (LTQ, Thermo Electron, San Jose, CA). Mobile phase A was 0.1% formic acid in water and B was 0.1% formic acid in acetonitrile. The peptide samples were injected and a gradient elution was performed under the following conditions: 2% B at 500 nL/min in 30 min; a linear increase of 2–42% B at 250 nL/min in 110 min; 42–98% in 30 min, consisting of the first 15 min at 250 nL/min and the second 15 min at 500nL/min; 98% at 500 nL/min for 10 min. The LIT-MS was operated in a data-dependent MS/MS mode where the five most abundant peptide molecular ions in every MS scan were sequentially selected for collision-induced dissociation (CID) using a normalized collision energy of 35%. Dynamic exclusion was applied to minimize repeated selection of peptides previously selected for CID. The capillary temperature and electrospray voltage were set to 160 °C and 1.5 kV, respectively.
Bioinformatic analysis
Tandem mass spectra were searched against the UniProt human proteomic database from the European Bioinformatics Institute (released in June 2005; http://www.ebi.ac.uk/) with SEQUEST operating on a 40 node Beowulf cluster. Peptides were searched using fully tryptic cleavage constraints. For a peptide to be considered legitimately identified, it had to achieve stringent charge state and cross correlation (Xcorr) scores of 1.9 for [M + H]1+, 2.2 for [M + 2H]2+, 3.1 for [M + 3H]3+, and a minimum delta correlation (ΔCn) of 0.08.
Proteomic categorization was performed on the basis of annotations from the Gene Ontology of the Cancer Genome Anatomy Project, National Cancer Institute, NIH (http://cgap.nci.nih.gov/Genes/BatchGeneFinder). The average molecular weight (MW) and theoretical isoelectric point (pI) of the proteins were calculated using online software from EXPASY Proteomics Server http://www.expasy.org/tools/pi_tool.html.
Statistical analysis
All experiments were performed at least three times, and representative results are presented. Independent-Samples T Test was used to compare the significance of differences in percentage of aggresome-containing cells using SPSS 10.0 for Windows (SPSS, Chicago, Illinois). P values equal to or less than 0.05 were considered statistically significant.
Results
Cd2+ is a potent aggresome inducer
Aggresome formation is thought to be a cellular response to high levels of undesirable and ubiquitinated proteins. Based on the finding that Cd2+ induces protein denaturation and ubiquitination, we hypothesized that Cd2+ may induce aggresome formation. We treated HEK293 cells with Cd2+ and then immunostained the cells with antisera specific to ubiquitin and γ-tubulin, which are concentrated in aggresomes (Johnston et al., 1998). In control cells, ubiquitinated proteins and γ-tubulin colocalize in the cytoplasm in a scattered pattern (Fig. 1A, a, b, and d). In contrast, in Cd2+-treated cells, high levels of ubiquitin and γ-tubulin are detected in prominent inclusion bodies (arrows in Fig. 1A, e, f, and h) that are close to the nucleus (Fig. 1A, g). The morphology and localization of these Cd2+-induced inclusion bodies resemble those of aggresomes. For further characterization, we analyzed Cd2+-treated cells by immunostaining with antisera specific to HDAC6 (Fig. 1B), dynein (Fig. 1C), and γ-tubulin, which are known components of the aggresomes. Fig. 1B and C shows colocalization of γ-tubulin with HDAC6 (Fig. 1B, a, b, d) and with dynein (Fig. 1C, a, b, d). These results suggest that Cd2+ induces the formation of aggresomes in HEK293 cells.
Fig. 1.
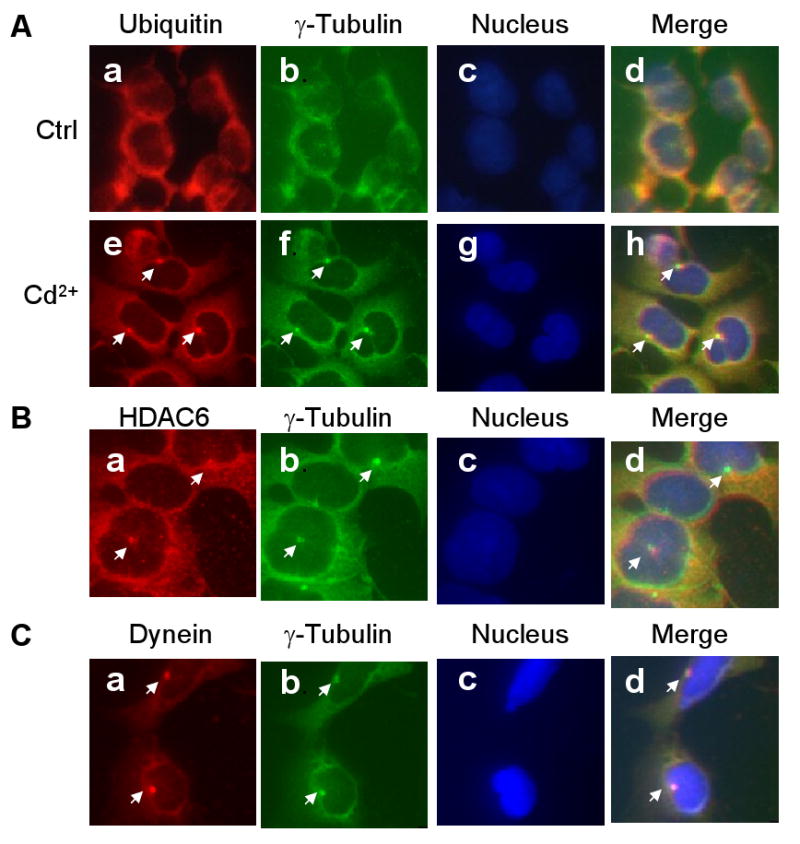
Cd2+-induced aggresome formation in HEK293 cells. HEK293 cells were cultured in the presence or absence of 20 μM Cd2+ for 6 h, then double-immunostained with antibodies against ubiquitin (Panel A, a, e; red) and γ-tubulin (Panel A, b, f; green). Panel B: Cd2+-treated cells immunostained with antibodies against HDAC6 (a, red) and γ-tubulin (b; green). Panel C: Cd2+-treated cells were immunostained with antibodies against dynein (a, red) and γ-tubulin (b, green). The nuclei (Panel A, c, g; Panel B, c; Panel C, c) were stained with DAPI (blue). Merged immunostaining images are shown in A, d, h; B, d; and C, d.
To examine whether aggresome formation induced by Cd2+ is a general phenomenon in mammalian cells, we treated cells derived from various species with Cd2+, followed by immunostaining with anti-VCP and anti-HDAC6 antibodies. Aggresomes were observed in human cervical cancer HeLa cells, Chinese hamster ovary CHO cells, and mouse embryonic fibroblast NIH3T3 cells (Fig. 2, left panel). In addition, in the PC-12 cell line, which is derived from rat pheochromocytoma and commonly used for the study of neuronal differentiation (Greene and Tischler, 1976), we also found aggresomes adjacent to nuclei after Cd2+ treatment (Fig. 2, upper right panel), suggesting the potential for Cd2+ to induce aggresome formation in neuronal cells. Similarly, Cd2+ also induced the formation of aggresomes in primary mouse liver cells (Fig. 2, lower right panel). Taken together, these results indicate that Cd2+ is capable of inducing aggresome formation in diverse mammalian cells.
Fig. 2.
