

Abstract
Life and death fate decisions allow cells to avoid massive apoptotic death in response to genotoxic stress. While the regulatory mechanisms and signaling pathways controlling DNA repair and apoptosis are well characterized, the precise molecular strategies that determine the ultimate choice of DNA repair and survival or apoptotic cell death remain incompletely understood. Here, we report that a protein tyrosine phosphatase, Eya, is involved in promoting efficient DNA repair rather than apoptosis in response to genotoxic stress in specific tissue/cell types by executing a damage-signal dependent dephosphorylation of an H2AX C-terminal tyrosine phosphate (Y142). This post-translational modification determines the relative recruitment of either DNA repair or pro-apoptotic factors to the tail of γH2AX and allows it to function as an active determinant of repair/survival versus apoptotic responses to DNA damage, revealing an additional phosphorylation-dependent mechanism that modulates survival/apoptotic decisions during mammalian organogenesis.
Keywords: Eya, H2AX, DNA repair, apoptosis
The developmentally regulated transcriptional co-factor Eya is a component of the retinal determination (RD) pathway that controls the development of various organ systems in metazoans, including the kidney [1–3]. The primary phenotypic consequence of loss of Eya activity is increased apoptotic cell death in early tissue primordium and subsequent agenesis of target tissues [3, 4]. Previous work by our lab and others identified a phosphatase enzymatic domain in mammalian Eya1-4 as well as the Drosophila homologue eyes absent (eya), and demonstrated that Eya is a functional phosphatase [6–8]. While early in-vitro phosphatase assays using synthetic phospho-peptides suggested that Eya might possess dual-specificity, subsequent data has indicated that, in-vivo, Eya primarily functions as a tyrosine phosphatase [9]. In this study, we demonstrate that increased apoptosis seen in the absence of Eya is at least in part due to persistent phosphorylation of H2AX Y142, a mark which is a component of the mechanisms that distinguish between apoptotic and repair responses to genotoxic stress.
Eya-H2AX interactions
We noticed that increased apoptosis and loss of renal tubules seen in the developing kidney of Eya1−/− mouse embryos coincided with increased immunostaining for serine139-phosphorylated H2AX (γH2AX) (Supplementary Fig. 1, Fig. 1a, b). Nuclear phosphorylation of the histone variant H2AX was recently shown to be a crucial component of apoptosis induced by the activation the JNK/SAPK stress response pathway[10], in addition to having a well-studied role in DNA damage repair [11–14].
Figure 1.
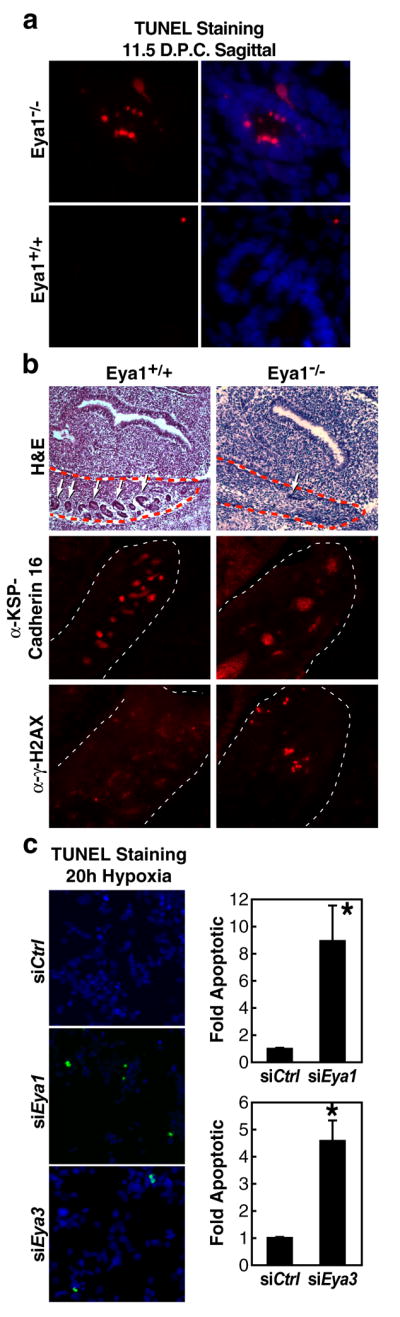
Loss of Eya leads to increased γH2AX-posititve apoptotic cells. (a) TUNEL staining reveals apoptotic cells within the developing kidney of Eya1−/− embryos at e11.5 not present in wild type littermates. (b) Abnormal morphology and loss of developing renal tubules (white arrows) within the urogenital ridge (red dotted line) in Eya1−/−embryos coincides with increased γH2AX-positive nuclei by immunostaining. (c) In culture, 293T human embryonic kidney cells depleted for Eya1/3 using siRNA displayed increased apoptotic response to hypoxia for 20hrs (2% O2). Cell counts were performed on TUNEL stained cells co-stained with DAPI in triplicate to identify the proportion of TUNEL-positive nuclei. Basal level of apoptosis under these conditions was 1.4% TUNEL-positive/total nuclei. Bar graphs represent mean +/− SEM of fold apoptotic cells normalized to control siRNA from triplicate samples. “*”p- <.05.
