

Abstract
A role for inflammation has been hypothesized in the etiology and progression of Parkinson’s disease (PD). In this study, we generated, characterized, and validated the first progressive PD-related mice model (C57/B6)-intrastriatal injection of lipopolysaccharide (LPS). We showed progressive and specific dopaminergic neurodegeneration in the substantia nigra, which is accompanied by striatal dopamine depletion and progressive behavioral impairment, which was alleviated by the use of the PD drug L-Dopa. We focused on the role of NO in inflammation-promoted cell death and suggest that the expression of the inducible nitric oxide synthase plays a role in the progressive loss of dopaminergic neurons but not the initial loss induced by LPS. With this model future research can be done in gene knockout mice to study other potential mechanisms of inflammation-induced neurodegeneration. In addition, this model can be used to screen therapeutics for PD at a more clinically relevant time (i.e., after LPS injection but before manifestation of PD-related behavioral impairment), as most PD drugs are screened in animal models where inhibitors are given pre-disease induction. Thus, this novel PD-related model should be further characterized and strongly considered as a tool for future drug studies.
Keywords: Inflammation, Neurodegeneration, Oxidative Stress, Nitric Oxide
Introduction
Parkinson’s disease (PD) is characterized by progressive loss of dopaminergic neurons in the substantia nigra (SN), striatal dopamine depletion (Hornykiewicz 1993), and motor impairments (Youdim and Riederer 1997). Evidence suggests that chronic inflammation (McGeer et al. 2001), oxidative stress (Beal 2003), and mitochondrial impairment (Parker et al. 1989) play important roles in PD neurodegeneration. It has been proposed that inflammation may be a driving factor in the progression of PD, especially since PD patients have increased proinflammatory molecules in their Cerebrospinal fluid and brain tissue (McRae-Degueurce et al. 1988; Mogi et al. 1994), and activated microglia are found clustered throughout the SN (McGeer et al. 1988).
Other supportive evidence for the role of chronic inflammation in PD includes a high correlation of PD occurring in post-encephalitis patients (Berger J.R. 2003) and a case report of Parkinsonism induced by accidental exposure to lipopolysaccharide (LPS) (Niehaus 2004). In addition, anti-inflammatory drugs have been shown to attenuate several of the toxin-induced PD models (Gao et al. 2002; He et al. 2001; Vijitruth et al. 2006).
In order to focus on the potential role of inflammation in PD, we and others have created several different LPS-induced parkinsonian models via intranigral (Arimoto and Bing 2003; Castano et al. 1998; Herrera et al. 2000), intrauterine (Ling et al. 2002), intrapallidal (Zhang et al. 2005), intrastriatal (Hunter et al. 2007b), and systemic LPS injection (Qin et al. 2007).
In these models, activated microglia mediate inflammation and induce what seems to be specific nigrostriatal dopaminergic neurodegeneration. In the intrastriatal model LPS induces a chronic inflammatory response that increases oxidative stress and impairs mitochondrial function prior to dopaminergic neurodegeneration (Hunter et al. 2007b). We propose that specific neurodegeneration occurs because dopaminergic neurons are under oxidative stress from dopamine metabolism (Jenner and Olanow 1998) and the SN has the highest concentration of microglia in the mouse brain (Lawson et al. 1990). In addition, the SN has one of the highest iron concentrations in the brain, and iron is a necessary substrate for the free radical chemistry reactions implicated in oxidative stress. Increased iron is found in the SN of PD patients and our lab has shown that intrastriatal LPS increases SN iron content (Hunter et al. 2008). Thus, the oxidative stress generated by chronic inflammation and mitochondrial impairment may exceed the available compensatory mechanisms of dopaminergic neurons, resulting in neuronal death.
In our rat study, anti-inflammatory drugs provide partial neuroprotection against intrastriatal LPS injection via attenuating inflammation, oxidative stress, and impaired mitochondria function (Hunter et al. 2007b). These novel findings suggest that intrastriatal LPS injection is a promising model because it is well suited to reveal the role of inflammation in PD-related neurodegeneration. We translated the intrastriatal LPS model into mice for further characterization and to provide a novel tool to study the precise mechanisms by which gene-mediated dosing can be used to determine the effects of various genes. In this study we focused on the role for inducible nitric oxide synthase (iNOS) derived NO in inflammation-promoted cell death, as overproduction of NO as a result of increased iNOS expression in LPS-activated microglia (Hunot et al. 1996; Knott et al. 2000) has been hypothesized to be detrimental to dopaminergic neurons via activation of more microglia and inhibition of mitochondrial respiration (Heales et al. 1999).
Materials and Methods
Animals
Three-month-old male C57/B6 mice were obtained from Harlan (Indianapolis, IN) as well as iNOS knockout (NOS2 tm1Lau/J) mice and strain control wild-type mice from Jackson Labs (Bar Harbor, ME). Mice were housed under a 12-h light–dark cycle with free access to food and water in the Division of Lab Animal Resources at the University of Kentucky. Experimental protocols involving the animals were in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of Kentucky.
