

Abstract
β-Adrenoceptor stimulation robustly increases cardiac L-type Ca2+ current (ICaL); yet the molecular mechanism of this effect is still not well understood. Previous reports have shown in vitro phosphorylation of a consensus protein kinase A site at serine 1928 on the carboxyl terminus of the α1C subunit; however, the functional role of this site has not been investigated in cardiac myocytes. Here, we examine the effects of truncating the distal carboxyl terminus of the α1C subunit at amino acid residue 1905 or mutating the putative protein kinase A site at serine 1928 to alanine in adult guinea pig myocytes, using novel dihydropyridine-insensitive α1C adenoviruses, coexpressed with β2 subunits. Expression of α1C truncated at 1905 dramatically attenuated the increase of peak ICaL induced by isoproterenol. However, the point mutation S1928A did not significantly attenuate the β-adrenergic response. The findings indicate that the distal carboxyl-terminus of α1C plays an important role in β-adrenergic upregulation of cardiac L-type Ca2+ channels, but that phosphorylation of serine 1928 is not required for this effect.
Keywords: protein kinase A, adenovirus, ion channel, calcium current, cAMP
Voltage-gated L-type (Cav1.2) Ca2+ channels play a central role in controlling cardiac function, providing the trigger for intracellular Ca2+ release during excitation-contraction coupling, contributing to the plateau phase of the cardiac action potential, and modulating pacemaker activity in the sinoatrial node.1-3 The L-type Ca2+ channel is a multiprotein complex, comprised of the pore-forming α1 subunit and the auxiliary β2 and α2δ subunits.4 In the heart, the molecular identity of the β subunit remains a subject of ongoing investigation.5,6
A number of receptor-mediated signal transduction pathways regulate Ca2+ influx via Cav1.2 channels, the most prominent of which is the β-adrenergic/cAMP signaling pathway. β-Adrenergic stimulation is the dominant mechanism of positive chronotropy, inotropy, and lusitropy in the heart.7 Activation of β-adrenergic receptors strongly enhances cardiac L-type Ca2+ current (ICaL) via cAMP generation and the activation of protein kinase A (PKA), presumably by phosphorylating specific sites on the channel protein.8,9 Nevertheless, the structural determinants of PKA action on the Ca2+ channel in native adult cardiomyocytes are unknown.
Previous biochemical studies have implicated the carboxyl terminus as a target of PKA-mediated phosphorylation: for example, a truncated form of the channel was reportedly not phosphorylated by PKA in vitro.10 The consensus phosphorylation site at serine 1928 was later identified as the main target of PKA.11,12 On the other hand, phosphorylation sites have also been mapped to the β subunit of the channel and localized to serines 478 and 479 of β2a.13,14
Mixed results have been obtained when attempts have been made to confirm the functional role of these sites in the regulation of Ca2+ channel activity, in part, because of the difficulty of reconstituting PKA-mediated channel activation in heterologous expression systems. Whereas some studies have observed PKA-mediated activation of the channel in heterologous systems,10,14 others have observed no such stimulation.15,16 Other groups have used PKA inhibitors to argue that Cav1.2 channels are phosphorylated at baseline when expressed in cultured cell lines.17-19
The failure to reproduce the strong upregulation of the L-type Ca2+ current in nonnative cells has been attributed to the lack of A-kinase anchoring proteins (AKAPs) in heterologous systems. By coexpressing AKAP 79, Gao et al20 were able to reconstitute forskolin-mediated upregulation of Ca2+ channels expressed in HEK 293 cells, and the authors concluded that S1928 was necessary to produce PKA-mediated activation of the L-type current. In contrast, a subsequent study by the same group reported PKA-mediated activation of Ca2+ current through L-type channels with a mutant α1C truncated at 1905 and provided evidence indicating that β-subunit phosphorylation was important.14 Additionally, other investigators have been unable to reproduce PKA-mediated upregulation of Ca2+ channels in HEK 293 cells, even in the presence of AKAP 79,21 and it has been suggested that the primary role of AKAP 79 is to facilitate α1 subunit trafficking.22
The equivocal findings in heterologous expression systems suggest that the actual site responsible for β-adrenergic regulation of L-type Ca2+ channel in cardiac myocytes remains undefined. In the present study, we use novel adenoviral gene transfer vectors to express mutant α1C subunits in native guinea pig cardiomyocytes to address this fundamental question of L-type Ca2+ channel physiology. The evidence supports a role for the distal carboxyl terminus of the α1 subunit in the β-adrenergic enhancement of ICaL. However, serine 1928 phosphorylation is not required.
Materials and Methods
Vector Construction
A full-length, dihydropyridine (DHP)-insensitive rabbit Cav1.2 (X15539) α1 subunit adenoviral shuttle vector pAdEcd-α1C(D-) was prepared as previously described.23 A serine to alanine substitution of α1C(D-) at S1928 (α1C(D-)-S1928A) was constructed by site-directed mutagenesis with overlapping primers, using the QuikChange XL kit (Stratagene). A carboxyl-terminal truncation mutant, pAdEcd-α1C(D-)Δ1905, in which the carboxyl terminus of the α1C was truncated after amino acid 1905, was also created. Hence, this construct also lacked the putative phosphorylation site at serine 1928. Mutations at Thr1066 (to Tyr) and Glu1089 (to Met) were also incorporated in this construct to make the channel DHP insensitive, as for α1C(D-) and α1C(D-)-S1928A, so that the current contributed by the mutated channels could be discriminated from that of native Cav1.2 channels.23 Concentration-response experiments with nitrendipine were performed on α1C(D-), α1C(D-)S1928A, and α1C(D-)Δ1905 in A549 cells as previously described23 to verify that each construct produced DHP-insensitive current. A full-length wild-type α1C fused to green fluorescent protein at its carboxyl terminus, α1C-GFP, was also constructed for the Western blot experiments shown in Figure 2.
Figure 2.
