

Abstract
Activated protein C (APC) is a protease with anticoagulant and cytoprotective activities. APC is neuroprotective in rodent models of stroke. But, an APC variant with reduced anticoagulant activity, 3K3A-APC, compared to wild type APC (wt-APC) shows greater neuroprotection with no risk for bleeding in stroke models. To determine whether 3K3A-APC exhibits species-dependent neuroprotection similar to that as seen with wt-APC, we studied murine and human recombinant 3K3A-APC mutants which show approximately 80% reduced anticoagulant activity. Murine 3K3A-APC (0.2 mg/kg i.v.) administered at 4 h after embolic stroke improved substantially functional outcome and reduced by 80% the infract volume 7 days after stroke. Human 3K3A-APC was neuroprotective after embolic stroke in mice, but at significantly higher concentrations (i.e., 2 mg/kg i.v.). Species-dependent neuroprotection, i.e., murine > human 3K3A-APC, was confirmed in a mouse model of permanent middle cerebral artery occlusion. Human 3K3A-APC had by 5-fold greater cytoprotective activity than murine 3K3A-APC in oxygen-glucose deprivation model in human brain endothelial cells, whereas murine 3K3A-APC was by 2.5-fold more potent than human 3K3A-APC in a mouse model of N-methyl-D-aspartate-induced neuronal apoptosis. Thus, 3K3A-APC exhibits species-dependent neuroprotection which should be taken into account when designing human trials for ischemic stroke with APC mutants.
INTRODUCTION
Activated protein C (APC) is an endogenous circulating serine protease with systemic anticoagulant activity that is mediated by irreversible proteolytic cleavage of coagulation factors Va and VIIIa with contributions by various cofactors (Mosnier et al., 2007a). Recent studies revealed that independent of its anticoagulant activity, APC exerts direct cellular activities including cytoprotective alterations of gene expression profiles, anti-inflammatory activity, anti-apoptotic activity and enhancement and protection of endothelial barriers (Joyce et al., 2001, Riewald et al., 2002, Cheng et al., 2003, Riewald and Ruff 2005, Feistritzer and Riewald, 2005, Finigan et al., 2005, Cheng et al., 2006). APC-mediated cleavage of protease activated receptor 1 (PAR1) results in anti-apoptotic and anti-inflammatory signaling in endothelial cells including brain endothelium of the blood-brain barrier (BBB), monocytes and tissue macrophages (Mosnier et al., 2007a), and neurons (Guo et al., 2004). Recent studies demonstrated that APC and its variants with reduced anticoagulant activity cross the BBB via endothelial protein C receptor (EPCR)-assisted transport (Deane et al., 2008).
APC is neuroprotective in different rodent models of stroke including transient ischemia and embolic stroke (Shibata et al., 2001, Cheng et al., 2003, Zlokovic et al., 2005), spinal cord injury (Taoka et al., 2000) and multiple sclerosis (Han et al., 2008). It also protects against diabetic endothelial and glomerular injury (Isermann et al., 2007), and reduces mortality from sepsis and prevents development of thrombosis in different animal models (Griffin et al., 2002). APC infusion has been approved by the FDA for use in patients with severe sepsis (Bernard et al., 2001). Currently, APC is being studied in patients with ischemic stroke (APCAST, The Activated Protein C in Acute Stroke Trial; http://clinicaltrials.gov).
A possible concern with APC therapy is adverse serious bleeding which might complicate its pharmacological utility (Bernard et al., 2001). Studies in non-human primates using APC at doses much higher than its therapeutic dose range currently used in sepsis or stroke (www.emea.europa.eu/humandocs/PDFs/EPAR/xigris/247102en6.pdf) revealed serious bleeding complications. Recently, APC mutants with reduced anticoagulant activity were shown to be equally beneficial or better than wild type APC (wt-APC) in animal models of sepsis (Kerschen et al., 2007) and stroke (Wang et al., 2008), respectively, while reducing the risk for bleeding. In models of stroke (Shibata et al., 2001; Cheng et al., 2003) and neuronal injury (Guo et al., 2004), wt-APC exhibits species-dependent differences which has importantly influenced design of dose escalation studies with APC in stroke patients (http://clinicaltrials.gov). Whether APC mutants with reduced anticoagulant activity exert species-dependent neuroprotection is not known. Here, we studied murine and human 3K3A-APC mutants with greatly reduced anticoagulant activities (Gale et al., 2002), but preserved cytoprotective activities (Mosnier et al., 2004), in two mouse models of stroke, oxygen/glucose deprivation (OGD) model in human brain endothelial cells (Cheng et al., 2006) and N-methyl-D-aspartate (NMDA) model of neuronal injury in mouse cortical cells (Guo et al., 2004).