Because the developing kidney is exposed to localized hypoxia during early development as the rapidly proliferating organ outgrows the local vasculature, potentially leading to activation of stress response pathways and increased generation of reactive oxygen species [15, 16], we considered the possibility that apoptosis induced in the absence of Eya might be related to altered DNA damage response pathways. To mimic the events in the Eya1−/− kidney in a cell model, we depleted endogenous Eya1 or Eya3 in 293T human embryonic kidney cells using specific siRNAs (Supplementary Fig. 2) and then subjected the cells to hypoxic conditions for 20 hours. Eya1 and Eya3 have been previously qualified as phosphatase enzymes [6–8] and both are expressed in 293T cells. Interestingly, knockdown of either Eya1 or Eya3 using specific siRNAs caused a significant increase in TUNEL-positive apoptotic nuclei in response to hypoxia (Fig. 1c). Analogous experiments directly inducing DNA damage with ionizing radiation resulted in a similar increase in sensitivity for Eya-depleted cells (Supplementary Fig. 3). Thus, in embryonic kidney cells, both in vivo and in culture, an increase in apoptotic cell death is observed in the absence of Eya1 that may be related to the cellular response to DNA damage, which involves γH2AX [11, 17].
We therefore investigated a potential interaction between Eya and H2AX by co-immunoprecipitation assays using 293T embryonic kidney cells before and after exposing the cells to ionizing radiation to induce DNA damage. We could detect interactions between H2AX and wild-type Eya1 or Eya3 only under DNA damage conditions both using transfected, tagged expression constructs for Eya1/3 and H2AX (Fig. 2a), and when examining endogenous Eya3 and H2AX proteins with specific antibodies (Fig. 2b). Eya was capable of interacting with H2AX in the context of chromatin, based on co-immunoprecipitation experiments using fixed sonicated chromatin from 293T cells as input (Fig. 2c). In response to IR-induced double stranded DNA breaks, H2AX is phosphorylated by ATM/ATR PI3K-family kinases on chromatin forming long stretches of serine phosphorylated γH2AX flanking the break visible as γH2AX immunostained foci [18]. Endogenous Eya3 co-immunoprecipitated γH2AX in 293T cells after IR treatment (Fig. 2b, lower panel), and immunostaining of transfected HA-tagged Eya1 or Eya3 protein in 293T embryonic kidney cells revealed a clear co-localization of Eya with γH2AX foci after treatment with IR (Fig. 2d, e). These results suggest that in response to damage, Eya is recruited to H2AX foci that mark DNA double-strand breaks. To formally test this, we utilized the estrogen receptor-I PpoI system [19, 20], in which 4-hydroxytamoxifen (4-OHT) is used to induce activation of the eukaryotic homing endonuclease I-PpoI which then generates double stranded breaks at defined genomic loci, including a site on chromosome 1 within an intron of the DAB1 locus. ChIP analysis following 4-OHT induction of I-PpoI in 293T cells revealed that γH2AX and Eya3 were present at a 6 hour time point at a 4kb region flanking the IPpoI cut site, which is in consistent with a direct role for Eya in the cellular response to genotoxic stress (Supplementary Fig. 4).
Figure 2.

Eya interacts with H2AX in a DNA-damage dependent manner. (a) HA-tagged Eya1 or Eya3 interacts with FLAG-tagged H2AX in 293T cells in response to IR (5Gy), but not under basal conditions. (b) Co-immunoprecipitation experiments examining endogenous Eya3 protein using a specific Eya3 antibody recapitulated that interaction data for the tagged proteins. (c) Using sonicated chromatin as input, co-immunoprecipitation experiments showed that HA-Eya1/3 interacts with H2AX on chromatin. Immunostaining of 293T cells demonstrates that transfected, HA-tagged Eya1 (d) or Eya3 (e) localizes to DNA-damage induced foci coincident with γH2AX specifically after treatment with IR (5Gy, 1hr). Representative examples of foci formation are shown.
Interestingly, we found that Eya3 is serine phosphorylated in 293T cells in response to genotoxic stress (Fig. 3a), consistent the recent identification of Eya3 as a potential substrate for the DNA damage-response protein kinases ATM and ATR [21–23]. Inhibition of ATM/ATR function, by pretreating cells with the PI3K inhibitor caffeine, blocked the interaction between Eya3 or Eya1 and H2AX in response to ionizing radiation (Fig. 3b). Serine 219 of Eya3 was identified by mass spectroscopy as a target residue for ATM/ATR phosphorylation [22], and a S219A Eya3 mutant failed to form damage-dependent nuclear foci or interact with H2AX after IR treatment (Fig. 3c, d), indicating that ATM/ATR phosphorylation of Eya3 on serine 219 is crucial for directing Eya-H2AX interactions. Because Eya1 and Eya3 are seen to interact in 293T embryonic kidney cells both before and after treatment with ionizing radiation (Fig. 3e), we suspect that regulation of Eya3 via damage-dependent phosphorylation at serine 219 is one cue that may direct both Eya1 and Eya3 to γH2AX, indicating that these covalent modifications of H2AX and Eya may act as sensors for the DNA damage response pathway.
Figure 3.