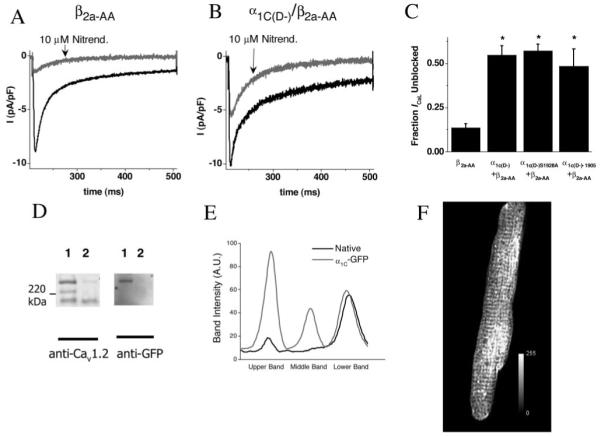
Overexpression of DHP-insensitive α1C enables pharmacological discrimination of native and exogenously expressed channels. Whole-cell current recordings were undertaken in adult myocytes transduced with α1C adenoviruses and β2a-AA adenoviruses. A repeated-pulsing protocol at 0.5 Hz to 0 mV was used to obtain a steady state, following which 10 μmol/L nitrendipine was added. Example current tracings are shown in myocytes transduced with β2a-AA (A) or α1C(D-)/β2a-AA (B) adenoviruses, before (black) and after (gray) the application of 10 μmol/L nitrendipine. In C, the fraction of ICaL left unblocked after the application of nitrendipine is shown following transduction with each of the DHP-insensitive α1C adenoviruses. D, Representative Western blot. Left, Lane 1 was transduced with α1C-GFP and AdVgRXR; lane 2, with AdVgRXR alone. Right, The same membrane was reprobed with anti-GFP antibody, confirming that the strong upper band was attributable to α1C-GFP expression. The absence of the middle band in lane 1 is indicative of carboxyl-terminal processing of α1C-GFP. In E, the anti-Cav1.2 signal intensity plot profile is shown for α1C-GFP and control cells, demonstrating the presence of novel peaks in upper and middle bands caused by α1C-GFP expression. F, Representative image of an α1C-GFP/AdVgRXR-transduced myocyte, stained with anti-GFP primary antibody, and an Alexa-Fluor 568 secondary antibody. *P<0.05 vs β2a-AA.
The adenoviral shuttle plasmid pAd-β2a, was created by subcloning the rat β2a cDNA (NM053851) into the EcoR1 site of the adenoviral shuttle vector pAdLox. To avoid potential confounding effects of β-subunit phosphorylation on the response, the S478A/S479A double mutation of β2a (pAd-β2a-S478/S479A; β2a-AA) was also created via site-directed mutagenesis with overlapping primers, using the QuikChange XL kit. pAd-VgRXR, a bicistronic vector containing the elements of the ecdysone receptor, was created as previously described.23,24
Adenoviral Construction
Adenoviruses were constructed by the Cre-lox technique, according to the methods of Hardy et al,25 with variations as previously described.23 The α1C adenovirus shuttle vectors pAdEcd-α1C(D-), pAdEcd-α1C(D-)S1928A, or pAdEcd-α1C(D-)Δ1905 were cotransfected onto CRE-FB cells, along with ψFB6 adenoviral DNA as previously described.23 Resulting viruses were expanded in 3 rounds and purified by plaque assay on 911-FB cells. The plaque purified virus was further expanded before purification on CsCl gradients, and dialyzed against 10 mmol/L Tris-HCl/1 mmol/L MgCl2. Viral titers were estimated by particle counts and calculated by plaque assay performed on 911-FB cells.
Ad-β2a-AA and AdVgRXR were grown by cotransfection of their respective shuttle plasmids with ψ5 adenoviral DNA,25 onto standard Cre-8 cells.25 These viruses were expanded in 3 rounds and purified on CsCl gradients, as described previously.23,26 Viral titers were estimated by particle counts and calculated by plaque assay performed on 911 cells.27
Adult Cardiac Myocyte Expression Experiments
Animal protocols used were in accordance with the Guide for Care and Use of Laboratory Animals, published by the US National Institutes of Health (NIH publication No. 85-23, revised 1996). Isolated guinea pig ventricular myocytes were obtained by enzymatic digestion as previously described.28 Freshly isolated myocytes (20 to 40×103/mL) were plated on laminin-coated coverslips and bathed in DMEM supplemented with 5% FBS and 1% penicillin-streptomycin. Virus was added after allowing cells to adhere to coverslips for 3 to 4 hours. For experiments involving overexpression of β2a-AA, cells were transduced with Ad-β2a-AA at ≈1×109 plaque-forming units (PFU).
For the DHP-insensitive α1C experiments, cells were transduced with Ad-VgRxR at 3×108 PFU/mL and Ad-β2a-AA at 1×109 PFU/mL, along with either Ad-α1C(D-), Ad-α1C(D-)S1928A or Ad-α1C(D-)Δ1905 at ≈5×109 PFU/mL. As we have described previously, coexpression of the α1C constructs with Ad-β2a-AA was necessary to enhance ICaL amplitude, which facilitates detection of the DHP-insensitive Ca2+ currents.23
After incubation with the viruses, the medium was replaced with fresh DMEM containing 10 μmol/L ponasterone A (H101-01, Invitrogen). Electrophysiological recordings were made at 36 to 48 hours following isolation.
Western Blot Experiments
The anti-Cav1.2 antibody used in these studies was an affinity purified rabbit polyclonal antibody, raised against a peptide made from amino acids 1 to 46 of the rabbit α1C subunit N terminus (Alomone Labs, Jerusalem, Israel). The anti-green fluorescence protein (GFP) antibody was a mouse monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, Calif). The lysate was prepared by scraping cultured cardiac myocytes into ice-cold RIPA buffer (1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate in PBS containing 5 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L β-glycerol phosphate, 1 mmol/L L-phenylalanine, 1 mmol/L sodium orthovanadate, 50 mmol/L sodium fluoride, and the following protease inhibitors: pepstatin A, 2 μg/mL; leupeptin, 10 μg/mL; aprotinin, 10 μg/mL; elastinal, 2 μg/mL; benzamidine, 0.5 mg/mL; calpain inhibitor peptide 10 μg/mL; and 1 mmol/L phenylmethane-sulfonyl fluoride, 0.4 mmol/L iodoacetic acid) and passed through a 25-gauge needle 15 times. This homogenate was lysed on ice for 1 hour, before centrifugation at 2000g for 20 minutes at 4°C.
