

Abstract
The cystic fibrosis transmembrane conductance regulator (CFTR) is a membrane protein that belongs to the same family as multidrug resistance-associated proteins whose main function is to expel xenobiotics and physiological organic anions from the cell interior. Despite the overall structural similarity with these membrane proteins, CFTR is not an active transporter but is instead a Cl− channel. We have tested the ability of known inhibitors of multidrug resistance-associated proteins to affect CFTR Cl− currents. We have found that sulfinpyrazone, probenecid, and benzbromarone are also inhibitors of CFTR activity, with a mechanism involving blockage of the channel pore.
Keywords: CFTR, multidrug resistance protein, chloride channel, channel blocker
1. Introduction
The cystic fibrosis transmembrane conductance regulator (CFTR) is a plasma membrane protein that belongs to the family of ATP-binding cassette (ABC) transporters (Dean et al., 2001; Schmitt and Tempe, 2002). Such membrane proteins share a similar architecture based on various transmembrane helices (from 12 to 17) and two nucleotide binding domains (NBD1 and 2). Interaction and hydrolysis of ATP at two sites in the NBDs induces conformational changes that drive active transport of various types of molecules across the plasma membrane (Dean et al., 2001; Schinkel and Jonker, 2003). CFTR is part of the subfamily C of ABC (ABCC) transporters which includes the multidrug resistance-associated proteins (Kruh and Belinsky, 2003). These proteins work as active transporters of endogenous substrates, like ABCC1 for LTC4 (Leier et al., 1994; Jedlitschky et al., 1994), and of exogenous substances, called xenobiotics. Such compounds are transported in their native state or as conjugates with glutathione (Ishikawa, 1992), glucunorate, or sulfates (Jedlitschky et al., 1996). In general, ABCC drug transporters have a preference for anionic compounds in contrast to the multidrug resistance protein 1, ABCB1, which is more selective for neutral or slightly basic compounds (Schinkel and Jonker, 2003). The wide spectrum of substances translocated by multidrug resistance proteins is beneficial because it provides protection against potentially toxic exogenous molecules (Leslie et al., 2001; Hipfner et al., 1999). However, many ABCC transporters, as well as ABCB1, are also responsible for the multidrug resistance shown by different types of human tumours (Grant et al., 1994; Kruh et al., 2001; Sawicka et al., 2004).
Among the ABCC subfamily, CFTR is the only protein that does not generate an active transport. In fact, CFTR is a plasma membrane Cl− channel (Anderson et al., 1991) in which the conformational changes generated by NBD/ATP interactions are not used for active transport but rather for the opening and closing of the pore (Sheppard et al., 1999). However, there are still some intriguing findings that suggest that multidrug resistance-associated proteins and CFTR have some similarities beyond the amino acid sequence homology. For example, it has been reported by some investigators that CFTR is also able to translocate glutathione as done by other ABCC proteins (although by passive diffusion and not by active transport) (Linsdell and Hanrahan, 1998). Furthermore, substrates of multidrug resistance-associated proteins inhibit CFTR Cl− currents by interacting with the CFTR pore from the cytosolic side (Linsdell and Hanrahan, 1999). This suggests a common mechanism of interaction at the level of the transmembrane portion of the proteins.
We have tested the ability of known ABCC inhibitors to affect CFTR Cl− currents. This is important to further explore the analogies between CFTR and ABCC drug transporters and, possibly, to develop novel CFTR blockers which could be useful for the treatment of secretory diarrhea (Verkman et al., 2006). Our data show that sulfinpyrazone, probenecid, and, particularly, benzbromarone are effective inhibitors of the CFTR channel through a probable block of the pore.
2. Materials and methods
2.1. Cell culture
Fischer rat thyroid (FRT) cells stably expressing human CFTR were cultured on plastic in Coon’s modified F12 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. T84 cells were cultured in DMEM/F12 plus 10% fetal bovine serum, L-glutamine and antibiotics (same concentrations as for FRT cells).