MATERIALS AND METHODS
Reagents
Murine and human recombinant 3K3A-APC (KKK191-193AAA) were prepared in HEK293 cells, as previously described (Gale et al., 2002; Mosnier et al., 2004). Human 3K3A-protein C (3K3A-PC) stable cell line was also generated in Chinese hamster ovary (CHO) cells. The cells were grown in suspension in CD OptiCHO medium (Invitrogen, Carlsbad, CA) containing 2 mM CaCl2, 10 μg/ml vitamin K, and 2 mM GlutaMAX (Invitrogen) in a 2 L Biowave bioreactor for production. A four-step purification procedure was used: capturing PC using FFQ resin (GE Health), purification of PC using Uno Q column (BioRad), activating with recombinant human thrombin (Zymogenetics, Seattle, WA), and removal of thrombin using Uno Q column. The purity of 3K3A-APC was determined by reduced SDS-PAGE/silver staining, the enzymatic activity by amidolytic assay, and the concentration by ELISA and A280 spectral analysis.
Measurement of in vitro human or mouse APC anticoagulant activity of wild type APC's and 3K3A-APC variants
A modified Activated Partial Thromboplastin time (APTT) clotting assay that was dependent primarily on inactivation of limiting amounts of mouse factor V was performed to compare the anticoagulant activity of human and mouse 3K3A-APC. Human Factor V deficient plasma containing 4 % mouse plasma as a source of factor V was incubated with the coagulation activator, Platelin LS (BioMerieux, Durham NC) and APC in a buffer containing 0.01 M Tris, 0.14 M NaCl, and 0.5 % bovine serum albumin, pH 7.4 for 180 sec at 37 °C and then 30 mM calcium chloride was added to initiate clotting.
Embolic stroke model
Surgical procedure
All procedures were done according to the NIH guidelines for animal care and approved by the Institutional Animal Care and Use Committee at the University of Rochester. Embolic stroke was induced by placing a single intact homologous clot at the origin of middle cerebral artery as reported (Zhang et al., 1997; Zlokovic et al., 2005). Studies were done in male C57BL/6 mice 8-12 months old (28-32 g) anesthetized with 100 mg/kg intraperitoneal (i.p.) ketamine and 10 mg/kg xylazine. Animals were allowed to breathe spontaneously. Rectal temperature was maintained at 37°C during surgical procedure. Blood gasses and pH were monitored.
Treatment
Mice were randomly assigned to the vehicle-treated group, murine 3K3A-APC-treated group and human 3K3A-APC-treated group. Murine recombinant 3K3A-APC (0.2 mg/kg), human 3K3A-APC (0.2 mg/kg, 2mg/kg) or vehicle were administrated via the right femoral vein 4 h after embolic stroke. 3K3A-APC was given as a 50% bolus/50% 30-minute infusion in total volume of 100 μL.
Functional tests
The following behavioral tests were performed in mice at 1, 2, 3, 4, 5, 6, and 7 days after stroke: (1) motor neurological score (no deficit, score 0; maximal deficit, score 5) (Cheng et al., 2003; Liu et al., 2004); (2) corner turn and forelimb use asymmetry test, for sensorimotor activity (Zlokovic et al., 2005); (3) foot-fault test, for locomotor assessment (Cheng et al., 2003; Liu et al., 2004; Gong et al., 2004).
Neuropathological analysis
For neuropathological analysis, mice were sacrificed at day 7 and brains isolated and prepared for analysis. Coronal brain sections (1-mm thick) were cut and incubated with 2% 2,3,5-triphenyltetrazolium chloride (TTC) solution in a phosphate buffered saline (PBS) for 5 min at 37° C, as reported (Wang et al., 1997). The injury volume (mm3) was calculated by multiplying the surfaces of all injured areas in mm2 by the thickness of brain sections. The infarction volume was obtained by subtracting the edema volume from the injury volume. Edema volume (tissue swelling) was calculated by subtracting the volume of the contralateral non-ischemic hemisphere from the volume of the ipsilateral ischemic hemisphere, as described (Wang et al., 1997).
Permanent distal middle cerebral artery occlusion (dMCAO)
Surgical procedure
All procedures were done according to the NIH guidelines for animal care and approved by the Institutional Animal Care and Use Committee at the University of Rochester. Male C57BL6 mice 6-8 weeks old (24-26 g) were anesthetized with ketamine (100 mg/kg i.p.) and xylazine (10 mg/kg i.p.). Animals were allowed to breathe spontaneously. Rectal temperature was maintained at 37.0°C using a feedback-controlled heating system. The right femoral artery was cannulated for monitoring of blood pressure and blood analysis. A modified permanent dMCAO technique was used as reported (Zhang et al., 2005). Under the surgical microscope, the left common carotid artery was isolated through a neck incision and ligated using a 5-0 silk. A skin incision was made between the right orbit and tragus. The zygomatic arch was removed and temporal muscle retracted laterally. The mandible was retracted downward. The MCA was visible through the temporal semi-translucent surface of the skull. Craniectomy was performed by drilling with a 0.9 mm round burr. The inner layer of the skull was removed with fine forceps. The dura was carefully opened, and the M1 branch of the MCA exposed and coagulated using a cauterizer, producing permanent dMCAO. The wound was sutured, and rectal temperature was controlled until mice regained full consciousness.