The concentration of the supernatant protein was measured with the Bio-Rad DC protein assay, according to the instructions of the manufacturer, and samples were divided into aliquots and frozen at -20°C. Samples were mixed with 5:1 Laemmli sample buffer and incubated at room temperature for 60 minutes, before gel loading.
Following separation by 7.5% SDS-PAGE, proteins were transferred to polyvinylidene sulfonyl fluoride membranes (Bio-Rad) in Tris/glycine transfer buffer containing 5% methanol and 0.05% sodium dodecyl sulfate. Membranes were blocked with 5% nonfat milk in PBS supplemented with 0.05% Tween 20 (PBS-T), for 1 hour at room temperature, and then incubated with primary antibody (1:50 to 1:200) overnight at 4°C. The membrane was washed 6 times for 5 minutes with PBS-T and incubated with horseradish peroxidase-conjugated donkey anti-rabbit antibody diluted 1:50 000 in PBS-T. Following 6 further washes in PBS-T, blots were visualized with the Lumi-light Plus blotting substrate (Roche), according to the instructions of the manufacturer.
Immunofluorescence
Immunofluorescence labeling experiments were undertaken using myocytes cultured with α1C-GFP using an anti-GFP antibody (Abcam-6556). Cultured myocytes were fixed for 20 minutes at room temperature in freshly prepared 3% paraformaldehyde in PBS. Fixed cells were permeabilized in 0.5% Triton X-100 in PBS/10 mmol/L glycine for 3 minutes. After 2 further washes in PBS/10 mmol/L glycine, myocytes were incubated for 1 hour in PBS/10% goat serum to block nonspecific binding. Following 2 washes in PBS/10 mmol/L glycine, the cells were incubated in primary antibody diluted 1:500 in PBS/10 mmol/L glycine supplemented with 4% goat serum, for 1 hour. The cells were then washed 2 times in PBS/10 mmol/L glycine, before a 1-hour incubation with secondary antibody, Alexa 568 goat anti-rabbit IgG (H+L) (Invitrogen), and diluted 1:500 in PBS/10 mmol/L glycine supplemented with 4% goat serum. After 2 final washes with PBS/10 mmol/L glycine, cells were mounted on coverslips. Experiments to determine nonspecific binding were performed with cells transduced with VgRXR alone, and control experiments were also carried out with secondary antibody alone.
Images were recorded using a 2-photon laser scanning microscope (Bio-Rad, MRC-1024MP) with excitation at 820 nm (Tsunami Ti:Sa laser, Spectra-Physics). The red emission of Alexa 568 was collected at 605±25 nm.
Electrophysiological Recordings
Electrophysiological recordings were performed using the whole-cell variation of the patch clamp technique. All experiments were performed at room temperature. Currents were recorded with an Axopatch 200B amplifier (Axon Instruments, Union City, Calif) and sampled at 5 kHz. The cells were superfused with an external solution comprised of (mmol/L): 140 NaCl, 5 CsCl, 1 MgCl2, 10 HEPES, 2 CaCl2, and 10 d-glucose. Patch pipettes (2.0 to 5.0 MΩ) were filled with an intracellular solution containing (mmol/L): 110 CsCl, 0.4 MgCl2, 5 d-glucose, 10 HEPES, 20 tetraethylammonium, and 5 BAPTA (1,2-bis[2-aminophenoxy]ethane-N,N,N’,N’-tetraacetic acid). In experiments involving the use of DHP-insensitive α1 subunits, 10 μmol/L nitrendipine (Sigma, St Louis, Mo) was washed in at the end of a subset of experiments to verify the expression of the exogenous α1C. Voltage pulses were applied and data recorded with custom software. Leak currents were subtracted by a P/4 leak subtraction protocol. Total series resistance after establishing the whole-cell configuration was typically 10 MΩ and was electronically compensated by ≈70%. Thus we estimate the voltage error for a 1-nA current (the amplitude of isoproterenol [ISO]-stimulated peak inward ICaL) to be <3 mV. ICaL was elicited by 300-ms pulses to different test potentials (-80 to +50mV), following a 200-ms prepulse to -40 mV, from a holding potential of -80 mV. ICaL measurements were normalized to cell capacitance, measured by integration of the area under an uncompensated -20 mV pulse from -80 mV. The current-density to voltage relationships (I-V) recorded were fitted to a modified Boltzmann equation:
where Gmax is the maximal conductance (nS/pF), Vrev is the reversal potential (mV), V1/2 is the midpoint voltage of activation (mV), and k is the slope factor.
Data analysis was performed offline, with custom software (Ionview, B.O’R.). Statistical analysis was performed with Origin 6.0 (Microcal). All data are presented as the mean±SEM. Statistical significance was calculated using an unpaired Student’s t test, and differences were considered significant at P<0.05.