Eya3 phosphorylation by ATM/ATR DNA-damage dependent kinases regulates interaction between Eya and H2AX. (a) Endogenous Eya3 was immunoprecipitated from 293T cells with a specific Eya3 antibody and western blotting was performed with an antibody specific to the phosphorylated target site of ATM/ATR, demonstrating phosphorylation of Eya3 in response to DNA damage (5Gy IR). (b) Eya1/3 interaction with H2AX is lost in the presence of a PI3K inhibitor (5 mM caffeine). (c) Mutation of the ATM/ATR phosphorylation site of Eya3 (S219) prevents formation of damage-induced Eya3 foci. Representative examples of foci formation are shown. (d) HA-Eya3 (S219A) fails to interact with FLAG-H2AX in response to DNA-damage (5Gy IR) by co-immunoprecipitation in 293T cells. (e) DNA damage-independent interaction of Eya3 and Eya1 was assessed by co-immunoprecipitation in 293T cells.
H2AX is an Eya tyrosine phosphatase substrate
We next tested whether the interaction between H2AX and Eya could represent a substrate-enzyme relationship. Because current evidence suggests that Eya is a tyrosine-specific phosphatase [6–8], we assessed its activity as a tyrosine phosphatase on γH2AX. H2AX purified either from 293T cells or from bovine histone fraction possesses tyrosine phosphorylation as seen using a phosphotyrosine-specific antibody (Supplementary Fig. 5). This tyrosine phosphorylation mark on H2AX decreased in response to DNA damage induced by ionizing radiation, the topoisomerase I inhibitor CPT, or hypoxia (Fig. 4a). To determine whether this H2AX phosphorylation mark might be a target of Eya phosphatase activity, we utilized an in-vitro phosphatase assay, mixing immuno-purified HA-tagged Eya1 or Eya3 with H2AX protein. Wild-type Eya effectively removed the phosphotyrosine mark from H2AX, while the phosphatase-inactive mutant Eya proteins (Eya1 D323A or Eya3 D246A) had little or no effect (Fig. 4b).
Figure 4.

Tyrosine phosphorylated H2AX is a substrate for Eya phosphatase. (a) IP-western of tyrosine phosphorylated H2AX in response to DNA-damage signals. Bars represent quantified western blot signals normalized to untreated cells. (b) In-vitro phosphatase assay using immunopurified wild type Eya1/3 or enzymatically-inactive mutant proteins (Eya1 D323A, Eya3D246A) and bovine histone. Bars represent quantified western blot signals normalized to input. Mean values +/− SEM from triplicate western blot experiments are shown. “**”p-value <.001. (c) siRNA knockdown of endogenous Eya1/3 in 293T cells (48h) and subsequent IP-western for tyrosine phosphorylated H2AX. (d) Rescue of Eya function by co-transfection of human siRNA and murine wild type or enzymatically inactive mutant Eya3 constructs in 293T human embryonic kidney cells reveals loss of H2AX phosphotyrosine mark dependent on Eya phosphatase activity. (e) Individual substitution mutations of four H2AX tyrosine residues followed by IP-western to detect phosphotyrosine. (f) In-vitro phosphatase assay using bacterially expressed Eya3 EYA domain, wild-type or D246A, with purified peptides of the H2AX tail (AA 128–142) phosphorylated at S139 (CTpep pS) or Y142 (CTpep pY) demonstrates that Eya phosphatase activity is specific for phosphotyrosine. The Km value for Eya dephosphorylation of CTpep pY was 0.38mM with a corresponding Kcat/Km value of 0.96M−1min−1. Bar graphs represent mean +/− SEM of nM PO4 released from triplicate phosphatase reactions.
To confirm this activity in a cellular context, 293T human embryonic kidney cells were transfected with siRNA against Eya1 or Eya3 or control siRNA and subsequently exposed to ionizing radiation. In contrast to untransfected cells or cells receiving control siRNA, which displayed a loss of γH2AX tyrosine phosphorylation in response to damage as seen previously, Eya siRNA-treated cells showed significantly increasedγH2AX tyrosine phosphorylation levels as assessed by western blot analysis (Fig. 4c). Knockdown of Eya1 or Eya3 had no effect on tyrosine phosphorylation of H2AX in 293T cells not exposed to ionizing radiation (Supplementary Fig. 6). Rescuing Eya function by expressing wild-type murine Eya3 (Fig. 4d) or Eya1 (Supplementary Fig. 7) constructs, not targeted by the siRNAs, into these siRNA-depleted cells reversed this increased H2AX phosphorylation, while a phosphatase-dead mutant Eya failed to rescue. The observation that depletion of either Eya1 or Eya3 alone proved to be sufficient to fully block H2AX tyrosine de-phosphorylation in these cells suggested a lack of compensatory activity by these two homologues. Because Eya1 and Eya3 co-purify in 293T cells before and after damage (Fig. 3e), we are tempted to suggest that, specifically in the context of this embryonic kidney cell line model, Eya1 and Eya3 may form a stable complex which exhibits tyrosine phosphatase activity toward γH2AX, with both components required for the overall stability of the enzymatic complex, although these factors may be non-redundant in-vivo.