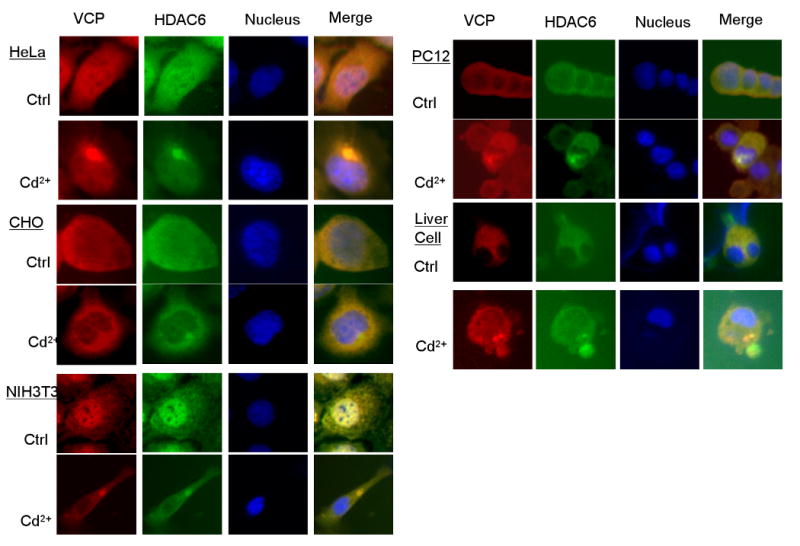
Cd2+-induced aggresome formation in various mammalian cells. Cells cultured in the presence or absence of 20 μM Cd2+ for 6 h, then double-immunostained with antibodies against VCP (red) and HDAC6 (green) The nuclei were stained with DAPI (blue), and merged immunostaining images are marked as Merge.
Enrichment of aggresomes in the lysates extracted with DB buffer
To characterize the composition of Cd2+-induced aggresomes, we treated HEK293 cells with 20μM Cd2+ for 6 h, then lysed cells with different lysis buffers, including PBS, DB buffer containing mild detergents, and SDS buffer containing the ionic detergent SDS. The lysates were separated into soluble (S) and insoluble (P) fractions by centrifugation. As shown in Fig. 3A, weight analyses show that 20.8 ± 4.2% (w/w) of the cellular proteins in the lysates of control cells were insoluble in PBS (PBS-P) and Cd2+ treatment increased this fraction to 40.2 ± 8.4%. Since the PBS-P also contained membrane fractions, we further lysed the cells with DB buffer, which contained non-ionic detergents for the membrane solubilization. As a result, the insoluble fractions (DB-P) detected after Cd2+ treatment increased from 4.7 ± 1.3% to 20.0 ± 4.3% of the total proteins. To analyze the protein composition, we re-solubilized PBS-P and DB-P with SDS buffer, resolved the contents on an SDS polyacrylamide gel, then performed Western blot analysis to determine the levels of ubiquitin, Hsp70, and Hsp40, which have been previously identified in aggresomes (Junn et al., 2002) (Fig. 3B). Hsp70 and Hsp40 both appeared in PBS-P but not in DB-P in the control cell lysates (Fig. 3B, lanes 2 and 4). In contrast, Hsp70 and Hsp40 were detected in both PBS-P and DB-P fractions of the Cd2+-treated cells (Fig. 3B, lane 8 and 10). In the Cd2+-treated cell fraction, the quantity of Hsp70 in DB-P was much lower than in PBS-P, while the difference in the quantity of Hsp40 was smaller. Likewise, the proportion of ubiquitinated proteins in the DB-P fraction of the Cd2+-treated cells was less than the PBS-P fraction (Fig. 3B, lane 8 and 10). Thus, re-suspending the PBS-insoluble fraction in DB may further dissolve some membrane components and moderately aggregated proteins.
Fig. 3.
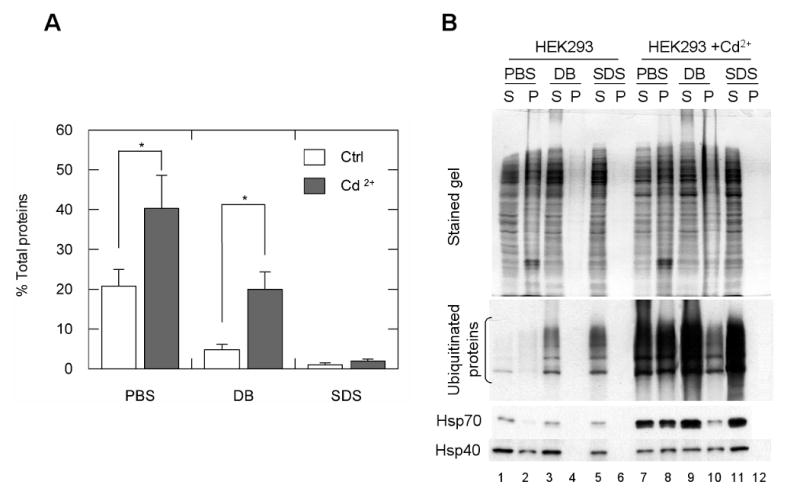
Aggresome enrichment in DB-P fractions. Untreated and treated (20 μM Cd2+) HEK293 cells were lysed in PBS, DB, or 1% SDS. The soluble fraction (S) and insoluble pellet (P) of the cell lysates were separated. (A) The percentage of the insoluble fraction of the cell lysate. The insoluble fractions of the cell lysates extracted with each buffer were weighed, and presented as a percentage of the weight of the total cell pellet. *, P<0.01, n = 4. (B) Ubiquitin, Hsp70, and Hsp40 were enriched in the DB-P fraction when the cells were treated with Cd2+. The soluble and insoluble fractions of the cell lysates were resolved on SDS-polyacrylamide gels, transferred to filters, and immunoblotted with antisera against ubiquitin, Hsp70, and Hsp40.
Mass spectrometry analysis of the aggresome-enriched DB-P fraction
To examine the composition of the aggresomes-enriched fraction, we analyzed the DB-P fraction using mass spectrometry (MS), which identified 145 proteins with increased quantity in DB-P insoluble fraction from Cd2+-treated cells as compared with untreated control cells. Among these proteins, 57 proteins, with observed peptide frequencies of each identified protein were greater than 14 and only appeared in extracts of Cd2+-treated cells; and the rest of 88 proteins in which the identified peptides of each protein in the sample of Cd2+-treated cells were 14 fold greater than that of the control cells. A comparison between the molecular weight of the proteins in the aggresome-enriched dataset and the whole human proteome dataset is shown in Fig. 4A. The peak MW distribution of the aggresome-enriched fraction is 120 kDa or greater, comprising 25% of the identified proteins, markedly higher than the 7% in the human proteome. In contrast, the MW distribution of the human proteome in the range of 20 kDa is 36%, significantly higher than the 8% in the aggresome-enriched fraction. These results suggest that the high molecular weight proteins tend to aggregate. Moreover, proteomic analyses revealed only one peak pI distribution of the DB-insoluble proteins at approximately 6 (Fig. 4B). This is in contrast to the two pI peak distribution at about 6 and 9 observed in the human proteome (Wu et al., 2006). Thus, the pI distribution of aggresome-enriched proteins seems to agree with the theory that proteins with a pI close to neutral pH have a higher tendency to aggregate (VanBogelen et al., 1999). Gene ontology analysis revealed that approximately 26% of these aggresome-enriched proteins were related to biosynthesis and protein translation, suggesting that Cd2+ treatment affects protein synthesis in the cells (Fig. 4C). In particular, we found 11 proteins, including tRNA synthetase, translation initiation factor, and ribosomal proteins, accumulated in the aggresome fraction when the cells were treated with Cd2+ (Table I). This suggests that Cd2+ may down-regulate protein translation by sequestering the components of the protein translation machinery into aggresomes. Also, a significant proportion of the identified proteins were classified as members of molecular chaperones (5%) and the ubiquitin proteasome system (8%) (Fig. 4C). These molecules eliminate misfolded or ubiquitinated proteins from cells by either refolding them into native forms or facilitating their degradation. In addition, 12% of the aggresome proteins are components of, or related to, the cytoskeleton, which may form the structure of aggresomes (Fig. 4C).