Treatment
Mice were randomly assigned to the vehicle-treated group, murine 3K3A-APC-treated group and human 3K3A-APC-treated group. Murine 3K3A-APC (0.2 mg/kg), human 3K3A-APC (0.2 mg/kg) or vehicle were administered by tail vein at 12 h, 1, 3, 5 and 7 days after dMCAO. Vehicle-treated group received saline.
Functional tests
At days 1, 3 and 7 after dMCAO the forelimb asymmetry test for sensory-motor activity (Schallert et al., 1984) and foot-fault test for locomotor assessment (Gong et al., 2004) were applied.
Neuropathological analysis
Mice were sacrificed after 7 days and the brains removed and rapidly frozen in CO2–snow. Brains were cut into serial 20 μm cryostat sections. Every tenth section was stained with cresyl-violet and the lesion area determined using an image analysis system (Image J). The infarct volume was calculated as explained above.
Human brain endothelial cell (BEC) cultures
Primary BECs were isolated from specimens (< 3h) from neurologically healthy, young individuals after surgery for epilepsy and were characterized and cultured, as we previously described (Cheng et al., 2003). Cells were maintained in serum-free Dulbecco's Modified Eagle Medium and exposed for 8 h to oxygen-glucose deprivation (OGD) (< 2% oxygen, no glucose). Hypoxia was induced using an anaerobic chamber (Forma Scientific, Holbrook, New York), as we described (Cheng et al., 2003). The levels of O2 were monitored by O2 Fyrite (Forma Scientific). Murine and human 3K3A-APC were added in the range from 3 to 100 nM at the time of OGD treatment. Hirudin was included to block any thrombin signaling. Cell survival was detected by water-soluble tetrazolium
WST-8 assay (Dojindo Molecular Technologies, Gaithersburg, ML). The WST-8 assay determines the amount of water-soluble formazan product in contrast to the MTT assay which determines the levels of water-insoluble formazan product (Isobe et al., 1999). The amount of WST-8 (yellow-color) is directly proportional to the number of living cells since it is produced by the activity of dehydrogenases in cells. Thus, the absorbance at 460 nm of WST formazan is directly proportional to the number of viable cells in the medium. Survival rate was represented as the viability percentage of non-treated cells.
Mouse neuronal cell cultures
Primary neuronal cultures were established as described (Bonfoco et al., 1995; Guo et al., 2004). In brief, cerebral cortex was dissected from fetal C57BL/6J mice at 16 days of gestation, treated with trypsin for 10 min at 37 °C, and dissociated by trituration. Dissociated cell suspensions were plated at 5 × 105 cells per well on 12-well tissue culture plates coated with poly-L-lysine, in serum-free Neurobasal medium plus B27 supplement (Gibco, Rockville, MA). Cultures were maintained in a humidified 5% CO2 incubator at 37 °C for 7 days before treatment. Medium was replaced every 3 days. In our earlier studies (Guo et al., 2004, Liu et al., 2004) and in the present study contamination of cortical neurons with astrocytes under present experimental conditions varied between 0.3% and 1%, and was comparable to that as previously reported using serum-free Neurobasal medium with B27 supplement (Brewer et al., 1993).
Cultures were exposed for 10 min to 300 μM NMDA/5 μM glycine in Mg2+-free Earle's balanced salt solution (EBSS) (Bonfoco et al., 1995) to induce neuronal apoptosis. Control cultures were exposed to EBSS alone. After the exposure, cultures were rinsed with EBSS, returned to the original culture medium and incubated with or without different concentrations of murine or human recombinant 3K3A-APC (2-20 nM). All experiments involving treatment with APC included hirudin (Sigma) to block any thrombin signaling (Riewald et al., 2002; Cheng et al., 2003; Riewald et al., 2005). Cell survival was detected by using WST-8 assay (Dojindo Molecular Technologies, Gaithersburg, ML).
Statistics
Data were presented as mean ± SEM. ANOVA followed by a post hoc Tukey test was used to determine statistically significant differences. The Bonferroni correction for multiple comparisons was used to control for a family wise type I error. P < 0.05 was considered statistically significant.
RESULTS
The major plasma substrate for APC's anticoagulant activity is factor V. Human 3K3A-APC has approximately 20 % of normal APC anticoagulant activity because the mutations decrease the ability of APC to cleave factor Va at Arg506 (Gale et al, 2002). To compare the anticoagulant action of murine 3K3A-APC to the human mutant, inactivation of murine factor V in human plasma was studied. The anticoagulant activities of human and murine 3K3A-APC in human factor V deficient plasma containing 4 % levels of mouse factor V were indistinguishable and were 16 to 26 % of respective wt-APC's (Fig. 1). Thus, the 3K3A mutation in both murine and human APC caused a similar loss of approximately 80% anticoagulant action on factor V.
Figure 1. Anticoagulant activity of human and mouse 3K3A-APC.