We next sought to identify precisely which tyrosine residue(s) on H2AX were phosphorylated. Mutagenesis of each of the four tyrosine residues in H2AX revealed that only mutation of tyrosine residue 142 blocked H2AX tyrosine phosphorylation as assessed by western blot analysis (Fig. 4e), indicating that Y142 was the only phosphorylated tyrosine.
To confirm the in-vitro tyrosine phosphatase function of Eya [6–8] and demonstrate specificity for tyrosine-phosphorylated H2AX, rather than serine, we generated a bacterially expressed construct representing the enzymatically active C-terminal Eya-Domain of Eya3 [6]. This Eya enzyme showed robust phosphatase activity when mixed with a synthetic phosphopeptide representing the C-terminal tail domain of H2AX (CT-pep) phosphorylated on tyrosine, but showed minimal activity toward a serine phosphorylated tail peptide (Fig. 4f). These data biochemically establish the ability of Eya to directly dephosphorylate H2AX phosphorylated on tyrosine 142.
Eya-dependent H2AX Y142 de-phosphorylation: function in apoptosis
To begin to evaluate a possible connection between Eya-mediated tyrosine dephosphorylation of H2AX Y142 and modulation of the apoptotic response, we examined the function of this phosphotyrsoine mark in the context of the DNA damage response. FLAG-tagged H2AX Y142F mutant was phosphorylated on S139 in response to damage, although at levels significantly lower than FLAG-tagged wild-type H2AX. (Fig. 5a) Time course anlysis of S139 phoshorylation of H2AX Y142F in response to 10Gy IR in 293T human embryonic kidney cells revealed consistently reduced levels compared to wild type between 1 and 8 hours (Supplementary Fig. 8). Thus, while Y142 phosphorylation does not function as a pre-requisite for S139 phosphorylation in the DNA damage response [24], it may play a significant role in promoting or maintaining serine phosphorylation by DNA-damage response kinases.
Figure 5.

H2AX Y142 phosphorylation discriminates between apoptotic and repair responses to DNA-damage. (a) S139 phosphorylation of H2AX Y142F is present but reduced in comparison to wild type H2AX after 5Gy IR. (b) Affinity purification performed on nuclear extract from irradiated 293T cells using synthetic peptides representing the C-terminal tail of H2AX bearing S139 phosphorylation +/− Y142 phosphorylation followed by western blot analysis. Co-immunoprecipitaiton confirms interaction between wild-type H2AX and JNK1 (c) or Fe65 (d) but not H2AX Y142F in 293T cells exposed to high-dose IR (50Gy). (e) Endogenous Fe65 interacts with JNK1 in 293T cells treated with etoposide (30uM). Figures 5d and 5e show individual bands from a single western blot exposure. (f) siRNA knockdown of Fe65 in 293T cells blocks the damage-dependent (50Gy) interaction of JNK1 and γH2AX by co-immunoprecipitation in cells transfected with Fe65 siRNA or control siRNA 48 hours prior to harvest. Results were confirmed with two separate siRNA sets for Fe65. (g) H2AX−/− MEF cells were transfected with wild type or mutant H2AX (Y142F) expression constructs and exposed to high-dose IR (100Gy). Apoptotic response among transfectants was assessed by γH2AX staining and TUNEL. Bar graphs represent mean +/− SEM of fold apoptotic values for triplicate or greater cell counts of transfected (green) nuclei. Basal level of apoptosis for WT-H2AX transfected cells under these conditions was 25.7% TUNEL positive/total transfeced nuclei. Values were normalized to WT-H2AX-transfected samples. “**”p <.001. (h) Proposed model for Y142 phosphorylation status of H2AX in regulation of apoptotic versus repair response.
It has been established that a key function of H2AX S139 phosphorylation is to provide a docking site for DNA repair factors near or at DNA double strand breaks [18]. These factors include Mediator of DNA Damage Checkpoint protein 1 (MDC1) which has been shown to bind directly to phosphorylated S139 of H2AX at the sites of double strand breaks [24] based on tandem BRCT1 repeats within the C-terminus of MDC1 [25]. MDC1 functions in the recruitment of a set of ancillary repair factors including MRE11, RAD50, NBS1 (the MRN complex), 53BP1 and BRCA1 [26, 27], although these factors are not wholly dependent on MDC1 and γH2AX for recruitment to breaks [28]. Because an intact H2AX COOH-terminal tyrosine has been found to be required for MDC1-H2AX interaction and productive DNA repair [24], it was of particular interest to determine whether persistent phosphorylation of Y142 in the absence of Eya could negatively impact MDC1 recruitment to the tail of γH2AX. We first generated peptides corresponding to the C-terminal tail of H2AX with phosphorylation of both S129 and Y142, or of S139 alone. Peptides lacking any phosphorylation marks or where tyrosine 142 was mutated to alanine failed to interact with MDC1, consistent with previously published reports (Supplementary Fig. 9) [24]. Affinity purification of nuclear extract from irradiated 293T cells with each peptide revealed that, in the absence of Y142 phosphorylation, a set of DNA repair factors including MDC1, MRE11 and Rad50 were bound to the S139 phosphorylated H2AX peptide (Fig. 5b). Intriguingly, when phosphorylated tyrosine 142 was present with phosphoserine 139, binding of these factors was greatly reduced; instead, the established pro-apoptotic factor JNK1 was now present (Fig. 5b). The stress-response kinase JNK1, activated by DNA damage and initiating a pro-apoptotic program, has been recently shown to translocate into the nucleus upon activation where it phosphorylates substrates including H2AX S139, an event critical for DNA degradation mediated by caspase-activated DNase (CAD) in apoptotic cells [10]. In agreement with our peptide purification experiments, we were able to detect a robust interaction between transfected wild-type H2AX and endogenous JNK1 in 293T cells in response to high-dose radiation; this interaction was markedly reduced in the case of the H2AX Y142F mutant (Fig. 5c).