Fig. 4.
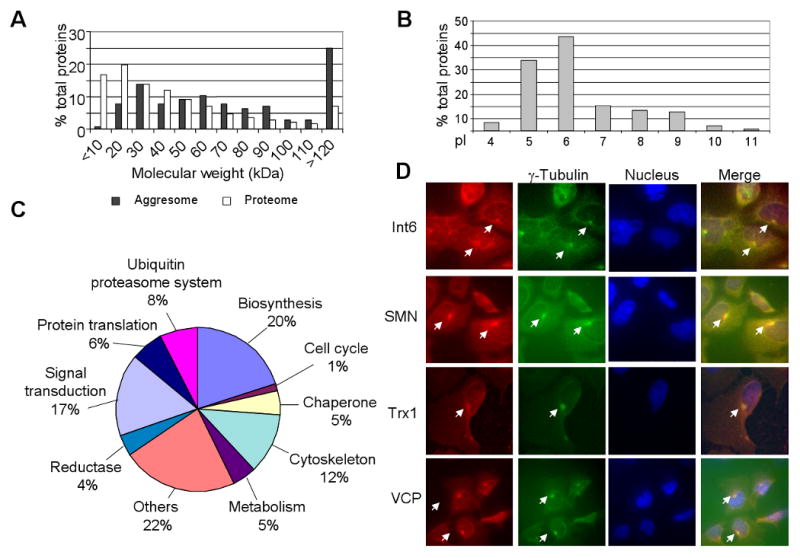
Molecular weight (MW), isoelectric point (pI), and functional classification of proteins identified in aggresome enriched fraction from cells treated with Cd2+. MW (A) and pI (B) of proteins were calculated using the pI/MW computation tool from ExPASy, and the MWs of the proteins identified in the aggresome enriched fraction were compared with 37,503 proteins in human proteome. Representative data from three independent experiments are presented. (C) Functional classification of proteins identified in the aggresome-enriched fraction of cells after exposure to Cd2+. Assignments were made on the basis of combined annotations from Gene Ontology of the Cancer Genome Anatomy Project database at the National Cancer Institute. The 145 proteins were chosen with the following criteria: 57 proteins exclusively in the aggresome enriched fraction, with at least fourteen peptides identified from each; 88 proteins highly abundant in the aggresome enriched fraction with the total number of peptides of each protein more than fourteen fold that of the control samples. (D) Immunostaining of eukaryotic translation initiation factor 3 subunit 6 (Int 6), survival of motor neuron 1 (SMN), thioredoxin I (Trx1), and VCP with their respective antibodies (red). The accumulation of these proteins in aggresomes is indicated by arrowheads. The centrosomes were indicated by immunostaining of γ- tubulin (green). The nucleus was stained with DAPI (blue).
Table I.
Protein translation-related proteins identified in the aggresome-enriched fraction of HEK293 cells exposed to Cd2+. Proteomic data of the DB-insoluble fraction of HEK293 cells cultured in the presence or absence of Cd2+ were compared. Listed here are the components of protein translation machinery. The ratio of identified peptides for each protein (Agg/Ctrl) between the Cd2+-treated sample (Agg _peptides) and the control (Ctrl_peptides) was calculated.
| Protein | Agg_peptides | Ctrl_peptides | Agg/Ctrl | Reference |
|---|---|---|---|---|
| Seryl-tRNA synthetase (EC 6.1.1.11) | 28 | 0 | / | SYS_HUMAN |
| Tryptophanyl-tRNA synthetase (EC 6.1.1.2) | 25 | 1 | 25 | SYW_HUMAN |
| Asparaginyl-tRNA synthetase, cytoplasmic (EC 6.1.1.22) | 40 | 2 | 20 | SYNC_HUMAN |
| Alanyl-tRNA synthetase (EC 6.1.1.7) | 93 | 5 | 19 | SYA_HUMAN |
| Tyrosyl-tRNA synthetase, cytoplasmic (EC 6.1.1.1) | 49 | 3 | 16 | SYYC_HUMAN |
| Eukaryotic translation initiation factor 3 subunit 6 (eIF-3 p48) (eIF3e) | 56 | 0 | / | IF36_HUMAN |
| Translation initiation factor eIF-2B delta subunit | 16 | 0 | / | EI2BD_HUMAN |
| Eukaryotic translation initiation factor 3 subunit 5 (eIF3f) | 20 | 1 | 20 | IF35_HUMAN |
| Eukaryotic translation initiation factor 5A (eIF-5A) | 135 | 7 | 19 | IF5A_HUMAN |
| 40S ribosomal protein S10 | 33 | 2 | 17 | RS10_HUMAN |
| 60S ribosomal protein L26 | 15 | 1 | 15 | RL26_HUMAN |
Proteins in the ubiquitin-proteasome system were also detected in the aggresome-enriched DB-P fraction. One E2 enzyme and two zinc finger-containing E3 enzymes were identified and, consistent with findings from a previous report (Johnston et al., 1998), four proteasome subunits accumulated in the aggresome-enriched DB-P fraction were also detected (Table II). It is of particular interest that four deubiquitinating enzymes and ubiquitin carboxyl-terminal hydrolases were also detected in DB-P (Table II). These results suggest that ubiquitin molecules, as tags for the to-be-degraded proteins, may be transported into aggresomes, then recycled by deubiquitinating enzymes within the aggresomes.
Table II.
Components of the ubiquitin proteasome system identified in the aggresome-enriched fraction of HEK293 cells exposed to Cd2+. Proteomic data of proteins reported to accumulate in aggresomes were compared by analyzing the DB-insoluble fraction of cells cultured in the presence or absence of Cd2+. The ratio of identified peptides for each protein (Agg/Ctrl) between the Cd2+-treated sample (Agg_peptides) and the control (Ctrl_peptides) was calculated.
| Protein | Agg_peptides | Ctrl_peptides | Agg/Ctrl | Reference |
|---|---|---|---|---|
| Ubiquitin-conjugating enzyme E2 M (EC 6.3.2.19) | 17 | 1 | 17 | UBE2M_HUMAN |
| Tripartite motif protein 25 (Zinc finger protein 147) | 22 | 0 | / | TRI25_HUMAN |
| Zinc finger protein 294 | 21 | 0 | / | ZN294_HUMAN |
| Proteasome subunit alpha type 5 (EC 3.4.25.1) | 19 | 0 | / | PSA5_HUMAN |
| Proteasome subunit beta type 1 (EC 3.4.25.1) | 19 | 0 | / | PSB1_HUMAN |
| 26S proteasome non-ATPase regulatory subunit 13 | 16 | 0 | / | PSD13_HUMAN |
| 26S proteasome non-ATPase regulatory subunit 6 | 21 | 1 | 21 | PSD6_HUMAN |
| Ubiquitin carboxyl-terminal hydrolase 14 (EC 3.1.2.15) | 22 | 0 | / | UBP14_HUMAN |
| Ubiquitin carboxyl-terminal hydrolase isozyme L3 (EC 3.4.19.12) | 16 | 0 | / | UCHL3_HUMAN |
| Ubiquitin carboxyl-terminal hydrolase 10 (EC 3.1.2.15) | 19 | 1 | 19 | UBP10_HUMAN |
| Ubiquitin carboxyl-terminal hydrolase 11 (EC 3.1.2.15) | 18 | 1 | 18 | UBP11_HUMAN |
The aggresomal localization of several proteins identified by proteomic analysis was confirmed by immunostaining in Cd2+-treated cells. Translation initiation factor 3, subunit 6 (Int 6), survival of motor neuron 1 (SMN), thioredoxin I, and VCP were highly abundant in aggresome-enriched DB-insoluble fractions. Immunostaining also revealed the colocalization of these proteins in aggresomes (Fig. 4D).