Results
β-Adrenergic Response in Adult Cardiomyocytes With β2a-AA Expression
The primary objective of this study was to investigate the role of the distal carboxyl terminus of α1C in the β-adrenergic response. In a prior study, we determined that replacement of the native Ca2+ channels with virally expressed mutants was most effectively achieved when β subunits were coexpressed,23 consistent with earlier reports that β-subunit expression is a limiting factor for Ca2+ channel membrane trafficking in myocytes.5 However, our initial experiments indicated that the expression of wild-type β2a subunits led to a marked hyperpolarizing shift (approximately -12.2 mV) in the midpoint of voltage-dependence for ICaL activation (V1/2) and a diminished ISO response, presumably attributable to the enhanced Ca2+ channel activity under basal conditions (data not shown). Mutation of the phosphorylation sites of β2a at S478 and S479 to alanine lessened the hyperpolarizing shift in Ca2+ channel activation, but preserved the ability of the β subunit to facilitate Ca2+ channel trafficking. Expression of β2a-AA subunits resulted in a marked increase in baseline L-type Ca2+ current density (uninfected myocytes -3.2±0.7 pA/pF versus β2a-AA expressing -9.2±1.6 pA/pF; P<0.05) and a hyperpolarizing shift in V1/2 of just -4.8 mV (Table). Importantly, a robust β-adrenergic response could be observed under these conditions. In Ad-β2a-AA-transduced myocytes, ISO (100 nmol/L) caused a 95% increase in peak ICaL (from -9.2±1.6 pA/pF to -17.9±1.4 pA/pF, P<0.05; Figure 1B and 1D) and a significant hyperpolarizing shift in V1/2 (Table). This was comparable to the ISO-induced increase in peak inward L-type Ca2+ currents in uninfected cultured myocytes (Figure 1A and 1C; ICaL baseline: -3.2±0.3 pA/pF versus 100 nmol/L ISO: -11.8±1.7 pA/pF). Thus, β2a-AA coexpression was used for the subsequent experiments with α1C mutants.
Voltage Dependence of Activation (V1/2) of L-Type Ca2+ Channels in Cardiac Myocytes.
| V1/2 | V1/2 (100 nmol/L ISO) | |
|---|---|---|
| Control | -7.9±0.1 | -20.4±2.2* |
| β2a-AA | -12.70±2.4 | -24.20±2.6† |
| α1C(D-)/β2a-AA | -16.95±1.9 | -25.21±1.9‡ |
| α1C(D-)1905/β2a-AA | -14.52±2.4 | -19.60±2.4§ |
| α1C(D-)S1928A/β2a-AA | -13.5±3.3 | -23.1±1.4¶ |
P<0.05 vs control
P<0.05 vs β2a-AA
P<0.05 vs α1C(D-)/β2a-AA
P=NS vs α1C(D-)Δ1905/β2a-AA
P<0.05 vs α1C(D-)S1928A/β2a-AA
Figure 1.
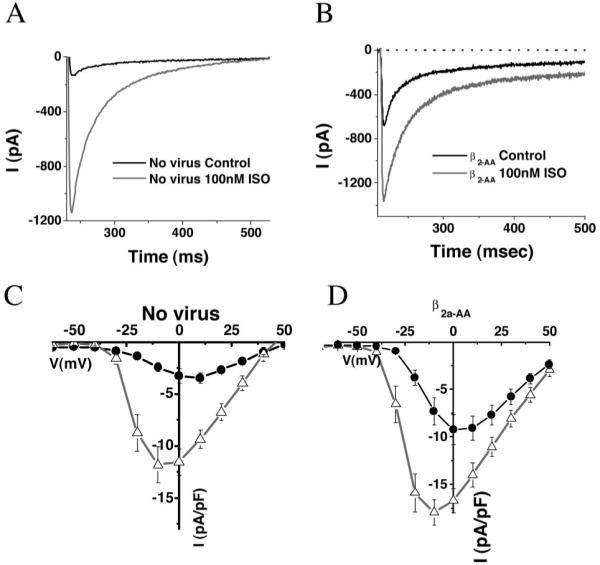
Isoproterenol response in adult myocytes transduced with adenoviruses overexpressing β2a-AA subunits. Whole-cell currents were recorded using voltage clamp pulses from a holding potential of -80 mV to test potentials, following a 200 ms prepulse to -40 mV, before and after the application of 100 nmol/L ISO. A and B, Example current tracings following pulses to 0 mV, before and after 100 nmol/L ISO. C and D, Current-voltage relationships: before (black) and after (gray) the application of 100 nmol/L ISO in uninfected myocytes (C) and myocytes transduced with β2a-AA (D). β2a-AA transduction caused a baseline increase in ICaL and a small hyperpolarizing shift in the voltage dependence of activation; however, the β-adrenergic response was preserved. Values are mean±SEM.
Functional Expression of DHP-Insensitive α1C Subunits in Cardiac Myocytes
Following a strategy similar to our previous study,23 α1C mutants were engineered to be dihydropyridine insensitive, to enable pharmacological discrimination of L-type Ca2+ current contributed by exogenously expressed channels from that of native Ca2+ channels. Consistent with our earlier results, a concentration of 10 μmol/L nitrendipine blocked more than 85% of ICaL in myocytes transduced with Ad-β2a-AA alone, whereas only ≈50% of the total ICaL was blocked in α1C(D-)+β2a-AA-transduced cells (Figure 2). Similar fractions of DHP-insensitive current were observed for myocytes expressing β2a-AA with either α1C(D-)S1928A or α1C(D-)Δ1905 (Figure 2C).
Significant expression of mutant α1C subunits was also confirmed in biochemical studies. A full-length α1C fused to green fluorescent protein at the carboxyl terminus was used to discriminate exogenous and native α1C subunits by molecular weight on Western blots and to provide a specific tag for the heterologous channels. Two intense high molecular mass bands appeared (>220 kDa) in samples from myocytes infected with Ad-α1C-GFP (Figure 2) using an anti-Cav1.2 antibody, in addition to the lowermost band representing native α1C. Densitometry confirmed that the signal from the uppermost bands in α1C-GFP cells represented 63±6% of total anti-Cav1.2 signal intensity (Figure 2). When reprobed with an anti-GFP antibody, only the uppermost band was evident, verifying the presence of the fusion protein and suggesting that the middle band was a carboxyl-terminal cleavage product of the Ca2+ channel.
Ad-α1C-GFP was next used to examine the spatial localization of the expressed mutant channels. Immunofluorescence staining of fixed cardiac myocytes with an anti-GFP antibody conjugated to the fluorescent dye Alexa 568 revealed a membrane distribution of expressed α1C subunits including strong transverse-bands consistent with t-tubular localization, in accord with the known distribution of α1C subunits in myocytes. Hence, expression of mutant α1C subunits was confirmed both functionally and biochemically.