To further confirm the specificity of these phosphorylation-dependent interactions we performed peptide competition assays. The H2AX tail peptide phosphorylated on S139 alone was able to effectively compete for binding of MDC1 in a peptide pull-down assay, while the free peptide bearing both S139 and Y142 phosphorylation marks competed away interaction with JNK1 (Supplementary Fig. 10).
Based on our previous data that loss of Eya phosphatase results in increased tyrosine phosphorylation of H2AX, we predicted that depleting Eya in 293T cells would result in decreased binding of MDC1 to H2AX in response to DNA damage. We knocked down Eya3 using specific siRNA and subsequently tested for MDC1-H2AX interaction by co-immunoprecipitation. As predicted, loss of Eya3 resulted in complete loss of this interaction in comparison to untransfected cells treated with 10Gy IR (Supplementary Fig. 11).
It was of particular interest to identify proteins containing SH2 and PTB phosphotyrosine-binding domains that could bind directly to H2AX phospho-tyrosine 142 under conditions of genotoxic stress. We tested a set of known nuclear proteins containing these domains for binding to tyrosine-phosphorylated H2AX (Supplementary Table 1, partial list) and found that, while most exhibited no interaction, the PTB-domain protein Fe65 [29], a co-factor for several cell-surface receptors that has been shown to translocate to the nucleus during DNA damage response and suggested to exert a pro-apoptotic role [30, 31], bound specifically to wild type γH2AX under DNA damage conditions, but not to the γH2AX Y142F mutant (Fig. 5d). Significantly, we found that Fe65 protein interacted with endogenous JNK1 by co-immunoprecipitation in 293T cells treated with the DNA-damage agent etoposide (Fig. 5e), consistent with the idea that Fe65 helps to mediate JNK1 recruitment to γH2AX. Co-immunoprecipitation experiments demonstrated that the second PTB domain on Fe65 may be key for the interaction between Fe65 and tyrosine phosphorylated H2AX (Supplementary Fig. 12a). GST pull-down assays using purified recombinant protein of Fe65 PTB domains 1 and 2 also revealed a direct interaction between PTB2 and the H2AX present in purified HeLa histones (Supplementary Fig. 12b). We postulated that Fe65 may function as an adaptor protein, binding directly to the phosphotyrosine reside on γH2AX via PTB2 and facilitating the recruitment of pro-apoptotic factors such as JNK1. To test this, we knocked down endogenous Fe65 in 293T cells using specific siRNAs (Supplementary Figure 2) and assessed the interaction between H2AX and JNK1 in response to genotoxic stress by co-immunoprecipitation. While control siRNA had no effect on the ability of H2AX to co-immunoprecipitate JNK1, knockdown of Fe65 strongly inhibited this interaction (Fig. 5f).
To confirm the function of tyrosine 142 phosphorylation in regulation of the apoptotic response, we transfected H2AX −/− mouse embryonic fibroblasts (MEFs) [32] with either wild type or Y142F H2AX expression constructs. When these cells were subjected to high-dose ionizing radiation, cells expressing H2AX Y142F displayed a reduced apoptotic response in comparison to cells expressing wild-type H2AX (~6-fold decrease) (Fig. 5g). These data suggested to us that lack of H2AX Y142 phosphorylation promotes a damage repair response instead of an apoptotic response to DNA damage, in part by promoting successful recruitment of MDC1 and associated repair factors. The presence of Y142 phosphorylation in wild type-H2AX transfected MEF cells is proposed to lead to the recruitment of pro-apoptotic factors such as JNK1 to H2AX, while inhibiting the recruitment of the damage repair complex, directly promoting apoptotic response to genotoxic stress.
Conclusions
Cells are confronted with DNA-damage resulting from a variety of stimuli under normal, physiological conditions and at each instance the cell must make fundamental decisions in the ratio of DNA repair and apoptotic response Our data suggest thatγH2AX serves as a component of the adjudication between the balance of these two outcomes, with a single post-translational modification, phosphorylation of tyrosine 142, being capable of influencing the recruitment to γH2AX of functional apoptotic or repair complexes. In the presence of Y142 phosphorylation, binding of repair factors to phosphorylated serine 139, which is mediated by MDC1, is inhibited (Fig. 5h), while recruitment of pro-apoptotic factors, including JNK1, is promoted.