Cd2+ promotes the accumulation of VCP in aggresomes
The colocalization of VCP with γ-tubulin in aggresomes suggests that VCP may be a component of the aggresome (Fig. 4D). Because of the dual-binding capability of VCP to HDAC6 and polyubiquitinated proteins, we studied the potential involvement of VCP in the process of aggresome formation. We constructed VCP-GFP fusion proteins to trace the accumulation of VCP in aggresomes in Cd2+-treated cells. Results showed colocalization of VCP-GFP with γ-tubulin (Fig. 5A) and HDAC6 (Fig. 5B), supporting the notion that VCP may participate in aggresome formation. In Fig. 5B, the core of the aggresome was surrounded by the intermediate fiber protein, vimentin. It is noted that the staining pattern of aggresomes in Cd2+-treated cells is similar to that of the aggresomes induced by proteasome inhibitors (Johnston et al., 1998; Kawaguchi et al., 2003).
Fig. 5.
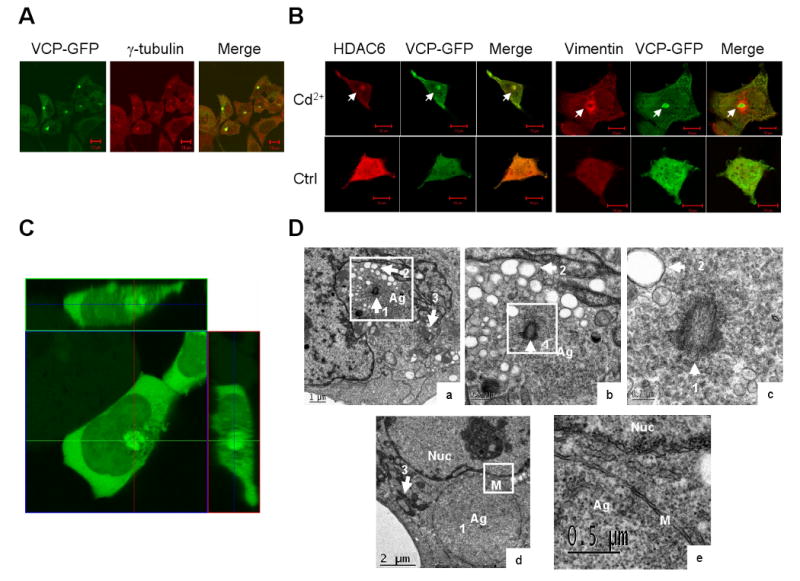
Cd2+ induces VCP accumulation in aggresomes. (A) HEK 293 cells stably expressing VCP-GFP were exposed to 20 μM Cd2+ for 6 h, and then the location of γ-tubulin, HDAC6, and vimentin (B) was detected by immunostaining with antibodies. Superimposed confocal microscopy images (Merge) demonstrate the colocalization of VCP-GFP (green) with aggresome markers, γ-tubulin (red) and HDAC6 (red). Accumulation of VCP-GFP is indicated by arrowheads. Scale bar = 10 μm. (C) HEK293 cells stably expressing VCP-GFP were treated with 20 μM Cd2+ for 6 h. The aggresome structure was indicated by the fluorescence of VCP-GFP in live cells. The confocal microscopy images of Z-stack, 0.5 μm interval, and orthogonal sections were shown on the upper and right sides. (D) Transmission EM of aggresomes. Ultrastructure of HEK293 12 h treatment with 30 μM Cd2+ (a) (d) and a higher-magnification view of the sections (b) bar = 0.5 μm, (c) bar = 0.2 μm, and (e) bar = 0.5 μm. Arrowhead 1, centriole; arrowhead 2, vacuoles; arrowhead 3, mitochondria; Ag, aggresome; Nuc, nucleus; M, membrane.
Use VCP-GFP fusion proteins offers a dynamic observation of the formation of aggresomes in live cells. The morphology of the aggresomes in live cells is shown in Fig. 5C, in which VCP-GFP is highly enriched in the sphere-shaped aggresomes adjacent to the nuclei and surrounded by structures with weak fluorescence, further supporting a role for VCP in promoting aggresome formation.
Electron micrographs of Cd2+-treated cells show that the aggresome contains a large number of fine granules located close to the centriole, which is surrounded by mitochondria and vacuoles (Fig. 5D a, b, c). Morphologically, these vacuoles are similar to those found in aggresomes formed by misfolded cystic fibrosis transmembrane conductance regulator (CFTR) aggregates (Johnston et al., 1998), in which no membrane encircles the aggresomes in the early stages. In addition, we found that later-stage aggresomes were enclosed by double membrane layers without surrounding vacuoles and mitochondria (Fig. 5D d, e, membrane indicated as M). Thus, the membrane envelope may serve to isolate protein aggregates from the cytoplasm.
The effect of Cd2+ on the accumulation of VCP into aggresomes was dose and time dependent. Aggresomes were formed during the 1 μM Cd2+ treatment for 6 h. The percentage of aggresome-containing cells increased to a maximum with the 20 μM Cd2+ treatment for 6 h (Fig. 6A). In the presence of 20 μM Cd2+, aggresomes started to form after 2 h (Fig. 6B).
Fig. 6.

The dose and time-course effect of Cd2+-induced aggresome formation. HEK293 cells transfected with VCP-GFP and cultured in 96-well plates were exposed to increased concentrations of Cd2+ for 6 h or with 20 μM Cd2+ for the indicated lengths of time. The green fluorescent aggresomes were observed using inverted fluorescence microscopy. Aggresome-containing cells from 150 randomly selected cells were counted. Standard errors were calculated from four independent experiments.
The N domain (#2–206) and C-terminal tail (#780–806) of VCP are essential for aggresome formation
To determine which VCP domain(s) might be required for aggresome formation, we transfected VCP-GFP variants with mutations in different regions into HEK293 cells. After treatment with Cd2+, aggresomes formed in the cells expressing wild-type VCP-GFP (#1–806, WT) (Fig. 7A, a). However, no aggresomes were observed in cells expressing N domain-deleted VCP-GFP (#207–806) (Fig. 7A, b), suggesting a requirement for the N domain. However, cells transfected with the N domain (#1–200) alone did not form aggresomes after treatment with Cd2+ (Fig. 7A, c), suggesting that the N domain of VCP by itself is not sufficient for the formation of aggresomes. We also tested the requirement of VCP ATPase activity for aggresome formation and found that the A1 (K251A) and A2 (K524A) mutants, which were defective in ATP-binding in the D1 and D2 domains, were still capable of mediating aggresome formation (Fig. 7A, d, e). Thus, the ATPase activity of VCP may be not required. VCP lacking the C-terminal 27 amino acids (#1–779 in Fig. 7A, f) failed to induce aggresomes, but those missing the C-terminal 21 (#1–785 in Fig. 7A, g) or 10 (#1–796 in Fig. 7A, h) residues maintained their aggresome formation capacities. These results suggest that the short fragment encompassed by VCP residues #779–785 plays a critical role in the effect of VCP on aggresome formation (Fig. 7B, C).
Fig. 7.