Distal Carboxyl Terminus of α1C Is Necessary for β-Adrenergic Regulation of Ca2+ Channels
To ensure that the observed responses could be exclusively attributed to the expressed α1C mutants, we first examined the effects of β-adrenergic stimulation on ICaL in the presence of 10 μmol/L nitrendipine. Under these conditions, 100 nmol/L ISO induced a 50±11% increase of peak ICaL in cells expressing α1C(D-)/β2a-AA (Figure 3A and 3D). Remarkably, the ISO response was almost completely eliminated (ICaL increased by only 3±7%) when the α1C carboxyl terminus was truncated at amino acid residue 1905 (α1C(D-)Δ1905/β2a-AA; Figure 3B and 3D). To determine whether the loss of responsiveness involved impairment of the pathway between the β-adrenergic receptor and PKA, we also repeated the experiments using the membrane permeable cAMP analogue 8-bromo-adenosine 3′5′-cyclic monophosphate (8-Br-cAMP) to directly activate the kinase. Similar results were obtained, with a substantial reduction in 8-Br-cAMP induced Ca2+ current augmentation with α1C(D-)Δ1905 compared with α1C(D-) (Figure 3E).
Figure 3.
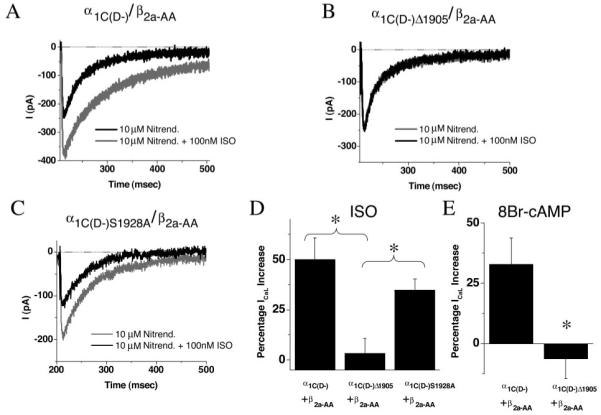
In the presence of nitrendipine, carboxyl-terminal truncation mutant shows diminished response to ISO, but S1928A remains responsive. Whole-cell current recordings were obtained in adult myocytes before and after 100 nmol/L ISO, for myocytes transduced with α1C(D-) (A), α1C(D-)Δ1905 (B), or α1C(D-)S1928A (C) viruses. Current records are for repeated pulses to 0 mV in the continuous presence of 10 μmol/L nitrendipine, to block ICaL carried by native channels. The fractional response to 100 nmol/L ISO is shown in D. ISO (100 nmol/L) elicited a robust increase in ICaL in α1C(D-) cells in the presence of the blocking dose of nitrendipine (A and D) but failed to elicit ICaL upregulation in α1C(D-)Δ1905 cells (B and D), indicating that the carboxyl terminus of α1C is necessary for β-adrenergic modulation of Cav1.2 channels in cardiac myocytes. ISO also elicited a robust increase in ICaL in α1C(D-)S1928A cells, indicating that S1928 phosphorylation is not essential for β-adrenergic modulation of L-type Ca2+ current. E, 8-Br-cAMP (1 mmol/L) elicited an increase in ICaL in α1C(D-) cells in the presence of 10 μmol/L nitrendipine but failed to elicit ICaL upregulation in α1C(D-)Δ1905 cells. Values are mean±SEM. *P<0.05, **P<0.01.
The ISO response was also examined in the absence of nitrendipine, to eliminate any possibility that the results were somehow dependent on the presence of concomitant Ca2+ channel block. ISO increased peak ICaL by 104±23% in myocytes expressing α1C(D-) and by only 25±16% in myocytes expressing α1C(D-)Δ1905. The small increase in the latter case is consistent with the presence of a small fraction of native L-type channels that would be available to be activated by ISO. In addition to the significant effect of the 1905 truncation to blunt the ISO effect on peak ICaL, it is notable that the hyperpolarizing shift in V½ was also markedly reduced in myocytes expressing α1C(D-)Δ1905 (Table).
S1928 Is Not Required for β-Adrenergic Regulation of Ca2+ Channels
In contrast to the effect of truncating the carboxyl terminus, the S1928A point mutation of the α1C did not significantly diminish the ISO response, either in the presence or in the absence of nitrendipine (Figures 3 and 4). In myocytes expressing α1C(D-)S1928A, ISO increased ICaL by 82±16% in the absence of nitrendipine (Figure 4) and a prominent leftward shift in V1/2 was still observed (Table).
Figure 4.
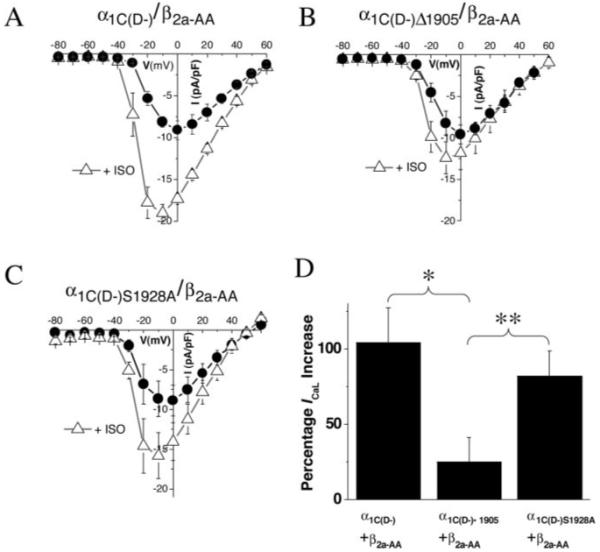
Carboxyl-terminal truncation mutant shows diminished response to ISO, but S1928A remains responsive. To ensure that the results were not a consequence of the presence of nitrendipine, whole-cell current recordings were obtained from myocytes before and after 100 nmol/L ISO in the absence of DHP. Current-voltage relationships before (black circles) and after (open triangles) 100 nmol/L ISO are plotted for myocytes transduced with α1C (D-)/β2a-AA (A), α1C(D-)Δ1905/β2a-AA (B), or α1C(D-)S1928A/β2a-AA (C) adenoviruses. The β-adrenergic response of ICaL was markedly attenuated in α1C(D-)Δ1905/β2a-AA cells (B and D), as it was in the presence of nitrendipine (Figure 3). However, the β-adrenergic enhancement of ICaL was preserved in α1C(D-)S1928A/β2a myocytes (C and D). The fractional increase in response to 100 nmol/L ISO of the peak ICaL is shown in D. Values are mean±SEM. *P<0.05, **P<0.025.