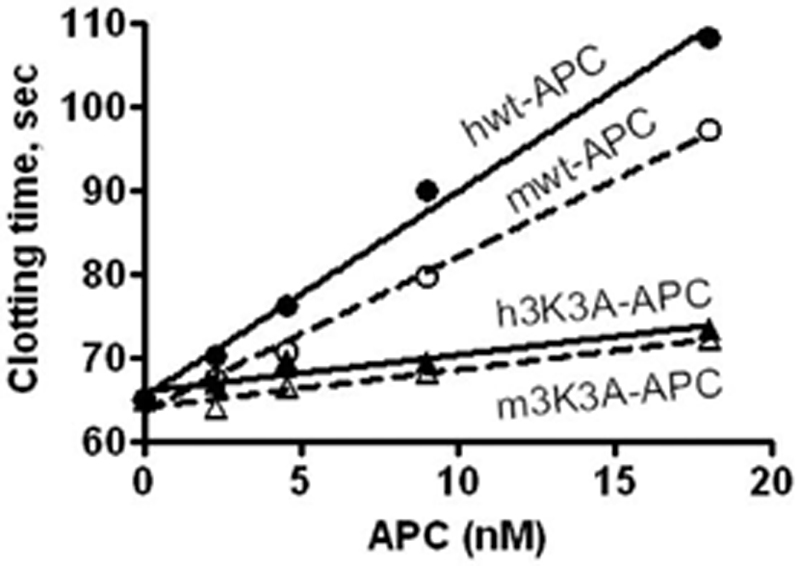
Modified APTT clotting assays were performed using human factor V deficient plasma containing 4% mouse plasma as a source of mouse factor V. Prolongation of clotting times were caused by human (—) or mouse (---) wild type wt-APC (circles) or 3K3A-APC mutants (triangles). APC concentrations indicate final amounts in the assay; h and m before APC denote human or murine APC, respectively.
Control mice receiving vehicle after embolic stroke developed a severe motor neurological deficit (Fig. 2A) and increases in corner test score (Fig. 2B), forelimb asymmetry (Fig. 2C) and foot fault test (Fig. 2D), associated with a significant infract lesion (Fig. 3A) and brain swelling (Fig. 3B) after 7 days of stroke, as previously described in this model. Murine 3K3A-APC (0.2 mg/kg) administered i.v. 4 h after embolic stroke substantially reduced motor neurological score and improved functional recovery in all behavioral tests (Fig. 2A-D), and reduced significantly the infarct volume by > 80% and the edema volume (Fig. 3A-B). The observed protection with murine 3K3A-APC was comparable, although somewhat better than that obtained with the wt-APC administered 4 h after embolic stroke (not shown), as we reported (Zlokovic et al., 2005). In contrast, human 3K3A-APC at 0.2 mg/kg did not have a significant effect on functional recovery or infract and/or edema volumes. But, significant neuroprotection was obtained with human 3K3A-APC at a higher dose of 2 mg/kg both with respect to functional recovery (Fig. 2A-D) and reductions in the infract lesion and edema volume (Fig. 3A-B), suggesting a possible species differences in murine and human 3K3A-APC preparations.
Figure 2. Murine and human recombinant 3K3A-APC given 4 h after embolic stroke improve functional outcome in mice within 7 days.

Motor neurological score (A), corner turn scores (B), forelimb use asymmetry test (C), and foot-fault test (D) in the presence and absence of murine 3K3A-APC (m3K3A-APC 0.2 mg/kg), HEK293-derived human 3K3A-APC (h3K3A-APC 0.2 and 2 mg/kg) or vehicle (V). Mean ± SEM, n = 6 mice per group. ap < 0.01, m3K3A-APC 0.2 mg/kg vs. h3K3A-APC 0.2 mg/kg; bp < 0.05, m3K3A-APC 0.2 mg/kg vs. h3K3A-APC 2 mg/kg; cp < 0.01 m3K3A-APC 0.2 mg/kg or h3K3A-APC 2 mg/kg vs. vehicle. 3K3A-APC was administered 50% as an i.v. bolus and 50% as a 30 min i.v. infusion. In graphs A-D, 0.2 denotes 0.2 mg/kg and 2 denote 2 mg/kg.
Figure 3. Murine and human recombinant 3K3A-APC given 4 h after embolic stroke control spread of ischemic lesion 7 days after stroke.

Infarct volume (A) and brain swelling (edema) (B) from experimental and control groups in Fig. 2. Mean ± SEM, n = 6 mice per group. m3K3A-APC is murine 3K3A-APC and h3K3A-APC is human 3K3A-APC. 3K3A-APC was administered 50% as an i.v. bolus and 50% as a 30 min i.v. infusion. In graphs A-B, 0.2 denotes 0.2 mg/kg and 2 denotes 2 mg/kg.