Eya binds to Six-class homeodomain transcription factors. While early in-vitro studies suggested that phosphatase activity was important for Eya-mediated transcriptional activation of certain Six-dependent reporter genes [6], recent studies in Drosophila suggest that the majority of Six/Eya transcriptional targets do not require phosphatase enzymatic activity for activation in-vivo [33]. Phosphatase activity of Eya may have a novel function in mammalian organogenesis, acting to block an improper apoptotic response to physiological levels of genotoxic stress by dephosphorylating H2AX on tyrosine.
Coincident with our studies, recently published work from Xiao, et. al. reported phosphorylation of H2AX on tyrosine 142 under basal conditions which decreases in response to DNA damage in mouse embryonic fibroblasts [34]. The relevant kinase was demonstrated to be WSTF (Williams-Beuren Syndrome Transcription Factor), which physically interacts with H2AX specifically in undamaged cells. The authors demonstrated that siRNA knockdown of WSTF results in loss of H2AX tyrosine 142 phosphorylation, which alters the kinetics of S139 phosphorylation in response to DNA damage. Thus, it appears that H2AX tyrosine phosphorylation is deposited by WSTF under basal conditions and, at least in the embryonic kidney cell model system, is removed by Eya in response to DNA damage.
The present study indicates that the phosphorylation of tyrosine 142 of H2AX prevents recruitment of repair complexes to phospho-serine 139 of γH2AX, although it is likely that there are many additional aspects that underlie the full molecular logic for the dual phosphorylation-mediated events. We hypothesize that the presence of both phosphorylated resides results in direct binding of the PTB-domain factor Fe65, which, at least in part, mediates the effective recruitment of other pro-apoptotic factors, including JNK1.
Methods Summary
Eya1 knockout mice were originally generated by the laboratory of Richard Maas (Harvard Medical School). 293T and H2AX −/− MEF cells were maintained in DMEM (Gibco) supplemented with 10% fetal calf serum (FCS; Gemini). Plasmids and siRNAs were transfected with Lipofectamine 2000 (Invitrogen) as directed. Specific antibodies for immuoprecipitation and immunostaining were obtained from Upstate (anti-γH2AX), Zymed (anti-phosphotyrosine), Cell Signaling Technology (anti-H2AX, anti-γH2AX), Abcam (anti-KSP-Cadherin 16, anti-MDC1), Sigma (anti-FLAG), and Santa Cruz Biotechnology (anti-RAD50, MRE11, JNK1). Purified peptides were obtained from Sigma Genosys, Abgent, and Anaspec.
Methods
Antibodies, Reagents and Cells
The following commercially available antibodies were used: anti-H2AX (Cell Signaling Technology and Abcam), anti-γH2AX (Cell Signaling Technology and Upstate), anti-phosphotyrosine (Zymed and Upstate), anti-KSP-Cadherin 16 (Abcam), anti-HA (Berkeley Antibody Company), anti-FLAG (Sigma), anti-MDC1 (Abcam and Bethyl laboratories), anti-RAD50, MRE11, JNK1 (Abcam and Santa Cruz Biotechnology). Antibodies to Eya3 were generated by immunizing guinea pigs with GST-purified peptides representing the amino-terminus of human EYA3 (AA 1-239). The following commercially available reagents were used: caffeine (Calbiochem). Eya1 and Eya3 siRNAs were purchased from Qiagen. H2AX−/−MEF was kindly provided by Drs. A. Nussenzweig (NCI), Y. Xu and H. Song (UCSD). Standard molecular cloning and tissue culture were performed as described by Sambrook and Russell (2001).
Animal Care and Immunohistochemistry
Eya1 knockout mice were originally generated by the laboratory of Dr. R. Mass (Harvard Medical School). Mouse embryos from E10.5 to E11.5 were fixed in 2% paraformaldehyde, penetrated with 24% sucrose in PBS, and embedded in OCT compound for cryo-sectioning. Serial 14um sections were blocked in 10% normal goat serum/PBS/0.1% Triton-X 100 and immunostained using antibodies to γH2AX or KSP-Cadherin16. Immunostaining was visualized using secondary antibodies conjugated to AlexaFluor-595 (Invitrogen) and sections were mounted using Vectashield mounting media plus DAPI (Vector Laboratories). Parallel sections were stained with Haematoxylin and Eosin as described (Li, et. al., 2003).
TUNEL Staining
TUNEL assay was performed using ApopTag In Situ Apoptosis Detection Kit (Chemicon). Tissue sections were post-fixed in ethanol:acetic acid 2:1 at −20°C for 5 minutes and incubated with TdT enzyme at 37°C for 1 hour. DIG incorporation was visualized using anti-digoxigenin-rhodamine secondary (Roche) and stained sections were mounted using Vectashield mounting media plus DAPI (Vector Laboratories).