Discussion
The 2 main findings of the study were (1) that the distal carboxyl terminus of α1C is required for the β-adrenergic stimulation of L-type Ca2+ channels in cardiac cells and (2) that serine 1928 phosphorylation is not required. This was most evident in the failure of Ca2+ currents to be enhanced by ISO in α1C(D-)Δ1905/β2a-AA-transduced cells in the presence of nitrendipine. The β-adrenergic response was also significantly blunted in α1C(D-)Δ1905/β2a-AA-transduced cells in the absence of nitrendipine, but the presence of ISO-responsive native L-type Ca2+ channels made the distinction less prominent. The ISO response was not significantly diminished in myocytes expressing the S1928A α1C point mutant.
The present study is the first to investigate the structural determinants of the α1C subunit involved in the β-adrenergic regulation of L-type Ca2+ channels in native adult myocytes. This was enabled by the efficient expression of CaV1.2 mutants using novel adenoviral constructs. Discrimination between the heterologous channels and native channels was accomplished by engineering the former to be DHP-insensitive. Both functional and biochemical data indicated that the majority of the L-type Ca2+ channels expressed were derived from the viral construct, so that significant differences in function could be readily observed, even in the absence of the dihydropyridine.
An important technical issue was how to optimize Ca2+ channel expression using β-subunit coexpression without introducing confounding effects caused by alterations in baseline Ca2+ channel activity by the β subunit. We found that mutation of the consensus phosphorylation sites of β2a prevented the pronounced hyperpolarization of the voltage dependence of L-type Ca2+ channel activation under basal conditions, permitting us to observe a robust ISO response. Beyond the technical advantage of this approach, the results with β2a-AA indicate that β subunit phosphorylation is not obligatory for the β-adrenergic response, a conclusion that differs from previous studies of Ca2+ channel regulation for channels heterologously expressed in cultured cell lines.14
The present results also provide evidence against the current dogma that PKA-mediated Ser1928 phosphorylation is required for the β-adrenergic stimulation of ICaL. Both the ISO-induced increase in peak inward ICaL and the left shift in the V1/2 of activation were preserved in cardiomyocytes expressing α1C(D-)S1928A. In this regard, the functional data supporting S1928 as the site of modulation have been equivocal. Gao et al20 reported that Ba2+ currents through expressed Ca2+ channels were upregulated by forskolin or 8-Br-cAMP in an AKAP 79 stably transfected HEK 293 cell line and that this response was absent for the S1928A α1C point mutation. However, only half of the control cells tested were clearly responsive to the effects of the PKA activators, suggesting a high degree of variability in this model system. In addition, forskolin increased Ba2+ current by only ≈50%, and 8-Br-cAMP by ≈15%, much less than that typically observed for PKA-mediated ICaL enhancement observed in cardiac myocytes. Moreover, a later study from the same laboratory14 reported that ICaL mediated by the 1905 α1C truncation mutant, lacking serine 1928, could be increased by PKA activation.
Other reports have also raised cautions about the conclusions reached in the earlier studies. Many investigators have failed to reproduce PKA modulation of L-type Ca2+ currents in heterologous expression systems. Zong et al16 tested 25 μmol/L PKA along with okadaic acid in CHO and HEK 293 cells expressing either α1C-a or α1C-b and were not able to observe PKA-mediated potentiation of Ba2+ currents. Mikala et al15 systematically substituted 5 potential PKA phosphorylation sites in human α1C, including the S1928 homolog, and found that modulation of baseline L-type Ca2+ current behavior could not account for the failure to observe PKA modulation. Altier et al22 also failed to observe PKA-mediated activation of Ba2+ currents in HEK 293 cells, even in the presence of AKAP 79 overexpression.
Although the functional effects of S1928 may be questionable, there is agreement that S1928 can be readily phosphorylated by PKA in vitro.11,12 However, according to a recent report, this site appears to be promiscuously phosphorylated by at least 3 isoforms of PKC (cPKCα, nPKCε, and aPKCζ) in addition to PKA.29 This raises an important question about the physiological relevance of this in vitro biochemical evidence, considering that the considerably divergent physiological effects of PKA and PKC activation on channel activity.8
The present data suggest that although S1928 is not critical for the β-adrenergic effect on Ca2+ channel function, the distal α1C carboxyl terminus beyond residue 1905 is required. One plausible explanation for this effect could be that the carboxyl terminus plays a central role in the assembly of a PKA signaling complex. For example, Hulme et al30 recently demonstrated an interaction between AKAP 15 and a leucine zipper motif in the distal carboxyl terminus of α1C using immunoprecipitation methods. Disruption of the interaction with competing AKAP 15 leucine zipper peptides substantially reduced ISO-induced upregulation of L-type Ca2+ currents in cardiac myocytes, suggesting that the AKAP 15-α1C interaction was necessary for efficient β-adrenergic regulation of Ca2+ currents in cardiac myocytes.30 Further investigation into the role of the leucine zipper motif in the response using the present strategy will be important.
The existing literature provides a number of potential alternative sites of PKA-mediated phosphorylation of α1C. For example, Leach et al,31 using phosphopeptide antibodies, reported that serines 1627 and 1700 were the primary sites of PKA-mediated phosphorylation of the cardiac Ca2+ channel α1C subunit, whereas no phosphopeptide products could be detected for sites S1575, S1848, or S1928 on the carboxyl terminus. Other sites at S182932and S114233 also have been proposed as possible sites of regulation. The present work reopens the issue and provides a new way to test potential sites.