To exclude the possibility that the observed differences in neuroprotection potency between murine and human 3K3A-APC are due to embolic stroke model, we employed a permanent dMCAO model and compared murine and human 3K3A-APC using a multi-dosing protocol (0.2 mg/kg) with the first dose administered i.v. at 12 h after the MCAO followed by the i.v. administration at 1, 3, 5 and 7 days. As shown in Fig. 4A, the injury after dMCAO in vehicle-treated mice is confined to cortex affecting the M1 primary cortex, S1FL and S1HL primary somatosensory cortex forelimb and hindlimb regions and S2 secondary somatosensory cortex, at the level of the optic chiasm. Human 3K3A-APC at 0.2 mg/kg multi-dosing did not exert a significant protection, in contrast to murine 3K3A-APC that spared several regions in the periphery of infarction including M1 and S1FL. Consistent with these results, murine 3K3A-APC reduced the infract volume by about 80% (Fig. 4B). In contrast, human 3K3A-APC at low 0.2 mg/kg dose was not protective (Fig 4B).
Figure 4. Effects of murine compared to human 3K3A-APC multi-dosing therapy beginning at 12 h after permanent distal MCAO on functional outcome and infarct volume in mice within 7 days.

(A) Cresyl-violet staining of brain coronal sections at the level of optic chiasm of ischemic mice treated with vehicle, h3K3A-APC 0.2 mg/kg or m3K3A-APC 0.2 mg/kg 7 days after dMCAO. 3K3A-APC was administered as multi-dosing intravenous therapy starting at 12 h after the dMCAO followed by administrations at 1, 3, 5 and 7 days. Affected regions in vehicle-treated mice include: M1, primary motor cortex; S1FL and S1HL, primary somatosensory cortex forelimb and hindlimb;S1BF, S1 cortex barrel field; S2, secondary somatosensory cortex. (B) Infarct volume in mice from experimental and control groups was determined after 7 days. Foot-fault test (C) and forelimb asymmetry test (D) in the presence and absence of murine 3K3A-APC (m3K3A-APC 0.2 mg/kg), CHO-derived human 3K3A-APC (h3K3A-APC 0.2 mg/kg) or vehicle. Mean ± SEM, n = 5-6 mice per group. ap < 0.01, m3K3A-APC 0.2 mg/kg vs. h3K3A-APC 0.2 mg/kg; bp < 0.01, m3K3A-APC 0.2 mg/kg vs. vehicle. 3K3A-APC was administered as multi-dosing therapy at 12 h, 1, 3, 5 and 7 days after dMCAO. In graphs A-C, 0.2 denotes 0.2 mg/kg.
Various behavioral tests can be performed to test functions of the affected regions after dMCAO; for M1 (open field, rotarod, foot fault), S1FL (adhesive removal, forelimb placing test, forelimb asymmetry), S1HL (hind limb placing test), S2 (elevated body swing), AuD and AuV, secondary auditory cortex dorsal and ventral (auditory startling) (Taguchi et al., 2004; Guegan et al., 2006; Leker et al., 2007; Wang et al., 2007). Here, we showed that murine 3K3A-APC improved substantially performance on foot-fault test (Fig. 4C) and forelimb asymmetry tests (Fig. 4D) within 7 days of dMCAO, reflecting functional improvements in M1 and S1FL regions, which correlates well with its neuroprotective effects seen in these corresponding zones of infarct (Fig. 4A).
Next, we tested a species difference using a human primary BEC OGD model, as reported (Cheng et al., 2006). The dose-response curve with human 3K3A-APC suggested a significantly higher cytoprotection compared to murine 3K3A-APC (Fig. 5A), and the corresponding IC50 values were 9.8 ± 1.47 and 47.27 ± 2.39 nM (Fig. 5B), indicating a 5-fold greater cytoprotective potency of human 3K3A-APC in human cells.
Figure 5. Human and murine 3K3A-APC protect human brain endothelial cells (BEC) from oxygen glucose deprivation (OGD).
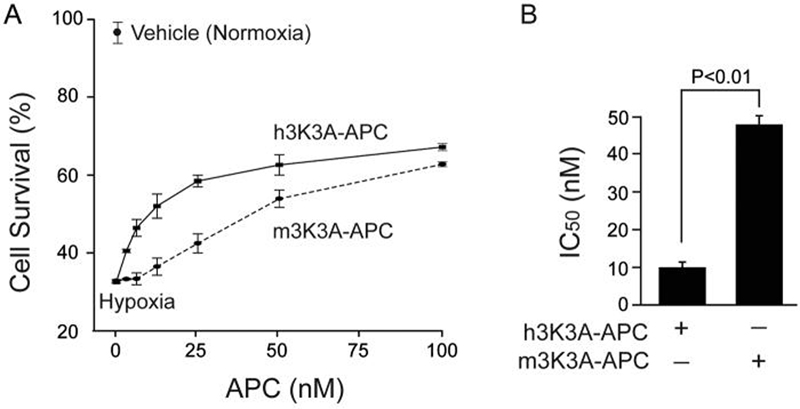
(A) Dose-dependent cytoprotection of human BEC subjected to 8 h of OGD by human CHO-derived h3K3A-APC and murine m3K3A-APC. Cell survival was quantified by WST-8 assay. (B) IC50 values for h3K3A-APC and m3K3A-APC calculated from experiments in Fig. 5A. Values are mean ± SEM, n = 3-4 independent experiments in triplicate.