Cell Treatment and Transfection/RNA interference
For hypoxia experiments, 293T cells were transferred to an 8% CO2, 2% O2 incubator and maintained for approximately 20 hours. Cells were immediately fixed or lysed upon removal from the hypoxia incubator. Gamma-irradiation of cultured cells was performed at the UCSD Medical Teaching Facility according to established protocols. The cells were gamma-irradiated approximately 36–48 hrs after transfection. Cells were transfected using Lipofectamine 2000 (Invitrogen). siRNA target sequences were as follows: EYA1- caggaaataattcactcacaa, EYA3- ccggaaagtgagagaaatcta, Fe65- ctgtattgatatcactaataa (Qiagen), cuacguagcucgugauaag, ggguagaugugauuaaugg, gaucaaguguuucgccgug, cgucagcucucuuaccaca (Dharmacon)
Immunoprecipitation/Western Blot Analysis
For immunoprecipitation and Western blotting, cells were rinsed in PBS, harvested, and lysed in Lysis buffer containing 10% glycerol, 0.5 mM EDTA, 25 mM Tris-HCl (pH8.0), 150 mM NaCl,, 1 mM Na2VO3, 10 mM β-glycerophosphate, 0.1% NP-40 and 1 mM DTT in presence of protease inhibitors (Roche) and 1 mM PMSF. The extracts were incubated with the specific antibody overnight at 4°C, followed by incubation with protein A/G agarose beads (Santa Cruz Biotech.), washed extensively, and separated by electrophoresis. Proteins were transferred onto nitrocellulose membranes (Bio-Rad) and Western blotting was performed following standard protocols.
Immunocytochemistry
Cells were fixed for 15 min with 2% paraformaldehyde in PBS and permeablized with 0.05% Triton X-100 in PBS for 30 min. After blocking with PGBA solution (0.1% BSA, 0.1% gelatine, 0.1% FBS), cells were incubated with specific antibodies for 2 hrs at RT. Antigen was detected with secondary antibodies conjugated to AlexaFluor-595 or AlexaFluor-488 (Invitrogen). Cells were coverslipped using Vectasheild mounting media plus DAPI (Vector Laboratories).
In vitro phosphatase assay
The HA-tagged Eya phosphatase was immunoprecipitated from gamma-irradiated 293T cells using anti-HA affinity resin (Roche). After extensive washing, Eya phosphatase was eluted with HA peptide. The reaction mixture containing purified Eya protein in 100 μl phosphatase buffer (50 mM Tris-HCl, pH 7.0, 5 mM MgCl2, 10% glycerol, 3mg/ml BSA) and bovine histone (Sigma) was incubated for 60–90 min at 30 °C. The H2AX was immuoprecipitated with anti-H2AX antibody and Western blotting was performed. GST-fusion proteins of Eya3 240-573 and Eya3 D246A 240-573 were expressed in BL21 bacterial cells and purified with glutathione-agarose beads (Sigma). Wild-type and mutant GST proteins were incubated with .2mg purified peptides of the H2AX tail bearing phosphorylation at either S139 or Y142 (Abgent) in phosphatase buffer for 1 hour and free phosphatase was detected using Malachite Green (BIOMOL).
Peptide affinity chromatography
Biotinylated synthetic peptides (hH2AX amino acid 128–142) were purchased from Sigma Genosys, Anaspec, and Abgent. For peptide affinity chromatography, biotinylated phosphopeptides and unphosphorylated peptides were coupled to streptavidin-coated Dynabeads M-280 (Invitrogen) for 2 hrs at RT. Beads were incubated with nuclear extract from 200 Gy-irradiated 293T cells and washed extensively with Tris buffered saline (pH 7.5) containing 0.5% Tween 20. The bound proteins were separated by SDS-PAGE using 4%–12% Bis-Tris NuPAGE gel (Invitrogen), followed by Western blot analysis.
Supplementary Material
Acknowledgments
We thank Victoria Lunyak (Buck Institute for Age Research), Jack Dixon (HHMI/UCSD), and Richard Koladner (UCSD) for review and discussions. We thank the laboratory of Randall S. Johnson (UCSD) for use of equipment and advice on hypoxia incubations, as well as Havilah Taylor for animal care assistance. We thank Drs. A. Nussenzweig (NCI), Y. Xu and H. Song (UCSD) for H2AX−/− MEF. We thank J. Hightower and M. Fisher for assistance with figure and manuscript preparation. We additionally thank Xue Li (Harvard Medical School), and Wen Liu (UCSD). MGR is an HHMI Investigator. This work was supported by grants from NIH and NCI to MGR, CKG. This work also was supported by the Sogang University Research Grant of 2008 to BJ.