LPS or Saline Injections and Drug Treatments
Mice were randomly selected and grouped for a study, to determine an effective dose of Salmonella minnesota LPS (Sigma-Aldrich, St Louis, MO) injection into the striatum, which would induce significant dopaminergic neuron loss. This method was previously described for rats (Hunter et al. 2007a; Hunter et al. 2007b) and has been modified in the stereotaxic coordinates and dose given for mice. Mice were anesthetized with sodium pentobarbital (Abbott Laboratories, Chicago, IL), positioned in a stereotax, and 1:2000 bupivacaine/epinephrine (Hospira, Lake Forest, IL) was administered as a local anesthetic. Four small openings were created in the skull using a dental trephine, and the stereotaxic coordinates, measured in millimeters, from Bregma were: anterior/posterior +1.18, medial/lateral +/− 1.5, and dorsal/ventral −3.5 as well as anterior/posterior −0.34, medial/lateral +/− 2.5, and dorsal/ventral −3.2 (Paxinos 2001). At each intrastriatal coordinate, either 1μl of sterile saline or 1μl of LPS (5, 7.5, and 10μg LPS/μl saline) was injected by using a 30 gauge 10μl Hamilton syringe. The rate of injection was 0.5μl/min and the needle was kept in place for five minutes post-injection before slowly withdrawing. Following surgery, the mice were kept on a heating pad, and subcutaneous sterile saline was given to aid in post-operative recovery. Saline injections continued until the animals become hydrated and ambulatory. Following surgery, a pain score rating and body condition scoring was used to ensure establishment of humane endpoints, so the animals did not suffer excessive pain. Animals were euthanized at one week post-injection. This LPS dose study showed significant loss of dopaminergic neurons occurred with the 5μg LPS/μl saline dose one week post-injections; thus, all subsequent studies used this dose, and mice were euthanized at four and twelve weeks post-injection.
L-dopa therapy, which replaces striatal dopamine levels to alleviate some of the motor dysfunctions observed in PD patients (Goetz 1999), was used to demonstrate the LPS-induced motor deficit of the denervated striatum at twelve weeks post-injection. This was done on the day following the determination of the 12th week’s behavior by subcutaneous injection of 2mg/kg carbidopa (Sigma), followed by 20mg/kg L-dopa (Sigma) 30 minutes later, and then by testing rotarod performance at 1, 1.5, and 2.5 hours post L-dopa injection to test peak levels and wear off time.
We also determined the optimal dose of the selective iNOS inhibitor, N6-(1-iminoethyl)-L-lysine (L-NIL, Cayman Chemical, Ann Arbor, Michigan) and the nonselective NOS inhibitor L-N(G)-nitro-arginine (L-NNA, Cayman Chemical) for attenuation of LPS-induced dopaminergic neuron loss. Mice were randomly selected, grouped, and pre-treated, 20 minutes prior to their intrastriatal LPS injections with either intraperitoneal injection of saline, one of three different doses of L-NIL (5, 10 and 15mg/kg), or one of two different doses of L-NNA (10 and 15mg/kg), as we previously used L-NNA in a rat study at 10mg/kg (Arimoto and Bing 2003). For a control, mice were intrastriatally injected with saline and received intraperitoneal saline injections. The saline, L-NIL, or L-NNA injections were continued once a day for six days, and on day seven treatments were given one hour before euthanasia for tissue collection. This study showed that L-NIL (10mg/kg) or L-NNA (10mg/kg) was optimal in attenuating LPS-induced dopaminergic neuron loss. In a subsequent study mice were randomly selected, grouped, and pre-treated, 20 minutes prior to their saline or LPS injections, with either intraperitoneal saline, L-NIL (10 mg/kg), or L-NNA (10 mg/kg) injection, and treatments were given once a day for up to one week, while the data was collected four weeks post-injection.
To validate the role of iNOS in LPS-induced dopaminergic neurodegeneration, iNOS knockout and strain control wild-type mice were injected with LPS as described above and dopaminergic neurodegeneration was measured two weeks post-LPS, as mortality seemed to be an issue in this strain of mice.
Tissue collection
Animals were anesthetized with CO2, killed by decapitation, and the brains were rapidly removed. For immunocytochemistry the lower half of the brain containing the SN was collected using a mice brain matrix and this tissue was immediately fixed in 4% paraformaldehyde, was stored at 4°C for two days, and was transferred to a 30% sucrose 1× PBS solution, which was stored at 4°C until the tissue was used for sectioning. For HPLC analysis, whole striatum were dissected out, snap frozen, placed into a pre-weighed Eppendorf tube, weighed, and stored at −80°C until assayed by HPLC. Mice used for the twelve week time-point were transcardially perfused with 4% PFA, brain was removed and stored in PFA at 4°C for two days, and was transferred to a 30% sucrose 1×PBS solution stored at 4°C until the tissue was used for sectioning.