Finally, we studied neuroprotective potency of mouse and human 3K3A-APC in a model of NMDA-induced excitotoxic injury in mouse cortical neurons. As suggested by the dose response curves, mouse 3K3A-APC exerted higher neuroprotective activity than its human isoform (Fig. 6A). Their respective IC50 values were 3.03 ± 0.32 and 7.01 ± 0.70 nM for the mouse and human isoform, suggesting that mouse 3K3A-APC has by about 2.5-fold greater neuroprotective acivity.
Figure 6. Protection of mouse cortical neurons from NMDA-induced injury by mouse and human 3K3A-APC.

(A) Dose-dependent neuroprotection by mouse and CHO-derived human 3K3A-APC at 24 h of NMDA. Cell survival was quantified with WST-8 assay. (B) IC50 values for mouse and human 3K3A-APC from dose-response curves shown in Fig. 6A. Values are mean ± SEM, n=3-5 independent experiments in triplicate. NMDA label at zero concentration APC on the abscissa shows cell survival of neurons treated with NMDA only without 3K3A-APC. Vehicle denotes the basal cell survival rate of neurons in the culture medium in the absence of NMDA.
DISCUSSION
This study shows that APC anticoagulant activity is not needed for APC-mediated protection after stroke in rodents. By using clinically relevant embolic model of stroke in mice we showed that murine 3K3A-APC non-anticoagulant mutant (0.2 mg/kg) administered intravenously 4 h after stroke improved substantially functional recovery and reduced the spread of ischemic lesion by > 80%, which was comparable to neuroprotection obtained by murine wt-APC in the same murine embolic stroke model (Zlokovic et al., 2005). Moreover, murine 3K3A-APC administered at 12 h, and 1, 3, 5 and 7 days after permanent dMCAO exhibited remarkable neuroprotective effects in behavioral tests and reduced by 80% the infract volume. It is of note, we used murine 3K3A-APC at a dose of 0.2 mg/kg based on previous studies with wt-APC in stroke models indicating that at this dose murine wt-APC provides maximal neuroprotection (Cheng et al., 2003; Liu et al., 2004; Zlokovic et al., 2005; Cheng et al., 2006).
Our functional tests as well as measurements of the infraction lesion volume have indicated that 3K3A-APC neuroprotective effects after embolic stroke and permanent MCAO are dose-dependent consistent with previous reports with wild-type recombinant APC after transient MCAO (Cheng et al., 2003; Cheng et al., 2006). Our results with an APC analog with significantly enhanced anti-coagulant activity revealed that the effect may also be time-dependent especially in regard to an increased risk for intracerebral bleeding (Wang et al., 2008), that is not seen with presently studied 3K3A-APC non-anticoagulant mutants.
Human 3K3A-APC was significantly less potent in mouse stroke models compared to murine 3K3A-APC, as indicated by our data demonstrating that comparable levels of improvements in functional recovery and control of the infarct volume were obtained with human 3K3A-APC at 2 mg/kg and murine 3K3A-APC at 0.2 mg/kg. This species specificity has been confirmed in human OGD model by showing that human 3K3A-APC was by 5-fold more potent than murine 3K3A-APC in protecting human cells from death. On the other hand, murine 3K3A-APC was by 2.5-fold more potent than human 3K3A-APC in protecting mouse neurons from NMDA injury.
The exact molecular mechanisms underlying the observed differences in neuroprotective potency between murine and human 3K3A-APC are not known at present. Similar differences have been described for murine wt-APC and human wt-APC after transient ischemia in mice (Shibata et al., 2001; Mosnier et al., 2003; Cheng et al., 2003). One possibility is that murine 3K3A-APC is more active than human 3K3A-APC. But, the fact that human 3K3A-APC protects human BEC from OGD-induced injury by about 5-fold higher potency rules out this possibility suggesting that the observed differences may indeed reflect the species-dependent differences. In other words murine 3K3A-APC interacts with murine cells and putative murine APC receptors, e.g., PAR1, PAR3 with a higher affinity than human isoforms (Guo et al., 2004), and vice versa, human 3K3A-APC interacts with human PAR1 and EPCR in human endothelial cells (Cheng et al., 2003) with much higher affinity than its murine isoform. Different post-translational modifications, such as differences in glycosilation profiles including different contents of negatively charged sialic acid and/or different distribution and branching of the N-linked glycan species may contribute to the observed differences which may create an evolutionary difference in protein C cellular system in mice and humans. Whatever the most relevant causes for the observed differences are, these important differences need to be taken into account when designing the dose regimens for possible safety and efficacy human trials for ischemic stroke with APC mutants, similar as it has been done with currently studied recombinant human wt-APC (APCAST, The Activated Protein C in Acute Stroke Trial; http://clinicaltrials.gov/ct2/show/NCT00533546?term=apc&rank=25).