There is also the possibility that phosphorylation of other Ca2+ channel-associated proteins could modify channel function. However, in contrast to previous reports,14 the present findings do not point toward the β subunit as the functionally important site of regulation in adult cardiac cells.
The results have demonstrated the potential for structure/function studies of the α1C subunit in native cells to complement investigations in heterologous systems. However, the potential limitations of this strategy need to be acknowledged. In this case, overexpressed β2 subunits may form complexes with native α1C subunits, or, conversely, overexpressed α1C may complex with native β subunits. However, although the possibilities for such channel subpopulations pose challenges for the interpretation of studies of molecular regulation, the experimental evidence in our study suggests that the overexpressed subunits dominated the phenotype of L-type Ca2+ currents. In the case of the β2 subunit, this was apparent with the changes in current density and the voltage dependence of activation. With α1C-subunit overexpression, this was evident with changes in DHP-sensitivity and the β-adrenergic response.
Conclusions
Our findings demonstrate the distal carboxyl terminus beyond amino acid 1905 of the α1C subunit is required for β-adrenergic enhancement of L-type Ca2+ currents in adult cardiomyocytes. However, our results do not support a functional role for phosphorylation of S1928. The present study also demonstrates that structure/function studies of the α1C subunit of the L-type Ca2+ channel are possible using adenoviral gene transfer methods.
Acknowledgments
This study was supported by NIH grant R01HL61711 (to B.O’R.), an American Heart Association Predoctoral Fellowship (0315387U, to A.N.G.), Deutsche Forschungsgemeinschaft MA 2528/1.1 (to C.M.), and NIH grant RO1-NS044541 (to D.C.J.). We thank Dr Henry Colecraft for useful discussions.
Footnotes
Under a licensing agreement between Excigen Inc and the Johns Hopkins University, D.C.J. is entitled to a share of royalty and milestone payments received by the University on sales of products described in this article. The terms of this agreement are being managed by the Johns Hopkins University in accordance with its conflict-of-interest policies.
This manuscript was sent to Harry A. Fozzard, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
References
- 1.Reuter H. Properties of two inward membrane currents in the heart. Annu Rev Physiol. 1979;41:413–424. doi: 10.1146/annurev.ph.41.030179.002213. [DOI] [PubMed] [Google Scholar]
- 2.Reuter H. The dependence of slow inward current in Purkinje fibres on the extracellular calcium-concentration. J Physiol. 1967;192:479–492. doi: 10.1113/jphysiol.1967.sp008310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 4.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 5.Colecraft HM, Alseikhan B, Takahashi SX, Chaudhuri D, Mittman S, Yegnasubramanian V, Alvania RS, Johns DC, Marban E, Yue DT. Novel functional properties of Ca(2+) channel beta subunits revealed by their expression in adult rat heart cells. J Physiol. 2002;541:435–452. doi: 10.1113/jphysiol.2002.018515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foell JD, Balijepalli RC, Delisle BP, Yunker AM, Robia SL, Walker JW, McEnery MW, January CT, Kamp TJ. Molecular heterogeneity of calcium channel beta-subunits in canine and human heart: evidence for differential subcellular localization. Physiol Genomics. 2004;17:183–200. doi: 10.1152/physiolgenomics.00207.2003. [DOI] [PubMed] [Google Scholar]
- 7.Xiao RP. Beta-adrenergic signaling in the heart: dual coupling of the beta2- adrenergic receptor to G(s) and G(i) proteins. Sci STKE. 2001;2001:RE15. doi: 10.1126/stke.2001.104.re15. [DOI] [PubMed] [Google Scholar]
- 8.Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res. 2000;87:1095–1102. doi: 10.1161/01.res.87.12.1095. [DOI] [PubMed] [Google Scholar]
- 9.Keef KD, Hume JR, Zhong J. Regulation of cardiac and smooth muscle Ca(2+) channels (Ca(V)1.2a,b) by protein kinases. Am J Physiol Cell Physiol. 2001;281:C1743–C1756. doi: 10.1152/ajpcell.2001.281.6.C1743. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida A, Takahashi M, Nishimura S, Takeshima H, Kokubun S. Cyclic AMP-dependent phosphorylation and regulation of the cardiac dihydropyridine sensitive Ca channel. FEBS Lett. 1992;309:343–349. doi: 10.1016/0014-5793(92)80804-p. [DOI] [PubMed] [Google Scholar]
- 11.Mitterdorfer J, Froschmayr M, Grabner M, Moebius FF, Glossmann H, Striessnig J. Identification of PK-A phosphorylation sites in the carboxyl terminus of L-type calcium channel alpha 1 subunits. Biochemistry. 1996;35:9400–9406. doi: 10.1021/bi960683o. [DOI] [PubMed] [Google Scholar]
- 12.De Jongh KS, Murphy BJ, Colvin AA, Hell JW, Takahashi M, Catterall WA. Specific phosphorylation of a site in the full-length form of the alpha 1 subunit of the cardiac L-type calcium channel by adenosine 3′,5′-cyclic monophosphate-dependent protein kinase. Biochemistry. 1996;35:10392–10402. doi: 10.1021/bi953023c. [DOI] [PubMed] [Google Scholar]
- 13.Haase H, Bartel S, Karczewski P, Morano I, Krause EG. In-vivo phosphorylation of the cardiac L-type calcium channel beta-subunit in response to catecholamines. Mol Cell Biochem. 1996;163-164:99–106. doi: 10.1007/BF00408645. [DOI] [PubMed] [Google Scholar]