Rotarod Behavioral Analysis
The ENV 575M rotarod treadmill (MED Associates, St. Albans, VT) was used to assess fine motor coordination and balance. Rotarod performance is measured on a rotating rod as the mice must walk forward to avoid falling off a continuously rotating cylinder (Carter et al. 1999; Jones and Roberts 1968). This was previously described with some minor modifications (Vijitruth et al. 2006). Pre-training prior to LPS injections consisted of three consecutive days at low rotational speeds (20, 24, 28 and 32 rpm) and on a rod that accelerates from 0–50 rpm the fourth day. Results obtained from day four pre-LPS are considered the baseline performance for time spent on the rod. Mice were tested once a week until euthanizing for tissue collection, where two trials were measured and the average time on the rod for each mouse was used for data analysis. Time on the rod was used to assess fine motor coordination and balance. In addition, the effects of L-Dopa/carbidopa on motor coordination and balance, was tested on the day following the collection of the twelve-week behavioral data, to demonstrate the LPS-induced motor deficit of the denervated striatum. We used subcutaneous administration of L-dopa/carbidopa (20/2mg/kg) (Yarkov et al. 2003), where carbidopa was given 30 minutes prior to L-Dopa and then rotarod performance was tested at 1, 1.5, and 2.5 hours post L-dopa injection.
Immunocytochemistry (ICC)
ICC, as previously described (Hunter et al. 2007b) for the dopaminergic neuronal marker tyrosine hydroxylase (TH; Pel Freez, Rogers, AR) was used to immunostain every sixth section using the ABC-peroxide method (Lu et al. 2000). Sections from the SN (30μm) were incubated overnight with primary antiserum (1:2000) at 4°C. Then, sections were washed and incubated with the appropriate secondary antibody (Vector Laboratories Inc., Burlingame, CA) before immunoreactive cells were visualized using the ABC Kit (Vector Labs) with DAB (Sigma) as the chromagen. All sections were mounted on glass slides, dehydrated and cover-slipped using Permount (Fisher Scientific, Fair Lawn, NJ). The number of TH-positive cells with clearly visualized cell bodies, within the outlines of the SN, were determined using the optical fractionator method of the Bioquant System (West 1993). This gave an estimate of the total number of dopaminergic neurons in the SN. Cell counts by an investigator that was blind to treatment groups were performed with unbiased stereology as previously described (Vijitruth et al. 2006). The outlines of the SN were determined in the TH-stained sections by the distribution of the dopaminergic neurons and by referencing well-established landmarks (Hagg and Varon 1993). The number of neurons in the saline-injected mice was used to calculate the percentage of surviving neurons in the LPS-injected mice. In addition, representative sections from the striatum were immunostained for dopaminergic neuron terminals and gamma aminobutyric acid (GABA)ergic neurons by using the TH or dopamine and cAMP-regulated phosphoprotein (DARP-32, Chemicon, Temecula, CA) antibodies, respectively.
HPLC analysis of striatal tissue levels of dopamine and metabolites
Tissue levels of dopamine, norepinephrine, and serotonin as well as dopamine’s primary metabolites dihydroxyphenylacetic acid and homovanillic acid, and the metabolite of serotonin 5-hydroxyindole acetic acid were measured using HPLC as previously described (Cass et al. 2003). Retention times of standards were used to identify peaks, and peak heights were used to calculate recovery of the internal standard (dihydroxybenzylamine) and the amount of monoamines and their metabolites. Results are expressed as ng/g wet weight of tissue.
Statistical analysis
Data are expressed as means +/− SEM. For all experiments animals were randomly selected and grouped. The Systat 10 software (SPSS Inc., Chicago, IL) was used to perform statistical analyses with either the Student’s t-test or ANOVA, which was followed by a protected least significant differences post hoc test when a significant F test result was obtained. Statistical significance was defined as a p value of less than 0.05.
Results
Dopaminergic neurodegeneration post-intrastriatal LPS is dose dependent
For the first part of this study we had to translate the intrastriatal LPS model into mice to validate and optimize the model. C57/B6 mice were injected with three different doses of LPS (5, 7.5, and 10μg/μl) and we examined TH-positive cell loss in the SN one week after injection as in our previous study in rats (Hunter et al. 2007b). The data shows that intrastriatal LPS injection has a dose dependent effect on TH-positive cell loss, where we saw 23, 45, and 61% cell loss one week-post-LPS injection (Figure 1). We chose to use the 5μg/μl LPS dose for the rest of the experiment because it was significant, it paralleled our previously published cell loss (21%) in rats (Hunter et al. 2007b), and it allowed the opportunity for a more progressive cell loss to occur.
Figure 1.

Dopaminergic neurodegeneration post-intrastriatal LPS is dose dependent. C57/B6 mice were injected with saline or three different doses of LPS and sections were immunostained for TH-positive cells. Unbiased stereological cell counts in the SN shows that intrastriatal LPS injection has a dose dependent effect on TH-positive cell loss, where one week post-injection of 5, 7.5, and 10μg/μl LPS cell loss was 23, 45, and 61%. There was n=3–6/group and *** = p ≤ 0.001 when comparing LPS vs. Saline. In addition, p was ≤ 0.001 when comparing the various doses of LPS.