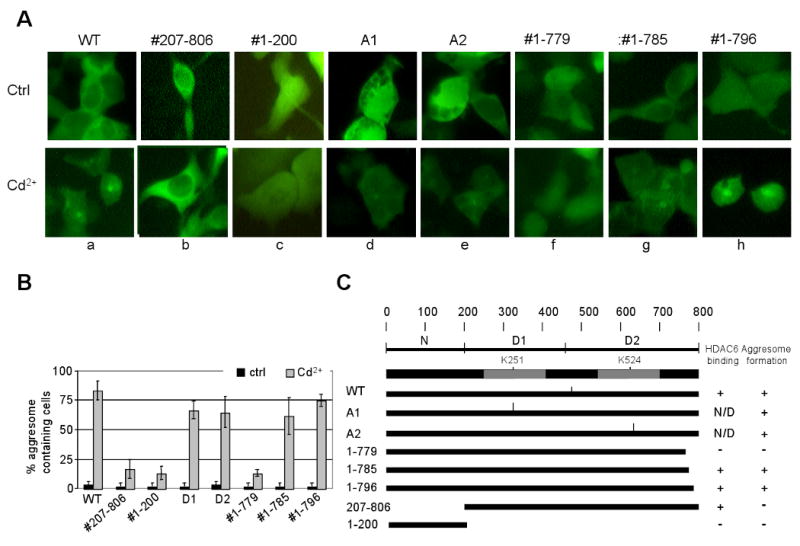
The requirement for VCP accumulation in aggresomes. HEK293 cells transfected with VCP-GFP variants were exposed to 20 μM Cd2+ for 6 h. The cell images were obtained using inverted fluorescence microscopy. (A) HEK293 cells were transfected with the following VCP-GFP variants: WT(a); the variant lacking the N domain, (b) #207–806; N domain only, (c) #1–200; the D1 or D2 ATPase site-specific mutants, (d, e) A1, A2; the C-terminal deletion mutants (f) #1–779, (g) #1–785, (h) #1–796. (B) Quantification (%) of cells containing aggresomes. Results are presented as the percentage of aggresome-containing cells per 150 randomly counted cells from three independent experiments. (C) A schematic diagram indicating the capacity of VCP variants to bind HDAC6 and to promote aggresome formation.
VCP interacts with HDAC6 through its C-terminal tail (#780–806)
It has been reported that HDAC6 transports ubiquitinated and misfolded proteins to aggresomes via a microtubule network (Kawaguchi et al., 2003). Consistent with this, we found that aggresome formation in Cd2+-treated cells was significantly suppressed by the histone deacetylase inhibitor scriptaid (Corcoran et al., 2004) and by the microtubulin depolarizing agent nocodazole (Johnston et al., 1998) (Fig. 8). Since previous studies indicate that HDAC6 not only associates with VCP (Seigneurin-Berny et al., 2001), but also bridges the interaction of polyubiquitinated and misfolded proteins with dynein (Kawaguchi et al., 2003), we examined whether VCP associates with HDAC6 in the same complex and found an increased association among VCP, HDAC6 and dynein after the cells were treated with Cd2+ (Fig. 9A). The VCP C-terminal deletion mutant #1–779, in contrast to #1–785 and #1–796, did not bind HDAC6, indicating that the fragment #779–785 is essential for VCP to interact with HDAC6 (Fig. 9B). To further verify the role of HDAC6 in VCP-mediated aggresome formation, we knocked down HDAC6 with shRNA, and then treated the cells with Cd2+ (Fig. 9C). The cells with HDAC6 shRNA showed significantly decreased aggresome formation efficiency (from 67.3±10.3% to 25.2±6.8%, Fig. 9D), supporting the notion that HDAC6 mediates the delivery of VCP into aggresomes. Based on these observations, we propose a model that VCP binds ubiquitinated proteins through its N domain and HDAC6 through its C-terminal region. In the meantime, HDAC6 interacts with a dynein motor to transport the complex of VCP and ubiquitinated proteins to aggresomes (Fig. 9E).
Fig. 8.
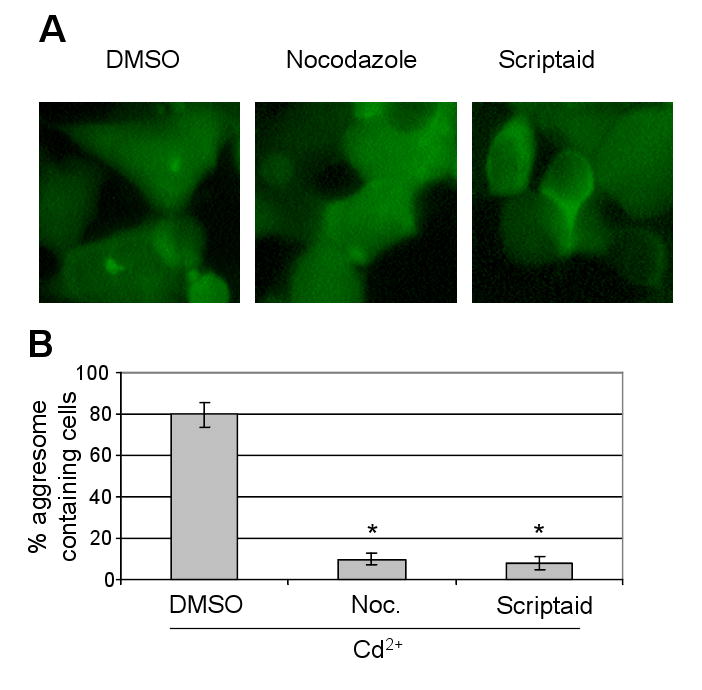
Accumulation of VCP-GFP in aggresomes via HDAC6 and microtubule network. HEK293 cells were exposed to 20 μM Cd2+ in the presence of DMSO, 0.6 μM nocodazole or 10 μM scriptaid for 6 h. (A) The cell images were obtained using inverted fluorescence microscopy. (B) The percentage of cells containing aggresomes. Results are presented as the percentage of aggresome-containing cells per 150 randomly counted cells from three independent experiments. *, significantly reduced aggresome formation is shown in the nocodozole and scriptaid treatment groups compared with DMSO-treated cells (P<0.01).
Fig. 9.
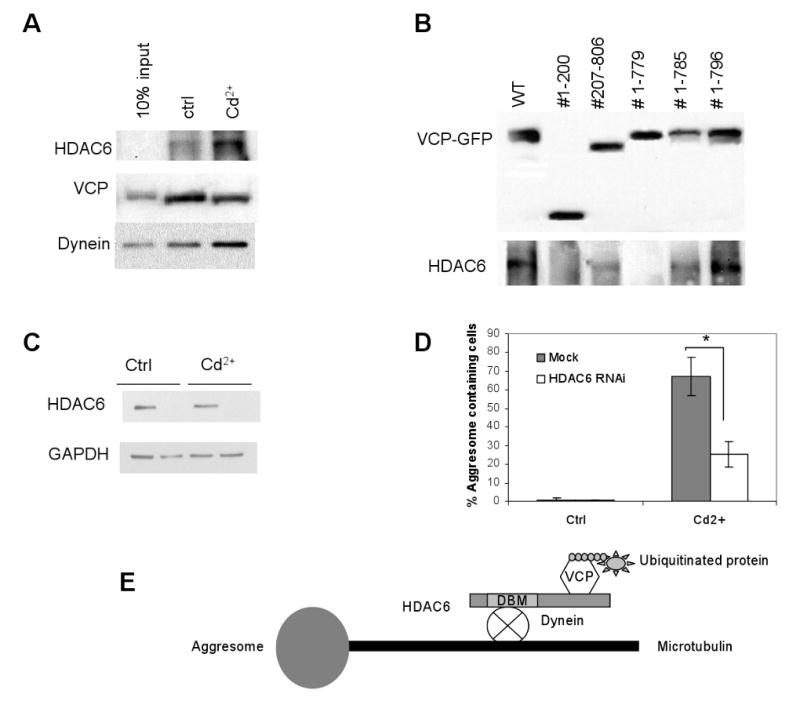
Requirement of C-terminal region in VCP-GFP for interaction with HDAC6. (A) HEK293 cells cultured in the presence or absence of 20 μM Cd2+ for 6 h were lysed in RIPA-NS buffer. VCP was immnunoprecipitated using an anti-VCP antibody, followed by Western blot analysis using anti-VCP, HDAC6, and dynein antibodies, respectively. (B) HEK293 cells expressing C-terminal deletion mutants of VCP were treated with 20 μM Cd2+ for 6 h, then lysed in RIPA-NS buffer. VCP-GFP variants were immunoprecipiated with an anti-GFP antibody. Bound proteins were separated by SDS-PAGE and detected by Western blot analysis using anti-VCP and anti-HDAC6 antibodies, respectively. (C) HEK293 cells transfected with HDAC6 shRNA for 5 days were examined for HDAC6 expression by Western blot analysis. (D) HEK293 cells transfected with HDAC6 shRNA for 5 days were treated with 20 μM Cd2+ for 6 h and the percentage of aggresome-containing cells was calculated from 150 randomly selected cells in four independent experiments. *, significantly reduced number of cells containing aggresomes as compared with mock-transfected cells (P<0.01). (E) Scheme illustrating the role of VCP in mediating aggresome formation through HDAC6.