2.2. Transepithelial Cl− currents
For short-circuit current measurements, cells were plated on Snapwell permeable supports (Corning-Costar) at 500,000 cells/Snapwell. After 7–9 days, when the cells had generated tight epithelia, the Snapwell supports were mounted in modified Ussing chambers. The basolateral solution contained (in mM): 130 NaCl, 2.7 KCl, 1.5 KH2PO4, 1 CaCl2, 0.5 MgCl2, 10 glucose, 10 Na-Hepes (pH 7.3). In the apical solution 65 mM NaCl was replaced by Na gluconate, and CaCl2 was increased to 2 mM. The basolateral membrane was permeabilized with 250 μg/ml amphotericin B. For T84 cells, apical and basolateral chambers contained (in mM): 126 NaCl, 0.38 KH2PO4, 2.1 K2HPO4, 1 MgSO4, 1 CaCl2, 24 NaHCO3 and 10 glucose (basolateral membrane not permeabilized). Solutions on both sides were bubbled with air (FRT) or 5% CO2 (T84) and temperature was kept at 37°C. Hemichambers were connected to a DVC-1000 voltage clamp (World Precision Instruments) via Ag/AgCl electrodes and 1 M KCl agar bridges for recording short-circuit current. All test compounds were added simultaneously to both sides of the chamber.
2.3. Patch-clamp recordings
Experiments were performed in the cell-attached and whole-cell configuration of the patch-clamp technique on FRT cells expressing human CFTR. For whole cell experiments, the bath solution contained (in mM): 150 NaCl, 1 CaCl2, 1 MgCl2, 10 glucose, 10 mannitol, 10 Na-Hepes (pH 7.4). For cell attached experiments the bath solution composition was instead (in mM): 130 KCl, 2 NaCl, 2 CaCl2, 2 MgCl2, 10 glucose, 20 mannitol, 10 Na-HEPES (pH 7.3). The pipette was filled with (in mM): 120 CsCl, 10 TEA-Cl, 0.5 EGTA, 1 MgCl2, 40 mannitol, 10 Cs-Hepes (pH 7.3). For whole-cell experiments the pipette solution also contained 1 mM ATP. Data were filtered at 500 Hz and digitized at 1000 Hz using an Instrutech ITC-16 AD/DA interface and the PULSE (Heka) software. Membrane potential values are reported by taking the extracellular solution as reference. Inhibitors were applied by extracellular perfusion.
3. Results
Fig. 1A shows the chemical structure of multidrug resistance-associated protein inhibitors compared to known CFTR blockers. It is evident that the compounds belong to different chemical classes. However, they have in common a negative electrical charge at physiological pH. The sensitivity of the CFTR channel towards inhibitors was determined by measuring transepithelial Cl−currents in transfected FRT cells. Maximal stimulation with the adenylyl cyclase activator, forskolin (20 μM), evoked large Cl− currents which were inhibited, in a dose-dependent way, by extracellular application of sulfinpyrazone and probenecid (Fig. 1A,B). The half-effective concentrations (Ki) were 191 ± 29 μM (n = 8) and 1244 ± 152 μM (n = 8) for sulfinpyrazone and probenecid, respectively (Fig. 1D). Given the low potency, probenecid caused only a partial CFTR inhibition at 2–3 mM (Fig. 1B). Higher concentrations were not tested because of limits in solubility. The residual current could be reduced to pre-stimulation levels with the potent and selective inhibitor, CFTRinh-172 (Fig. 1B). Interestingly, benzbromarone, an uricosuric agent also able to block multidrug resistance-associated proteins (Schinkel and Jonker, 2003), inhibited CFTR currents at concentrations much lower than sulfinpyrazone and probenecid (Fig. 1C,D), with a Ki of 11.5 ± 3.1 μM (n = 10). In null FRT cells lacking expression of CFTR, forskolin and inhibitors had no effect on transepithelial currents (data not shown).
Fig. 1.