Intrastriatal LPS induces progressive SN dopaminergic neurodegeneration
Following intrastriatal LPS injection (5μg/μl) there was a subsequent progressive dopaminergic neuron death in the SN. ICC coupled with unbiased stereological cell counts was used to determine the number of TH-positive cells within the SN pars compacta region. Specifically, TH-positive cell loss progressed from 26% at one week, to 72% at four weeks, and reached 81% by twelve weeks (Figure 2A). In addition, ICC shows TH-positive fibers are significantly decreased in the striatum, but no apparent loss of GABAergic neurons occurs, three months after LPS injection (Figure 2B). These findings support our hypothesis that intrastriatal LPS induces specific and progressive SN dopaminergic neurodegeneration.
Figure 2.

Intrastriatal LPS induces progressive SN dopaminergic neurodegeneration. (A) A graph of the data collected from unbiased stereological cell counts of TH-positive cells within the SN shows LPS induces progressive dopaminergic neurodegeneration. TH-positive cell loss progressed from 26% at one week, to 72% at four weeks, and reached 81% by twelve weeks. There was n=3–6/group and *** = p ≤ 0.001 when comparing LPS vs. Saline at each time point. In addition, when comparing the 4th and 12th week LPS vs. the 1st week LPS p was ≤ 0.001, but when comparing the 4th week LPS vs. the12th week LPS p was = 0.004. (B) Representative ICC from the twelve week time-point shows TH-positive cell loss in the SN, as indicated in the circled area. In addition, ICC shows TH-positive fibers are significantly decreased in the striatum (ST), but no loss of GABAergic neurons (DARP-32) occurs post LPS.
PD-related behavioral impairment and a decrease in striatal dopamine was observed following intrastriatal LPS injection
The automated rotarod test, is a well-established behavioral test for evaluation of overall motor deficits in PD models, as it is used for the assessment of akinetic symptoms (Rozas et al. 1997). Our data show that intrastriatal saline injection has no effect on rotarod performance in C57/B6 mice. However, mice injected with LPS have a significant decrease in their duration on the rotarod beginning two weeks after LPS injection, and their performance decreases progressively with time for up to four weeks post-LPS (Figure 3A). Our data also shows that the rotarod performance of mice injected with intrastriatal LPS is still significantly decreased relative to saline injected mice by three months post-injection. In addition, we show that subcutaneous administration of L-dopa/carbidopa significantly improves rotarod performance relative to baseline (LPS-damaged) performance at 1 and 1.5 hours post injection. In addition, the effects of L-dopa/carbidopa wear off by 2.5 hours and rotarod performance returns to the LPS damaged baseline levels (Figure 3B). This data clearly demonstrates the denervated striatum and the ability of the PD drug L-dopa to provide functional recovery. We also show that intrastriatal LPS injection significantly decreases by 42% (p= 0.014) the levels of striatal dopamine at four weeks post-injection, which supports the rotarod data showing maximal impairment at four weeks (Figure 4). A 21% decrease in striatal serotonin levels (data not shown) was also observed.
Figure 3.

Progressive behavioral impairment indicative of a denervated striatum is evident post-intrastriatal LPS. (A) A graph of the average time spent on a rotarod shows, mice injected with LPS have a significant decrease in their duration on the rotarod beginning two weeks after LPS injection, and their performance decreases progressively with time (n=6 animals/group, where ** = p ≤ 0.001). (B) A graph of rotarod data from animals tested three months post-intrastriatal LPS or Saline injection, shows persistent behavioral impairment post-LPS. In addition, a subcutaneous injection of L-dopa/carbidopa (20/2mg/kg), prior to rotarod testing, significantly improves rotarod performance relative to baseline performance at 1 and 1.5 hours post-injection, where the effects of L-dopa/carbidopa wear off by 2.5 hours, and performance returns to the LPS damaged baseline levels (n=3/group, where ** = p ≤ 0.01 when comparing LPS vs. Saline). When comparing L-dopa/carbidopa effects in LPS mice vs. their twelve week impairment, p was = 0.029 and 0.044 for 1 and 1.5 hour time-points.
Figure 4.
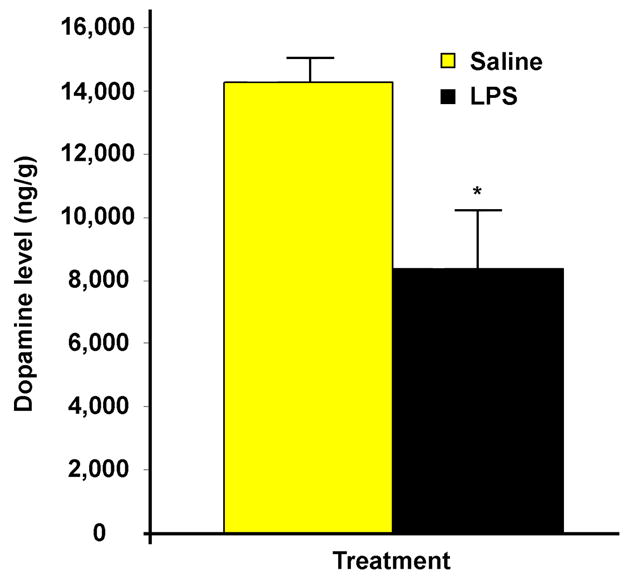
A decrease in striatal dopamine was observed post-intrastriatal LPS injection. HPLC analysis reveals that intrastriatal LPS injection significantly decreases by 42% the levels of striatal dopamine at four weeks post-injection (n=6/group, where * = p ≤ 0.05 when comparing LPS vs. Saline).