Discussion
In this study, we identified Cd2+ as an aggresome inducer in diverse types of mammalian cells exposed to this toxic metal ion. In addition, we found that VCP plays a critical role in Cd2+-induced aggresome formation. The potential for Cd2+ to induce aggresome formation is supported by the observations that Cd2+ causes broad protein misfolding and ubiquitination (Jungmann et al., 1993; Nies, 1999; Hall, 2002). Cd2+ induces protein misfolding mainly through ion substitution and oxidation. As a divalent ion with its high binding affinity to metal-sensitive groups, such as thiol or histidyl moieties, Cd2+ inactivates metal-containing enzymes by replacing the biologically functional ions, such as Ca2+, Fe2+, Cu2+, and Zn2+, from vital cellular components (Nies, 1999). Cd2+ also causes oxidative stress by promoting the formation of free radicals and depleting glutathione, and by binding to sulfhydryl groups of the proteins (Hall, 2002). In addition, Cd2+ treatment has been shown to enhance the level of ubiquitinated proteins in mammalian and yeast cells (Jungmann et al., 1993; Figueiredo-Pereira and Cohen, 1999) through suppressing both ATP-dependent and -independent protein degradation, but not the protease activities of the 20S proteasome (Figueiredo-Pereira and Cohen, 1999). However, the mechanisms by which Cd2+ inhibits protein degradation remain to be elucidated.
Aggresomes are known to collect ubiquitinated and misfolded proteins. The proteomic composition of aggresomes is still unknown. To examine the aggresome's composition, we used sucrose density centrifugation to attempt aggresome purification; however, this proved to be difficult due to variations in their size and density. We hypothesized that aggresomes may have low solubility in detergent buffer and may be enriched in detergent-insoluble fractions. Our examination of aggresome solubility in detergent buffer revealed that, compared to untreated control cells, lysates of the Cd2+ -treated cells retain significant amounts of insoluble proteins, including ubiquitinated and heat shock proteins. This finding suggested that aggresomes could be enriched in the detergent-insoluble fraction. Our proteomic analyses of DB-P from Cd2+-treated cells identified 145 proteins in this aggresome-enriched fraction, which contained a large proportion (25%) of molecules above 120 kDa, as compared to 7% in the whole proteome. This observation indicates that high-molecular weight proteins tend to aggregate under stress conditions, while low-molecular weight proteins tend to remain in the soluble fraction (Mogk et al., 1999). We also found that the molecules involved in protein translation, such as tRNA synthetase, translation initiation factors, and ribosomal proteins, accumulate in the DB-P fraction (Table I). This may explain why the levels of soluble ribosomal proteins increased in the cytosol after the cells were exposed to Cd2+ for 1 h, followed by a decrease in their levels after exposure for 12 h (Bae and Chen, 2004).
Proteomic data also suggest a mechanism for the ubiquitin recycling that occurs during aggresome formation. In the ubiquitin proteasome system, a pool of free ubiquitin molecules is critical for the ubiquitination of misfolded proteins (Wu et al., 1981). The cellular level of free ubiquintin is generated and maintained by proteasomal degradation of ubiquitinated proteins and deubiquitinating enzyme activity (Amerik and Hochstrasser, 2004). Proteasomes degrade ubiquitinated proteins and release the ubiquitin chain, while deubiquitinating enzymes directly remove ubiquitin from ubiquitinated proteins through their hydrolase activity. The level of free ubiquitin molecules in cells will decrease if ubiquitinated proteins are not properly degraded due to a loss of proteasome activity, which will decrease the levels of free ubiquitin in the cytoplasm and consequently the ubiquitination of misfolded proteins. In the DB-P fraction, we identified four deubiquitinating enzymes: (1) ubiquitin carboxyl-terminal hydrolase 14 (EC 3.1.2.15); (2) ubiquitin carboxyl-terminal hydrolase isozyme L3 (EC 3.4.19.12); (3) ubiquitin carboxyl-terminal hydrolase 10 (EC 3.1.2.15); and (4) ubiquitin carboxyl-terminal hydrolase 11 (EC 3.1.2.15). These enzymes may play an important role in the continuous formation of aggresomes by recycling ubiquitin proteins from aggresomes to the cytosolic pool.
The capacity of Cd2+ to promote aggresome formation is rapid and efficient. In our study, proteins, including ubiquitin, HDAC6, and dynein, accumulated into aggresomes in HEK293 cells treated with 20 μM Cd2+ for 6 h (Fig. 1A e, 1B a, 1C a). The kinetics of aggresome formation were visualized with confocal microscopy, using VCP-GFP in live HEK293 cells, which showed that smaller aggregates merged and became microscopically visible within 2 h (unpublished results). The efficacy Cd2+-induced aggresome formation was shown by the observation that other divalent heavy metal ions, such as Pb2+, Cu2+, Co2+, Ni2+, Zn2+, and Mn2+, failed to induce aggresomes even at concentrations as high as 80 μM (unpublished results), while Cd2+ induced aggresomes at concentrations as low as 1 μM.
Nevertheless, other divalent ions have been reported to also induce inclusion body formation at higher concentrations. For example, Moore and Goyer reported that feeding rats a diet mixed with 1% lead induced inclusion body formation in cells (Moore and Goyer, 1974). Romero et al. also reported that 1.38 mM and 1.04 mM lead nitrate induced inclusion body formation in Buffalo Green Monkey (BGM) and VERO cells (Romero et al., 2004). The reason we did not observe aggresome formation induced by other ions may be due to differences in the ion concentrations and cell types that we used. Whether all heavy metal ions could induce inclusion body formation at higher concentrations through mechanisms similar to those used by Cd2+ requires further examination.
Aggresome formation is a general response to Cd2+ in mammalian cells. In addition to HEK293 cells, we observed that Cd2+ induced aggresomes with similar efficiency in cell lines derived from different species: HeLa, CHO, and NIH3T3 cells. Mouse primary liver cells also responded to Cd2+ by forming aggresomes. The observation that Cd2+ induces aggresome formation in the PC-12 neuronal cell line derived from rat pheochromocytoma suggests that the aggresome induction property of Cd2+ may correlate with its ability to induce and exacerbate neurodegenerative diseases.