- 14.Bunemann M, Gerhardstein BL, Gao T, Hosey MM. Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the beta(2) subunit. J Biol Chem. 1999;274:33851–33854. doi: 10.1074/jbc.274.48.33851. [DOI] [PubMed] [Google Scholar]
- 15.Mikala G, Klockner U, Varadi M, Eisfeld J, Schwartz A, Varadi G. cAMP-dependent phosphorylation sites and macroscopic activity of recombinant cardiac L-type calcium channels. Mol Cell Biochem. 1998;185:95–109. doi: 10.1023/a:1006878106672. [DOI] [PubMed] [Google Scholar]
- 16.Zong X, Schreieck J, Mehrke G, Welling A, Schuster A, Bosse E, Flockerzi V, Hofmann F. On the regulation of the expressed L-type calcium channel by cAMP-dependent phosphorylation. Pflugers Arch. 1995;430:340–347. doi: 10.1007/BF00373908. [DOI] [PubMed] [Google Scholar]
- 17.Perets T, Blumenstein Y, Shistik E, Lotan I, Dascal N. A potential site of functional modulation by protein kinase A in the cardiac Ca2+ channel alpha 1C subunit. FEBS Lett. 1996;384:189–192. doi: 10.1016/0014-5793(96)00303-1. [DOI] [PubMed] [Google Scholar]
- 18.Singer-Lahat D, Lotan I, Biel M, Flockerzi V, Hofmann F, Dascal N. Cardiac calcium channels expressed in Xenopus oocytes are modulated by dephosphorylation but not by cAMP-dependent phosphorylation. Receptors Channels. 1994;2:215–226. [PubMed] [Google Scholar]
- 19.Perez-Reyes E, Yuan W, Wei X, Bers DM. Regulation of the cloned L-type cardiac calcium channel by cyclic-AMP-dependent protein kinase. FEBS Lett. 1994;342:119–123. doi: 10.1016/0014-5793(94)80484-2. [DOI] [PubMed] [Google Scholar]
- 20.Gao T, Yatani A, Dell’Acqua ML, Sako H, Green SA, Dascal N, Scott JD, Hosey MM. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. 1997;19:185–196. doi: 10.1016/s0896-6273(00)80358-x. [DOI] [PubMed] [Google Scholar]
- 21.Dai S, Klugbauer N, Zong X, Seisenberger C, Hofmann F. The role of subunit composition on prepulse facilitation of the cardiac L-type calcium channel. FEBS Lett. 1999;442:70–74. doi: 10.1016/s0014-5793(98)01632-9. [DOI] [PubMed] [Google Scholar]
- 22.Altier C, Dubel SJ, Barrere C, Jarvis SE, Stotz SC, Spaetgens RL, Scott JD, Cornet V, De Waard M, Zamponi GW, Nargeot J, Bourinet E. Trafficking of L-type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J Biol Chem. 2002;277:33598–33603. doi: 10.1074/jbc.M202476200. [DOI] [PubMed] [Google Scholar]
- 23.Ganesan AN, O’Rourke B, Maack C, Colecraft H, Sidor A, Johns DC. Reverse engineering the L-type Ca(2+) channel alpha(1c) subunit in adult cardiac myocytes using novel adenoviral vectors. Biochem Biophys Res Commun. 2005;329:749–754. doi: 10.1016/j.bbrc.2005.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoppe UC, Marban E, Johns DC. Adenovirus-mediated inducible gene expression in vivo by a hybrid ecdysone receptor. Mol Ther. 2000;1:159–164. doi: 10.1006/mthe.1999.0023. [DOI] [PubMed] [Google Scholar]
- 25.Hardy S, Kitamura M, Harris-Stansil T, Dai Y, Phipps ML. Construction of adenovirus vectors through Cre-lox recombination. J Virol. 1997;71:1842–1849. doi: 10.1128/jvi.71.3.1842-1849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maack C, Ganesan A, Sidor A, O’Rourke B. Cardiac sodium-calcium exchanger is regulated by allosteric calcium and exchanger inhibitory peptide at distinct sites. Circ Res. 2005;96:91–99. doi: 10.1161/01.RES.0000151334.48676.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fallaux FJ, Kranenburg O, Cramer SJ, Houweling A, Van Ormondt H, Hoeben RC, Van Der Eb AJ. Characterization of 911: a new helper cell line for the titration and propagation of early region 1-deleted adenoviral vectors. Hum Gene Ther. 1996;7:215–222. doi: 10.1089/hum.1996.7.2-215. [DOI] [PubMed] [Google Scholar]
- 28.O’Rourke B, Ramza BM, Marban E. Oscillations of membrane current and excitability driven by metabolic oscillations in heart cells. Science. 1994;265:962–966. doi: 10.1126/science.8052856. [DOI] [PubMed] [Google Scholar]
- 29.Yang L, Liu G, Zakharov SI, Morrow JP, Rybin VO, Steinberg SF, Marx SO. Ser1928 is a common site for Cav1.2 phosphorylation by protein kinase C isoforms. J Biol Chem. 2005;280:207–214. doi: 10.1074/jbc.M410509200. [DOI] [PubMed] [Google Scholar]
- 30.Hulme JT, Lin TW, Westenbroek RE, Scheuer T, Catterall WA. Beta-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc Natl Acad Sci U S A. 2003;100:13093–13098. doi: 10.1073/pnas.2135335100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leach RN, Brickley K, Norman RI. Cyclic AMP-dependent protein kinase phosphorylates residues in the C-terminal domain of the cardiac L-type calcium channel alpha1 subunit. Biochim Biophys Acta. 1996;1281:205–212. doi: 10.1016/0005-2736(96)00013-2. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi E, Fukuda K, Miyoshi S, Murata M, Kato T, Ita M, Tanabe T, Ogawa S. Leukemia inhibitory factor activates cardiac L-Type Ca2+ channels via phosphorylation of serine 1829 in the rabbit Cav1.2 subunit. Circ Res. 2004;94:1242–1248. doi: 10.1161/01.RES.0000126405.38858.BC. [DOI] [PubMed] [Google Scholar]
- 33.Erxleben C, Gomez-Alegria C, Darden T, Mori Y, Birnbaumer L, Armstrong DL. Modulation of cardiac Ca(V)1.2 channels by dihydropyridine and phosphatase inhibitor requires Ser-1142 in the domain III pore loop. Proc Natl Acad Sci U S A. 2003;100:2929–2934. doi: 10.1073/pnas.2628046100. [DOI] [PMC free article] [PubMed] [Google Scholar]