TH-positive cell loss is attenuated when using inhibitors of nitric oxide synthase
In a previous study using intranigral LPS injection in rats we showed that the NOS inhibitor L-NNA attenuates LPS-induced TH-positive cell loss (Arimoto and Bing 2003). However, L-NNA is considered a non-specific NOS inhibitor, and for this study we wanted to test the effects of a specific iNOS inhibitor, L-NIL. First, we tested a dose response for L-NIL as well as L-NNA. The data showed that 10 and 15mg/kg but not 5mg/kg L-NIL attenuated TH-positive cell loss to the same degree against intrastriatal LPS one week post-injection, and the same was true for L-NNA (data not shown). Thus, we chose the 10mg/kg dose for both inhibitors for further study. At four weeks post-injection both L-NIL and L-NNA attenuated the LPS-induced TH-positive cell loss with L-NIL being superior in protection. However, all groups still had significant cell loss of 49% or more compared to the saline control (Figure 5). Thus, neither drug prevented the decrease in striatal dopamine levels or behavioral impairment induced by LPS (data not shown). L-NIL did prevent the decrease in striatal serotonin (p= 0.011), while L-NNA only showed a trend (p= 0.074) for preventing the decrease in striatal serotonin (data not shown).
Figure 5.

TH-positive cell loss is attenuated by using NOS inhibitors. A graph of the data collected from unbiased stereological cell counts of TH-positive cells within the SN shows LPS induces significant dopaminergic neurodegeneration (72% loss) at four weeks post-injection. In addition, both L-NIL and L-NNA attenuated the LPS-induced TH-positive cell loss with L-NIL being superior in protection (51% vs. 61% loss). However, all groups still had significant cell loss compared to the saline control. There was n=4–6/group and *** = p ≤ 0.001 when comparing any LPS treated group vs. the Saline group. In addition, when comparing any LPS treated group p was ≤ 0.001.
Mice deficient in iNOS are partially protected against intrastriatal LPS injection
To determine the effects of iNOS deficiency on LPS-induced neurodegeneration, iNOS knockout (NOS2 tm1Lau/J) and wild-type mice were injected with intrastriatal LPS. ICC coupled with unbiased stereological cell counts of the TH-positive cells within the SN, showed iNOS deficient mice had 30% more TH-positive cells than wild-type mice at two weeks after injection (Figure 6A), which demonstrates the protective effects of iNOS deficiency against dopaminergic neurodegeneration in the intrastriatal LPS-induced PD model. These mice were also accessed for rotarod performance, and the data shows that LPS induces a significant decrease in duration on the rotarod in both wild-type and iNOS knockout mice one week post-LPS injection when compared to their pre-injection performance. However, two weeks after LPS injection, there was a progressive decrease in the performance of only the wild-type mice, which implies that iNOS knockout mice were protected against the progressive cell loss induced by LPS (Figure 6B).
Figure 6.

Mice deficient in iNOS are partially protected against intrastriatal LPS injection, where progressive behavioral impairment in iNOS knockout mice is attenuated. (A) A graph of the data collected from unbiased stereological cell counts of TH-positive cells within the SN shows iNOS knockout mice have 30% more dopaminergic neurons compared to wild-type mice, following intrastriatal LPS (n=6–7/group, where *** = p ≤ 0.001 when comparing iNOS knockout vs. wild-type mice). (B) Rotarod performance shows that LPS induces a parallel one week post-injection decrease in duration on the rotarod in both wild-type and iNOS knockout mice when compared to their baseline pre-injection performance (# and @ = p ≤ 0.05 when comparing post-injection vs. baseline rotarod performance for iNOS knockout or wild-type mice, respectively). Two weeks after LPS injection, the rotarod performance of the wild-type mice progressed (@@ = p ≤ 0.01 when comparing post-injection vs. baseline rotarod performance for wild-type mice) and the behavioral impairment was a significantly decreased compared to the iNOS knockout mice, where * = p ≤ 0.05 when comparing iNOS knockout vs. wild-type mice.
Discussion
The present study supports our hypothesis that injection of LPS into the striatum is a promising new model to reveal the potential role of inflammation in PD-related neurodegeneration. It appears that we successfully translated the intrastriatal LPS model into C57/B6 mice and provided a novel tool to study the role of inflammation-induced dopaminergic neurodegeneration. In summary we showed a dose dependent effect of LPS-induced inflammation on SN dopaminergic neurons, so that when a low enough dose is used, one can induce a parkinsonian-like model that exhibits progressive loss of dopaminergic neurons. In conjunction with the neurodegeneration there is progressive PD-related behavioral impairment that is accompanied by the loss of striatal dopamine, and this impairment can be relieved by treatment with L-Dopa/carbidopa. To determine the role of iNOS derived NO in this inflammation driven model we used two specific NOS inhibitors and iNOS knockout mice. We found that NO plays a partial role in protection against inflammation-induced neurodegeneration but it is not responsible for the process, as it appeared to only protect against the progressive loss of dopaminergic neurons.