References
- 1.Kumar JP. Signalling pathways in Drosophila and vertebrate retinal development. Nat Rev Genet. 2001;2(11):846–57. doi: 10.1038/35098564. [DOI] [PubMed] [Google Scholar]
- 2.Silver SJ, Rebay I. Signaling circuitries in development: insights from the retinal determination gene network. Development. 2005;132(1):3–13. doi: 10.1242/dev.01539. [DOI] [PubMed] [Google Scholar]
- 3.Pignoni F, et al. The eye-specification proteins So and Eya form a complex and regulate multiple steps in Drosophila eye development. Cell. 1997;91(7):881–91. doi: 10.1016/s0092-8674(00)80480-8. [DOI] [PubMed] [Google Scholar]
- 4.Bonini NM, Leiserson WM, Benzer S. The eyes absent gene: genetic control of cell survival and differentiation in the developing Drosophila eye. Cell. 1993;72(3):379–95. doi: 10.1016/0092-8674(93)90115-7. [DOI] [PubMed] [Google Scholar]
- 5.Xu PX, et al. Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat Genet. 1999;23(1):113–7. doi: 10.1038/12722. [DOI] [PubMed] [Google Scholar]
- 6.Li X, et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. 2003;426(6964):247–54. doi: 10.1038/nature02083. [DOI] [PubMed] [Google Scholar]
- 7.Rayapureddi JP, et al. Eyes absent represents a class of protein tyrosine phosphatases. Nature. 2003;426(6964):295–8. doi: 10.1038/nature02093. [DOI] [PubMed] [Google Scholar]
- 8.Tootle TL, et al. The transcription factor Eyes absent is a protein tyrosine phosphatase. Nature. 2003;426(6964):299–302. doi: 10.1038/nature02097. [DOI] [PubMed] [Google Scholar]
- 9.Rayapureddi JP, et al. Characterization of a plant, tyrosine-specific phosphatase of the aspartyl class. Biochemistry. 2005;44(2):751–8. doi: 10.1021/bi0481794. [DOI] [PubMed] [Google Scholar]
- 10.Lu C, et al. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol Cell. 2006;23(1):121–32. doi: 10.1016/j.molcel.2006.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bassing CH, Alt FW. The cellular response to general and programmed DNA double strand breaks. DNA Repair (Amst) 2004;3(8–9):781–96. doi: 10.1016/j.dnarep.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Bassing CH, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci U S A. 2002;99(12):8173–8. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karagiannis TC, El-Osta A. Chromatin modifications and DNA double-strand breaks: the current state of play. Leukemia. 2007;21(2):195–200. doi: 10.1038/sj.leu.2404478. [DOI] [PubMed] [Google Scholar]
- 14.van Attikum H, Gasser SM. The histone code at DNA breaks: a guide to repair? Nat Rev Mol Cell Biol. 2005;6(10):757–65. doi: 10.1038/nrm1737. [DOI] [PubMed] [Google Scholar]
- 15.Lee YM, et al. Determination of hypoxic region by hypoxia marker in developing mouse embryos in vivo: a possible signal for vessel development. Dev Dyn. 2001;220(2):175–86. doi: 10.1002/1097-0177(20010201)220:2<175::AID-DVDY1101>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 16.Haase VH. Hypoxia-inducible factors in the kidney. Am J Physiol Renal Physiol. 2006;291(2):F271–81. doi: 10.1152/ajprenal.00071.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez-Capetillo O, et al. H2AX: the histone guardian of the genome. DNA Repair (Amst) 2004;3(8–9):959–67. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 18.Rogakou EP, et al. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146(5):905–16. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berkovich E, Monnat RJ, Jr, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9(6):683–90. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- 20.Berkovich E, Monnat RJ, Jr, Kastan MB. Assessment of protein dynamics and DNA repair following generation of DNA double-strand breaks at defined genomic sites. Nat Protoc. 2008;3(5):915–22. doi: 10.1038/nprot.2008.54. [DOI] [PubMed] [Google Scholar]
- 21.Matsuoka S, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 22.Stokes MP, et al. Profiling of UV-induced ATM/ATR signaling pathways. Proc Natl Acad Sci U S A. 2007;104(50):19855–60. doi: 10.1073/pnas.0707579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6(8):931–42. doi: 10.4161/cc.6.8.4180. [DOI] [PubMed] [Google Scholar]
- 24.Stucki M, et al. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123(7):1213–26. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 25.Lee MS, et al. Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the gamma-H2AX histone tail. J Biol Chem. 2005;280(37):32053–6. doi: 10.1074/jbc.C500273200. [DOI] [PubMed] [Google Scholar]
- 26.Kim JE, Minter-Dykhouse K, Chen J. Signaling networks controlled by the MRN complex and MDC1 during early DNA damage responses. Mol Carcinog. 2006;45(6):403–8. doi: 10.1002/mc.20221. [DOI] [PubMed] [Google Scholar]
- 27.Wu X, et al. ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature. 2000;405(6785):477–82. doi: 10.1038/35013089. [DOI] [PubMed] [Google Scholar]
- 28.Celeste A, et al. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. 2003;5(7):675–9. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 29.Duilio A, et al. A rat brain mRNA encoding a transcriptional activator homologous to the DNA binding domain of retroviral integrases. Nucleic Acids Res. 1991;19(19):5269–74. doi: 10.1093/nar/19.19.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minopoli G, et al. Essential roles for Fe65, Alzheimer amyloid precursor-binding protein, in the cellular response to DNA damage. J Biol Chem. 2007;282(2):831–5. doi: 10.1074/jbc.C600276200. [DOI] [PubMed] [Google Scholar]
- 31.Nakaya T, Kawai T, Suzuki T. Regulation of FE65 Nuclear Translocation and Function by Amyloid {beta}-Protein Precursor in Osmotically Stressed Cells. J Biol Chem. 2008;283(27):19119–31. doi: 10.1074/jbc.M801827200. [DOI] [PubMed] [Google Scholar]
- 32.Celeste A, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296(5569):922–7. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jemc J, Rebay I. Identification of transcriptional targets of the dual-function transcription factor/phosphatase eyes absent. Dev Biol. 2007;310(2):416–29. doi: 10.1016/j.ydbio.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiao A, et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature. 2009;457(7225):57–62. doi: 10.1038/nature07668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.