The anticoagulant action of APC is mediated by its binding to factor Va followed by proteolytic cleavage of factor Va at Arg506 resulting in inactivation of factor Va (Mosnier et al., 2007a). Factor Va binding to APC is mediated by positively charged residues in the surface loops on APC's protease domains including loop 37 (residues 190-193), the Ca2+-binding loop (residues 225-235), and the autolysis loop (residues 301-316). Substitution of positively charged lysine residues with 3 alanine residues in loop 37 (KKK191-193AAA) decreases APC's anticoagulant activity by > 80% in murine and human APCs, which reduces the risk for bleeding through reductions in both APC's anticoagulant and APC-dependent profibrinolytic activities (Mosnier et al., 2007b), as confirmed in vivo in different models of sepsis (Kerschen et al., 2007) and stroke (Wang et al., 2008). This mutation leaves intact the N-terminal Gla domain in 3K3A-APC which mediates APC binding to EPCR on the endothelial cells, and does not alter serine-protease catalytic site which activates cell-signaling through PAR1, resulting in completely preserved cytoprotective activity (Mosnier et al., 2007b).
The present study shows species-dependent neuroprotection by 3K3A-APC in mouse stroke models. By extension, these findings may suggest that non-anticoagulant 3K3A-APC mutants might also be suitable for treating acute ischemic stroke in humans given that wt-APC is currently being studied. Because of species differences, it is likely that lower dosages of human 3K3A-APC than those used in a mouse stroke model will be neuroprotective in stroke patients. Our data showing that human 3K3A-APC is directly vasculoprotective in a model of ischemia in human brain endothelium further supports this point. Importantly, the use of non-anticoagulant APC mutants compared to wt-APC might potentially increase the therapeutic dose of APC in stroke patients that otherwise may not be possible with wt-APC due to the risk for severe adverse bleeding complications including intracerebral hemorrhage.
Acknowledgments
This work was supported by the NIH grants HL63290 and HL81528 to BVZ. ZZ Biotech LLC provided CHO-derived human 3K3A-APC mutant. We want to thank Dr. J. H. Griffin for carefully reading the manuscript and for his valuable suggestions. Dr. B.V. Zlokovic is a Chief Scientific Officer of ZZ Biotech which is committed to develop APC mutants for stroke.
REFERENCES
- Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci U S A. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Cheng T, Liu D, Griffin JH, Fernandez JA, Castellino F, Rosen ED, Fukudome K, Zlokovic BV. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. 2003;9:338–342. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- Cheng T, Petraglia AL, Li Z, et al. Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med. 2006;12:1278–1285. doi: 10.1038/nm1498. [DOI] [PubMed] [Google Scholar]
- Deane R, LaRue B, Sagare AP, Castellino FJ, Zhong Z, Zlokovic BV. Endothelial protein C receptor-assisted transport of activated protein C across the mouse blood-brain barrier. J Cereb Blood Flow Metab. 2008 doi: 10.1038/jcbfm.2008.117. DOI:10.1038/jcbfm.2008.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105:3178–3184. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- Finigan JH, Dudek SM, Singleton PA, Chiang ET, Jacobson JR, Camp SM, Ye SQ, Garcia JG. Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J Biol Chem. 2005;280:17286–17293. doi: 10.1074/jbc.M412427200. [DOI] [PubMed] [Google Scholar]
- Gale AJ, Tsavaler A, Griffin JH. Molecular characterization of an extended binding site for coagulation factor Va in the positive exosite of activated protein C. J Biol Chem. 2002;277:28836–28840. doi: 10.1074/jbc.M204363200. [DOI] [PubMed] [Google Scholar]
- Gong Y, Hua Y, Keep RF, Hoff JT, Xi G. Intracerebral hemorrhage: effects of aging on brain edema and neurological deficits. Stroke. 2004;35:2571–2575. doi: 10.1161/01.STR.0000145485.67827.d0. [DOI] [PubMed] [Google Scholar]
- Griffin JH, Zlokovic B, Fernandez JA. Activated protein C: potential therapy for severe sepsis, thrombosis, and stroke. Semin Hematol. 2002;39:197–205. doi: 10.1053/shem.2002.34093. [DOI] [PubMed] [Google Scholar]
- Guegan C, Braudeau J, Couriaud C, Dietz GP, Lacombe P, Bahr M, Nosten-Bertrand M, Onteniente B. PTD-XIAP protects against cerebral ischemia by anti-apoptotic and transcriptional regulatory mechanisms. Neurobiol Dis. 2006;22:177–186. doi: 10.1016/j.nbd.2005.10.014. [DOI] [PubMed] [Google Scholar]
- Guo H, Liu D, Gelbard H, Cheng T, Insalaco R, Fernandez JA, Griffin JH, Zlokovic BV. Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Neuron. 2004;41:563–572. doi: 10.1016/s0896-6273(04)00019-4. [DOI] [PubMed] [Google Scholar]
- Han MH, Hwang SI, Roy DB, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451:1076–1081. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- Isermann B, Vinnikov IA, Madhusudhan T, et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13:1349–1358. doi: 10.1038/nm1667. [DOI] [PubMed] [Google Scholar]
- Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW. Gene expression profile of antithrombotic protein c defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. 2001;276:11199–11203. doi: 10.1074/jbc.C100017200. [DOI] [PubMed] [Google Scholar]
- Kerschen EJ, Fernandez JA, Cooley BC, et al. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007;204:2439–2448. doi: 10.1084/jem.20070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leker RR, Soldner F, Velasco I, Gavin DK, Androutsellis-Theotokis A, McKay RD. Long-lasting regeneration after ischemia in the cerebral cortex. Stroke. 2007;38:153–161. doi: 10.1161/01.STR.0000252156.65953.a9. [DOI] [PubMed] [Google Scholar]
- Liu D, Cheng T, Guo H, Fernandez JA, Griffin JH, Song X, Zlokovic BV. Tissue plasminogen activator neurovascular toxicity is controlled by activated protein C. Nat Med. 2004;10:1379–1383. doi: 10.1038/nm1122. [DOI] [PubMed] [Google Scholar]
- Mosnier LO, Gale AJ, Yegneswaran S, Griffin JH. Activated protein C variants with normal cytoprotective but reduced anticoagulant activity. Blood. 2004;104:1740–1744. doi: 10.1182/blood-2004-01-0110. [DOI] [PubMed] [Google Scholar]
- Mosnier LO, Griffin JH. Inhibition of staurosporine-induced apoptosis of endothelial cells by activated protein C requires protease-activated receptor-1 and endothelial cell protein C receptor. Biochem J. 2003;373:65–70. doi: 10.1042/BJ20030341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosnier LO, Yang XV, Griffin JH. Activated protein C mutant with minimal anticoagulant activity, normal cytoprotective activity, and preservation of thrombin activable fibrinolysis inhibitor-dependent cytoprotective functions. J Biol Chem. 2007a;282:33022–33033. doi: 10.1074/jbc.M705824200. [DOI] [PubMed] [Google Scholar]
- Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007b;109:3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–1882. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- Riewald M, Ruf W. Protease-activated receptor-1 signaling by activated protein C in cytokine-perturbed endothelial cells is distinct from thrombin signaling. J Biol Chem. 2005;280:19808–19814. doi: 10.1074/jbc.M500747200. [DOI] [PubMed] [Google Scholar]
- Schallert T, Whishaw IQ. Bilateral cutaneous stimulation of the somatosensory system in hemidecorticate rats. Behav Neurosci. 1984;98:518–540. doi: 10.1037//0735-7044.98.3.518. [DOI] [PubMed] [Google Scholar]
- Shibata M, Kumar SR, Amar A, Fernandez JA, Hofman F, Griffin JH, Zlokovic BV. Anti-inflammatory, antithrombotic, and neuroprotective effects of activated protein C in a murine model of focal ischemic stroke. Circulation. 2001;103:1799–1805. doi: 10.1161/01.cir.103.13.1799. [DOI] [PubMed] [Google Scholar]
- Taguchi A, Soma T, Tanaka H, et al. Administration of CD34+ cells after stroke enhances neurogenesis via angiogenesis in a mouse model. J Clin Invest. 2004;114:330–338. doi: 10.1172/JCI20622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taoka Y, Schlag MG, Hopf R, Redl H. The long-term effects of pre-treatment with activated protein C in a rat model of compression-induced spinal cord injury. Spinal Cord. 2000;38:754–761. doi: 10.1038/sj.sc.3101096. [DOI] [PubMed] [Google Scholar]
- Wang L, Kittaka M, Sun N, Schreiber SS, Zlokovic BV. Chronic nicotine treatment enhances focal ischemic brain injury and depletes free pool of brain microvascular tissue plasminogen activator in rats. J Cereb Blood Flow Metab. 1997;17:136–146. doi: 10.1097/00004647-199702000-00002. [DOI] [PubMed] [Google Scholar]
- Wang Y, Jin K, Mao XO, Xie L, Banwait S, Marti HH, Greenberg DA. VEGF-overexpressing transgenic mice show enhanced post-ischemic neurogenesis and neuromigration. J Neurosci Res. 2007;85:740–747. doi: 10.1002/jnr.21169. [DOI] [PubMed] [Google Scholar]
- Wang Y, Thiyagarajan M, Chow N, Singh I, Guo H, Davis TP, Zlokovic BV. Differential Neuroprotection and Risk for Bleeding From Activated Protein C With Varying Degrees of Anticoagulant Activity. Stroke. 2008 doi: 10.1161/STROKEAHA.108.536680. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Potrovita I, Tarabin V, Herrmann O, Beer V, Weih F, Schneider A, Schwaninger M. Neuronal activation of NF-kappaB contributes to cell death in cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:30–40. doi: 10.1038/sj.jcbfm.9600004. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chopp M, Zhang RL, Goussev A. A mouse model of embolic focal cerebral ischemia. J Cereb Blood Flow Metab. 1997;17:1081–1088. doi: 10.1097/00004647-199710000-00010. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, Zhang C, Liu D, Fernandez J, Griffin JH, Chopp M. Functional recovery after embolic stroke in rodents by activated protein C. Ann Neurol. 2005;58:474–477. doi: 10.1002/ana.20602. [DOI] [PubMed] [Google Scholar]