Our data showed a dose dependent effect for intrastriatal LPS injection, which appeared to be specific for only dopaminergic neurons, as GABAergic staining in the striatum appeared to be unaltered three months post-LPS injection. In a previous study in rats we showed a dose dependent effect of intrastriatal LPS injection on TH-positive SN neurons (Hunter et al. 2007b). In addition, LPS did not affect GABAergic neurons in the striatum or SN (Herrera et al. 2000; Hunter et al. 2007b). Thus, our study further supports the hypothesis that dopaminergic neurons are preferentially susceptible to inflammation-induced neurodegeneration. We were also able to reproduce our model as our 5μg of LPS dose showed 23% TH-positive cell loss in our LPS dose study and our progressive cell loss study showed 26% cell loss at one week post-injection.
In this study we showed for the first time that intrastriatal LPS injection leads to progressive dopaminergic neurodegeneration, which is accompanied by increased behavioral impairment as well as a decreased striatal dopamine level. Several LPS models have already shown that LPS-induced dopaminergic neurodegeneration is accompanied by a loss of striatal dopamine (Herrera et al. 2000; Hsieh et al. 2002; Hunter et al. 2007c). However, no study to our knowledge has ever used of a PD drug such as L-dopa, which replaces striatal dopamine levels to alleviate some of the motor dysfunctions observed in PD patients (Goetz 1999), to validated the denervation of the striatum. Thus, we report for the first time that the administration of L-Dopa/carbidopa relieved the LPS-induced impairment of rotarod performance. In fact, the rotarod performance gradually improved almost to control levels during the “on time”, peaked, and then the effect wore off, which is similar to the effects of L-dopa in a PD patient. This data suggests that our LPS-model is a valid tool for PD-related research.
In our study we also examined a potential mechanism, as we used two NOS inhibitors and iNOS knockout mice to determine the role of iNOS derived NO in inflammation-induced dopaminergic neurodegeneration. Our data showed that inhibition of NOS only partially protected against LPS-induced dopaminergic neurodegeneration. Support for our results comes from studies that demonstrated partial protection when using NOS inhibitors in the rat intranigral LPS-induced PD model (Arimoto and Bing 2003; Iravani et al. 2002). Our data also showed that L-NIL the iNOS specific inhibitor prevented more TH-positive cell loss than the non-specific NOS inhibitor L-NNA. Support for this comes from the hypothesized role for overproduction of NO as a result of increased iNOS expression in activated microglia, which is then detrimental to dopaminergic neurons (Hunot et al. 1996; Knott et al. 2000). In addition, we showed iNOS deficient mice had 30% more TH-positive cells than strain control wild-type mice at two weeks after injection, which demonstrated the protective effects of iNOS deficiency against dopaminergic neurodegeneration in the intrastriatal LPS-induced PD model. Although we did not inject saline into iNOS knockout mice or their strain-matched wild-type controls, others have shown no difference in the number of TH-positive cells within the SN (Liberatore et al. 1999).
We also observed behavioral impairment in both the wild-type and iNOS knockout mice. However, the impairment of rotarod performance in the iNOS knockout mice reached a plateau after the first week, while the behavioral impairment for the wild-type mice was worse two weeks post-injection. These wild-type mice had 55% TH-positive cell loss at two weeks post-LPS, which is between the 26% we saw at one week and the 72% at four weeks in our progressive cell loss study. Overall, this data supports the hypothesis that iNOS derived NO is important in the progression of dopaminergic neurodegeneration induced by LPS. However, this data also suggests that inhibition of iNOS does not protect against the initial cell loss induced by LPS. This is in contrast to the rat intranigral LPS-induced PD model studies that demonstrated partial protection when using NOS inhibitors (Arimoto and Bing 2003; Iravani et al. 2002). However, these studies have maximal TH cell loss within less than a week, which leaves little time to observe protection against progressive cell loss when there is such acute damage. We speculate that in our inhibitor study that we did not see attenuation of dopamine loss or behavioral impairment, as we only treated animals for one week following LPS injection. Thus, we missed the real effect of inhibition of NOS on the progression of LPS-induced TH-positive cell loss in this portion of the study, and future studies should be done to validate this hypothesis.
Although, it should be noted that the role of inflammation in the pathogenesis of PD remains controversial, this study did validate the potential for using the intrastriatal LPS injection model for PD-related research, as we showed progressive dopaminergic neurodegeneration in the SN, which is accompanied by progressive behavioral impairment and striatal dopamine depletion. In addition, the behavioral impairment can be alleviated by the use of the PD drug L-Dopa. We also suggest that iNOS plays a role in the progressive loss of dopaminergic neurons but not the initial loss induced by LPS. With this model future research can be done in gene knockout mice to study other potential mechanisms of LPS-induced neurodegeneration. In addition, the availability of a progressive PD-related model was lacking until now. Thus, this model can be used to screen therapeutics for PD at a more clinically relevant time (i.e., after LPS injection but before manifestation of PD-related behavioral impairment), as most PD drugs are screened in animal models where inhibitors are given pre-disease induction. The rotenone, 6-hydroxydopamine, and MPTP models of PD do not take this long to develop their versions of parkinsonism, yet they are routinely used to screen PD drugs, and with the exception of MPTP, none have been proven to induce parkinsonism, let alone PD. We previously suggested the use of this model for early intervention drug screening in a study that looked at intrastriatal LPS-induced iron accumulation with the thought that magnetic resonance imaging technology can be coupled with treatment intervention (Hunter et al. 2008). Thus, this novel PD-related model should be further characterized and strongly considered as a tool for future drug studies.