Aggresomes share common markers with inclusion bodies in neurodegenerative diseases. HDAC6 and ubiquitin both accumulate in aggresomes and in Lewy bodies, a hallmark inclusion body seen in Parkinson's disease (Kawaguchi et al., 2003). Moreover, many inclusion body proteins, such as dentatorubral-pallidoluysian atrophy protein with polyglutamine expansion (Shimohata et al., 2002), peripheral myelin protein 22 (PMP22) protein mutants (Ryan et al., 2002), alpha-synuclein (Lee and Lee, 2002), presenilin (Namekata et al., 2002), and parkin (Junn et al., 2002), are capable of forming aggresomes. Degeneration of these proteins is associated with the death of specific neuronal cell populations in a number of neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), Alzheimer's disease (AD), Parkinson's disease, Huntington disease, and other polyglutamine expansion disorders. Therefore, chemicals with the capacity to induce aggresome formation, like Cd2+, may promote the progression of neurodegenerative diseases. This hypothesis has been supported by observations in both animals and human. For instance, Wistar rats developed peripheral polyneuropathy after long-term exposure to Cd2+ from drinking water (Sato et al., 1978). In humans, Cd2+ concentrations are significantly elevated in the brains of AD (Lui et al., 1990) and ALS (Vinceti et al., 1997) patients. Also, occupational exposure to Cd2+ increases the risk of AD (Bar-Sela et al., 2001), ALS, and brain atrophy (Bar-Sela et al., 1992). Exposure to Cd2+ can cause the development of parkinsonism three months after recovery from acute Cd2+ toxicity (Okuda et al., 1997). Based on the similarities between the aggresomes and inclusion bodies found in these diseases, it is plausible that the capacity of Cd2+ to induce aggresomes and inclusion bodies contributes to the progression of neurodegenerative diseases.
In studying the molecular mechanism of Cd2+-induced aggresome formation, we found that VCP is present in aggresomes, consistent with the report that decreased expression of VCP protein suppresses aggresome formation (Wojcik et al., 2004). More recently, Kitami et al. reported that VCP is a component of aggresomes induced by proteasome inhibitors (Kitami et al., 2006). These observations support a key role of VCP in the formation of aggresomes.
VCP interacts with HDAC6, which, as a central regulator, directs the transport of ubiquitinated proteins to aggresomes. Since the binding of the ZnF-UBP motif of HDAC6 to free ubiquitin blocks the interaction of VCP with HDAC6 (Seigneurin-Berny et al., 2001), the free ubiquitin concentration in cells could play a regulatory role in such interactions. When the degradation of polyubiquitinated proteins is suppressed, the accumulation of these proteins decreases the level of free ubiquitin in the cell, causing the release of free ubiquitin from the ZnF-UBP binding site of HDAC6. This enables VCP/polyubiquitinated proteins to form complexes with HDAC6 and to load onto the dynein motor. The dynein motor then transports the VCP/polyubiquitin/HDAC6 complex via a microtubule track to the aggresomes. Our model suggests that the interaction between HDAC6 and VCP may act as a sensor for regulating the transport of polyubiquitinated proteins to the aggresomes. It is interesting to note that VCP proteins bearing mutations at their ATP binding sites still retain their capacity to promote aggresome formation in Cd2+-exposed cells. This differs from a previous report showing that the ATPase activity of the VCP D2 domain is required for the induction of aggresomes by MG132, a proteasome inhibitor (Kitami et al., 2006). The reason for this discrepancy is not known and requires further investigation.
In summary, we have identified Cd2+ as a novel aggresome inducer and suggest a mechanistic basis for the capacity of this heavy metal to act as an etiologic factor for neurodegenerative diseases. In addition, we have discovered a critical role for VCP in Cd2+-induced aggresome formation. Our findings may shed light on the development of therapeutics for Cd2+ poisoning and associated diseases.
Acknowledgments
The authors thank Alma C Arnold, Michael Jason de la Cruz, Wanghua Gong and Dr. Jian Huang for technical assistance in confocal, electron microscopy, cell culture and RNA interference respectively, and thank Dr. Joost J. Oppenheim for reviewing the manuscript. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amerik AY, Hochstrasser M. Mechanism and function of deubiquitinating enzymes. Biochim Biophys Acta. 2004;1695:189–207. doi: 10.1016/j.bbamcr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Bae W, Chen X. Proteomic study for the cellular responses to Cd2+ in Schizosaccharomyces pombe through amino acid-coded mass tagging and liquid chromatography tandem mass spectrometry. Mol Cell Proteomics. 2004;3:596–607. doi: 10.1074/mcp.M300122-MCP200. [DOI] [PubMed] [Google Scholar]
- Bar-Sela S, Levy M, Westin JB, Laster R, Richter ED. Medical findings in nickel-cadmium battery workers. Isr J Med Sci. 1992;28:578–583. [PubMed] [Google Scholar]
- Bar-Sela S, Reingold S, Richter ED. Amyotrophic lateral sclerosis in a battery-factory worker exposed to cadmium. Int J Occup Environ Health. 2001;7:109–112. doi: 10.1179/107735201800339470. [DOI] [PubMed] [Google Scholar]
- Corcoran LJ, Mitchison TJ, Liu Q. A novel action of histone deacetylase inhibitors in a protein aggresome disease model. Curr Biol. 2004;14:488–492. doi: 10.1016/j.cub.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Dai RM, Chen E, Longo DL, Gorbea CM, Li CC. Involvement of valosin-containing protein, an ATPase Co-purified with IkappaBalpha and 26 S proteasome, in ubiquitin-proteasome-mediated degradation of IkappaBalpha. J Biol Chem. 1998;273:3562–3573. doi: 10.1074/jbc.273.6.3562. [DOI] [PubMed] [Google Scholar]
- Dai RM, Li CC. Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat Cell Biol. 2001a;3:740–744. doi: 10.1038/35087056. [DOI] [PubMed] [Google Scholar]
- Dai RM, Li CC. Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat Cell Biol. 2001b;3:740–744. doi: 10.1038/35087056. [DOI] [PubMed] [Google Scholar]
- Elinder CG, Lind B, Kjellstrom T, Linnman L, Friberg L. Cadmium in kidney cortex, liver, and pancreas from Swedish autopsies. Estimation of biological half time in kidney cortex, considering calorie intake and smoking habits. Arch Environ Health. 1976;31:292–302. doi: 10.1080/00039896.1976.10667239. [DOI] [PubMed] [Google Scholar]
- Figueiredo-Pereira ME, Cohen G. The ubiquitin/proteasome pathway: friend or foe in zinc-, cadmium-, and H2O2-induced neuronal oxidative stress. Mol Biol Rep. 1999;26:65–69. doi: 10.1023/a:1006909918866. [DOI] [PubMed] [Google Scholar]
- Garcia-Mata R, Bebok Z, Sorscher EJ, Sztul ES. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J Cell Biol. 1999;146:1239–1254. doi: 10.1083/jcb.146.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonda MA, Aaronson SA, Ellmore N, Zeve VH, Nagashima K. Ultrastructural studies of surface features of human normal and tumor cells in tissue culture by scanning and transmission electron microscopy. J Natl Cancer Inst. 1976;56:245–263. doi: 10.1093/jnci/56.2.245. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JL. Cellular mechanisms for heavy metal detoxification and tolerance. J Exp Bot. 2002;53:1–11. [PubMed] [Google Scholar]
- Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Illing ME, Kopito RR. Cytoplasmic dynein/dynactin mediates the assembly of aggresomes. Cell Motil Cytoskeleton. 2002;53:26–38. doi: 10.1002/cm.10057. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmann J, Reins HA, Schobert C, Jentsch S. Resistance to cadmium mediated by ubiquitin-dependent proteolysis. Nature. 1993;361:369–371. doi: 10.1038/361369a0. [DOI] [PubMed] [Google Scholar]
- Junn E, Lee SS, Suhr UT, Mouradian MM. Parkin accumulation in aggresomes due to proteasome impairment. J Biol Chem. 2002;277:47870–47877. doi: 10.1074/jbc.M203159200. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Kitami MI, Kitami T, Nagahama M, Tagaya M, Hori S, Kakizuka A, Mizuno Y, Hattori N. Dominant-negative effect of mutant valosin-containing protein in aggresome formation. FEBS Lett. 2006;580:474–478. doi: 10.1016/j.febslet.2005.12.044. [DOI] [PubMed] [Google Scholar]
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Kopito RR. The missing linker: an unexpected role for a histone deacetylase. Mol Cell. 2003;12:1349–1351. doi: 10.1016/s1097-2765(03)00498-2. [DOI] [PubMed] [Google Scholar]
- Le Y, Iribarren P, Zhou Y, Gong W, Hu J, Zhang X, Wang JM. Silencing the formylpeptide receptor FPR by short-interfering RNA. Mol Pharmacol. 2004;66:1022–1028. doi: 10.1124/mol.104.001982. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Lee SJ. Characterization of cytoplasmic alpha-synuclein aggregates, Fibril formation is tightly linked to the inclusion-forming process in cells. J Biol Chem. 2002;277:48976–48983. doi: 10.1074/jbc.M208192200. [DOI] [PubMed] [Google Scholar]
- Li Z, Arnaud L, Rockwell P, Figueiredo-Pereira ME. A single amino acid substitution in a proteasome subunit triggers aggregation of ubiquitinated proteins in stressed neuronal cells. J Neurochem. 2004;90:19–28. doi: 10.1111/j.1471-4159.2004.02456.x. [DOI] [PubMed] [Google Scholar]
- Lui E, Fisman M, Wong C, Diaz F. Metals and the liver in Alzheimer's disease: an investigation of hepatic zinc, copper, cadmium, and metallothionein. J Am Geriatr Soc. 1990;38:633–639. doi: 10.1111/j.1532-5415.1990.tb01421.x. [DOI] [PubMed] [Google Scholar]
- Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G. A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. Embo J. 2000;19:2181–2192. doi: 10.1093/emboj/19.10.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogk A, Tomoyasu T, Goloubinoff P, Rudiger S, Roder D, Langen H, Bukau B. Identification of thermolabile Escherichia coli proteins: prevention and reversion of aggregation by DnaK and ClpB. Embo J. 1999;18:6934–6949. doi: 10.1093/emboj/18.24.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JF, Goyer RA. Lead-induced inclusion bodies: composition and probable role in lead metabolism. Environ Health Perspect. 1974;7:121–7. doi: 10.1289/ehp.747121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekata K, Nishimura N, Kimura H. Presenilin-binding protein forms aggresomes in monkey kidney COS-7 cells. J Neurochem. 2002;82:819–827. doi: 10.1046/j.1471-4159.2002.01039.x. [DOI] [PubMed] [Google Scholar]
- Nies DH. Microbial heavy-metal resistance. Appl Microbiol Biotechnol. 1999;51:730–750. doi: 10.1007/s002530051457. [DOI] [PubMed] [Google Scholar]
- Nordberg GF. Cadmium and health in the 21st century – historical remarks and trends for the future. Biometals. 2004;17:485–489. doi: 10.1023/b:biom.0000045726.75367.85. [DOI] [PubMed] [Google Scholar]
- Okuda B, Iwamoto Y, Tachibana H, Sugita M. Parkinsonism after acute cadmium poisoning. Clin Neurol Neurosurg. 1997;99:263–265. doi: 10.1016/s0303-8467(97)00090-5. [DOI] [PubMed] [Google Scholar]
- Peters JM, Walsh MJ, Franke WW. An abundant and ubiquitous homo-oligomeric ring-shaped ATPase particle related to the putative vesicle fusion proteins Sec18p and NSF. EMBO J. 1990;9:1757–1767. doi: 10.1002/j.1460-2075.1990.tb08300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero D, Gomez-Zapata M, Luna A, Garcia-Fernandez AJ. Morphological characterization of renal cell lines (BGM and VERO) exposed to low doses of lead nitrate. Histol Histopathol. 2004;19:69–76. doi: 10.14670/HH-19.69. [DOI] [PubMed] [Google Scholar]
- Ryan MC, Shooter EM, Notterpek L. Aggresome formation in neuropathy models based on peripheral myelin protein 22 mutations. Neurobiol Dis. 2002;10:109–118. doi: 10.1006/nbdi.2002.0500. [DOI] [PubMed] [Google Scholar]
- Sato K, Iwamasa T, Tsuru T, Takeuchi T. An ultrastructural study of chronic cadmium chloride-induced neuropathy. Acta Neuropathol (Berl) 1978;41:185–190. doi: 10.1007/BF00690433. [DOI] [PubMed] [Google Scholar]
- Seigneurin-Berny D, Verdel A, Curtet S, Lemercier C, Garin J, Rousseaux S, Khochbin S. Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways. Mol Cell Biol. 2001;21:8035–8044. doi: 10.1128/MCB.21.23.8035-8044.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih CM, Ko WC, Wu JS, Wei YH, Wang LF, Chang EE, Lo TY, Cheng HH, Chen CT. Mediating of caspase-independent apoptosis by cadmium through the mitochondrisa-ROS pathway in MRC-5 fibroblasts. J Cell Biochem. 2004;91:384–397. doi: 10.1002/jcb.10761. [DOI] [PubMed] [Google Scholar]
- Shimohata T, Sato A, Burke JR, Strittmatter WJ, Tsuji S, Onodera O. Expanded polyglutamine stretches form an ‘aggresome’. Neurosci Lett. 2002;323:215–218. doi: 10.1016/s0304-3940(02)00162-3. [DOI] [PubMed] [Google Scholar]
- VanBogelen RA, Schiller EE, Thomas JD, Neidhardt FC. Diagnosis of cellular states of microbial organisms using proteomics. Electrophoresis. 1999;20:2149–2159. doi: 10.1002/(SICI)1522-2683(19990801)20:11<2149::AID-ELPS2149>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Vinceti M, Guidetti D, Bergomi M, Caselgrandi E, Vivoli R, Olmi M, Rinaldi L, Rovesti S, Solime F. Lead, cadmium, and selenium in the blood of patients with sporadic amyotrophic lateral sclerosis. Ital J Neurol Sci. 1997;18:87–92. doi: 10.1007/BF01999568. [DOI] [PubMed] [Google Scholar]
- Wang Q, Song C, Li CC. Molecular perspectives on p97-VCP: progress in understanding its structure and diverse biological functions. J Struct Biol. 2004;146:44–57. doi: 10.1016/j.jsb.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Wojcik C, Fabunmi R, Demartino GN. Modulation of gene expression by RNAi methods. Mol Med. 2004;108:381–394. doi: 10.1385/1-59259-850-1:381. [DOI] [PubMed] [Google Scholar]
- Wojcik C, Schroeter D, Wilk S, Lamprecht J, Paweletz N. Ubiquitin-mediated proteolysis centers in HeLa cells: indication from studies of an inhibitor of the chymotrypsin-like activity of the proteasome. Eur J Cell Biol. 1996;71:311–318. [PubMed] [Google Scholar]
- Wu RS, Kohn KW, Bonner WM. Metabolism of ubiquitinated histones. J Biol Chem. 1981;256:5916–5920. [PubMed] [Google Scholar]
- Wu S, Wan P, Li J, Li D, Zhu Y, He F. Multi-modality of pI distribution in whole proteome. Proteomics. 2006;6:449–455. doi: 10.1002/pmic.200500221. [DOI] [PubMed] [Google Scholar]