Acknowledgments
Thank you to the National Institute on Aging and the National Institute of Neurological Disorders and Stroke for their funding: AG017963 (to WAC) and NS044157 (to GYB). In addition, we thank Laura Peters for her assistance with the HPLC.
References
- Arimoto T, Bing G. Up-regulation of inducible nitric oxide synthase in the substantia nigra by lipopolysaccharide causes microglial activation and neurodegeneration. Neurobiol Dis. 2003;12(1):35–45. doi: 10.1016/s0969-9961(02)00017-7. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:120–131. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- Berger JR. GIC. Von’s Economo’s Encephalitis. In: Nath AB JR, editor. Clinical Neurovirology. New York: Marcel Dekker; 2003. pp. 523–542. [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J Neurosci. 1999;19(8):3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cass WA, Harned ME, Peters LE, Nath A, Maragos WF. HIV-1 protein Tat potentiation of methamphetamine-induced decreases in evoked overflow of dopamine in the striatum of the rat. Brain Res. 2003;984(1–2):133–142. doi: 10.1016/s0006-8993(03)03122-6. [DOI] [PubMed] [Google Scholar]
- Castano A, Herrera AJ, Cano J, Machado A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J Neurochem. 1998;70(4):1584–1592. doi: 10.1046/j.1471-4159.1998.70041584.x. [DOI] [PubMed] [Google Scholar]
- Gao HM, Hong JS, Zhang W, Liu B. Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci. 2002;22(3):782–790. doi: 10.1523/JNEUROSCI.22-03-00782.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz CG. Textbook of Clinical Neurology. Philadelphia, PA: W.B. Saunders Co; 1999. [Google Scholar]
- Hagg T, Varon S. Ciliary neurotrophic factor prevents degeneration of adult rat substantia nigra dopaminergic neurons in vivo. Proc Natl Acad Sci U S A. 1993;90(13):6315–6319. doi: 10.1073/pnas.90.13.6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Appel S, Le W. Minocycline inhibits microglial activation and protects nigral cells after 6-hydroxydopamine injection into mouse striatum. Brain Res. 2001;909(1–2):187–193. doi: 10.1016/s0006-8993(01)02681-6. [DOI] [PubMed] [Google Scholar]
- Heales SJ, Bolanos JP, Stewart VC, Brookes PS, Land JM, Clark JB. Nitric oxide, mitochondria and neurological disease. Biochim Biophys Acta. 1999;1410(2):215–228. doi: 10.1016/s0005-2728(98)00168-6. [DOI] [PubMed] [Google Scholar]
- Herrera AJ, Castano A, Venero JL, Cano J, Machado A. The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol Dis. 2000;7(4):429–447. doi: 10.1006/nbdi.2000.0289. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O. Parkinson’s disease and the adaptive capacity of the nigrostriatal dopamine system: possible neurochemical mechanisms. Adv Neurol. 1993;60:140–147. [PubMed] [Google Scholar]
- Hsieh PF, Chia LG, Ni DR, Cheng LJ, Ho YP, Tzeng SF, Chang MH, Hong JS. Behavior, neurochemistry and histology after intranigral lipopolysaccharide injection. Neuroreport. 2002;13(3):277–280. doi: 10.1097/00001756-200203040-00006. [DOI] [PubMed] [Google Scholar]
- Hunot S, Boissiere F, Faucheux B, Brugg B, Mouatt-Prigent A, Agid Y, Hirsch EC. Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience. 1996;72(2):355–363. doi: 10.1016/0306-4522(95)00578-1. [DOI] [PubMed] [Google Scholar]
- Hunter RL, Choi DY, Kincer JF, Cass WA, Bing G, Gash DM. Fenbendazole treatment may influence lipopolysaccharide effects in rat brain. Comp Med. 2007a;57(5):487–492. [PubMed] [Google Scholar]
- Hunter RL, Dragicevic N, Seifert K, Choi DY, Liu M, Kim HC, Cass WA, Sullivan PG, Bing G. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J Neurochem. 2007b;100(5):1375–1386. doi: 10.1111/j.1471-4159.2006.04327.x. [DOI] [PubMed] [Google Scholar]
- Hunter RL, Liu M, Choi DY, Cass WA, Bing G. Inflammation and age-related iron accumulation in F344 rats. Current Aging Science 1. 2008 doi: 10.2174/1874609810801020112. In Press. [DOI] [PubMed] [Google Scholar]
- Hunter RL, Liu M, Choi DY, Hardy PA, Bing G. Society For Neuroscience. CA: San Diego; 2007c. Inflammation and age related iron accumulation in F344 rats. [DOI] [PubMed] [Google Scholar]
- Iravani MM, Kashefi K, Mander P, Rose S, Jenner P. Involvement of inducible nitric oxide synthase in inflammation-induced dopaminergic neurodegeneration. Neuroscience. 2002;110(1):49–58. doi: 10.1016/s0306-4522(01)00562-0. [DOI] [PubMed] [Google Scholar]
- Jenner P, Olanow CW. Understanding cell death in Parkinson’s disease. Ann Neurol. 1998;44(3 Suppl 1):S72–84. doi: 10.1002/ana.410440712. [DOI] [PubMed] [Google Scholar]
- Jones BJ, Roberts DJ. A rotarod suitable for quantitative measurements of motor incoordination in naive mice. Naunyn Schmiedebergs Arch Exp Pathol Pharmakol. 1968;259(2):211. doi: 10.1007/BF00537801. [DOI] [PubMed] [Google Scholar]
- Knott C, Stern G, Wilkin GP. Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol Cell Neurosci. 2000;16(6):724–739. doi: 10.1006/mcne.2000.0914. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39(1):151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5(12):1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- Ling Z, Gayle DA, Ma SY, Lipton JW, Tong CW, Hong JS, Carvey PM. In utero bacterial endotoxin exposure causes loss of tyrosine hydroxylase neurons in the postnatal rat midbrain. Mov Disord. 2002;17(1):116–124. doi: 10.1002/mds.10078. [DOI] [PubMed] [Google Scholar]
- Lu X, Bing G, Hagg T. Naloxone prevents microglia-induced degeneration of dopaminergic substantia nigra neurons in adult rats. Neuroscience. 2000;97(2):285–291. doi: 10.1016/s0306-4522(00)00033-6. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Akiyama H, McGeer EG. Rate of cell death in parkinsonism indicates active neuropathological process. Ann Neurol. 1988;24(4):574–576. doi: 10.1002/ana.410240415. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Yasojima K, McGeer EG. Inflammation in Parkinson’s disease. Adv Neurol. 2001;86:83–89. [PubMed] [Google Scholar]
- McRae-Degueurce A, Rosengren L, Haglid K, Booj S, Gottfries CG, Granerus AC, Dahlstrom A. Immunocytochemical investigations on the presence of neuron-specific antibodies in the CSF of Parkinson’s disease cases. Neurochem Res. 1988;13(7):679–684. doi: 10.1007/BF00973287. [DOI] [PubMed] [Google Scholar]
- Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. 1994;180(2):147–150. doi: 10.1016/0304-3940(94)90508-8. [DOI] [PubMed] [Google Scholar]
- Niehaus I. Lipopolysaccharides induce inflammation-mediated neurodegeneration in the substantia nigra and cerebral cortex (a case report) In: Hanin IFA, Cacabelos R, editors. New Trends in Alzheimer and Parkinson Related Disorders. Bologna: Monduzzi Editore; 2004. pp. 36–39. [Google Scholar]
- Parker WD, Jr, Boyson SJ, Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann Neurol. 1989;26(6):719–723. doi: 10.1002/ana.410260606. [DOI] [PubMed] [Google Scholar]
- Paxinos GaF, K BJ. The Mouse Brain in Stereotaxic Coordinates. 2. San Diego: Academic Press; 2001. [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55(5):453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas G, Guerra MJ, Labandeira-Garcia JL. An automated rotarod method for quantitative drug-free evaluation of overall motor deficits in rat models of parkinsonism. Brain Res Brain Res Protoc. 1997;2(1):75–84. doi: 10.1016/s1385-299x(97)00034-2. [DOI] [PubMed] [Google Scholar]
- Vijitruth R, Liu M, Choi DY, Nguyen XV, Hunter RL, Bing G. Cyclooxygenase-2 mediates microglial activation and secondary dopaminergic cell death in the mouse MPTP model of Parkinson’s disease. J Neuroinflammation. 2006;3:6. doi: 10.1186/1742-2094-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ. New stereological methods for counting neurons. Neurobiol Aging. 1993;14(4):275–285. doi: 10.1016/0197-4580(93)90112-o. [DOI] [PubMed] [Google Scholar]
- Yarkov AV, Hanger D, Reploge M, Joyce JN. Behavioral effects of dopamine agonists and antagonists in MPTP-lesioned D3 receptor knockout mice. Pharmacol Biochem Behav. 2003;76(3–4):551–562. doi: 10.1016/j.pbb.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Youdim MB, Riederer P. Understanding Parkinson’s disease. Sci Am. 1997;276(1):52–59. doi: 10.1038/scientificamerican0197-52. [DOI] [PubMed] [Google Scholar]
- Zhang J, Stanton DM, Nguyen XV, Liu M, Zhang Z, Gash D, Bing G. Intrapallidal lipopolysaccharide injection increases iron and ferritin levels in glia of the rat substantia nigra and induces locomotor deficits. Neuroscience. 2005;135(3):829–838. doi: 10.1016/j.neuroscience.2005.06.049. [DOI] [PubMed] [Google Scholar]