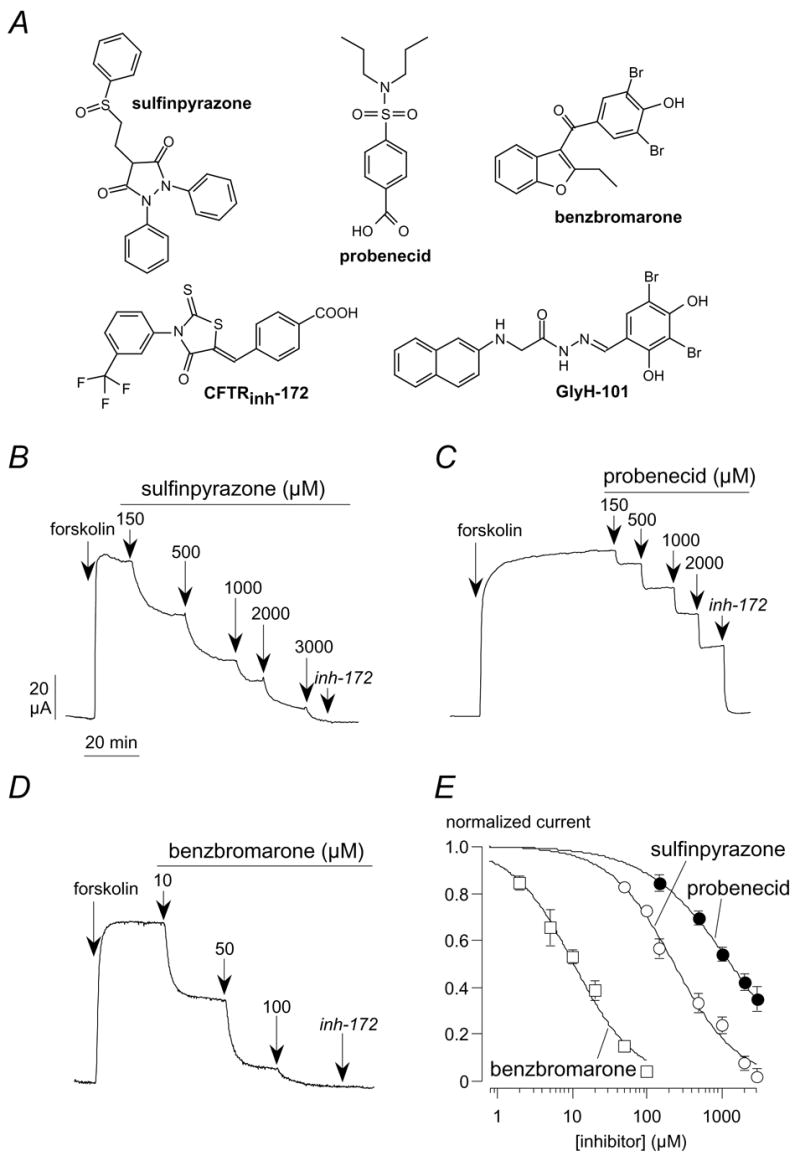
Block of CFTR Cl− currents by inhibitors of multidrug resistance-associated proteins. (A) Chemical structure of sulfinpyrazone, probenecid, and benzbromarone compared to known CFTR blockers. (B–D) Representative recordings of transepithelial Cl− currents in FRT cells expressing human CFTR. After full activation with forskolin (20 μM), CFTR currents were inhibited with increasing concentrations of sulfinpyrazone, probenecid, and benzbromarone. The CFTRinh-172 (10 μM) was added at the end of the experiment to block residual CFTR activity. (E) Normalized dose-responses. Each point is the mean ± SEM of 8–10 experiments.
Inhibition of Cl− transport was also confirmed in T84 cells, which derive from the human colon epithelium and possess CFTR-dependent Cl− secretion (Bell and Quinton, 1992). In short-circuit current experiments, T84 cells were stimulated with forskolin to maximally activate CFTR currents. Subsequent application of sulfinpyrazone, probenecid, or benzbromarone caused a dose-dependent decrease in Cl− secretion (not shown). The order of potency was similar to that observed for FRT cells, with benzbromarone and probenecid being the most and least potent blocker, respectively.
The mechanism of CFTR block by sulfinpyrazone, probenecid, and benzbromarone was investigated in whole-cell patch clamp experiments. Stimulation with forskolin alone caused the appearance of large membrane currents at all membrane voltages (Fig. 2). The shape of the current-voltage relationship was linear and no relaxations were observed upon application of voltage steps as expected for CFTR channels. In the presence of inhibitors the aspect of currents changed significantly. A marked reduction of the membrane current amplitude was observed mainly at negative voltages. In particular, negative voltage steps evoked a time-dependent deactivation of the current. The initial current measured at the beginning of the voltage step decreased rapidly to a value significantly smaller than that measured in the absence of the inhibitor (Fig. 2A–C). At steady-state, the shape of the current-voltage relationship in the presence of inhibitors was curved, with the percentage of block increasing progressively at more negative membrane potentials (Fig. 2D). Accordingly, the estimated Ki changed with the applied membrane potential. For example, the benzbromarone Ki was 8.8 μM and 19.8 μM at–60 and +40 mV, respectively (not shown).
Fig. 2.
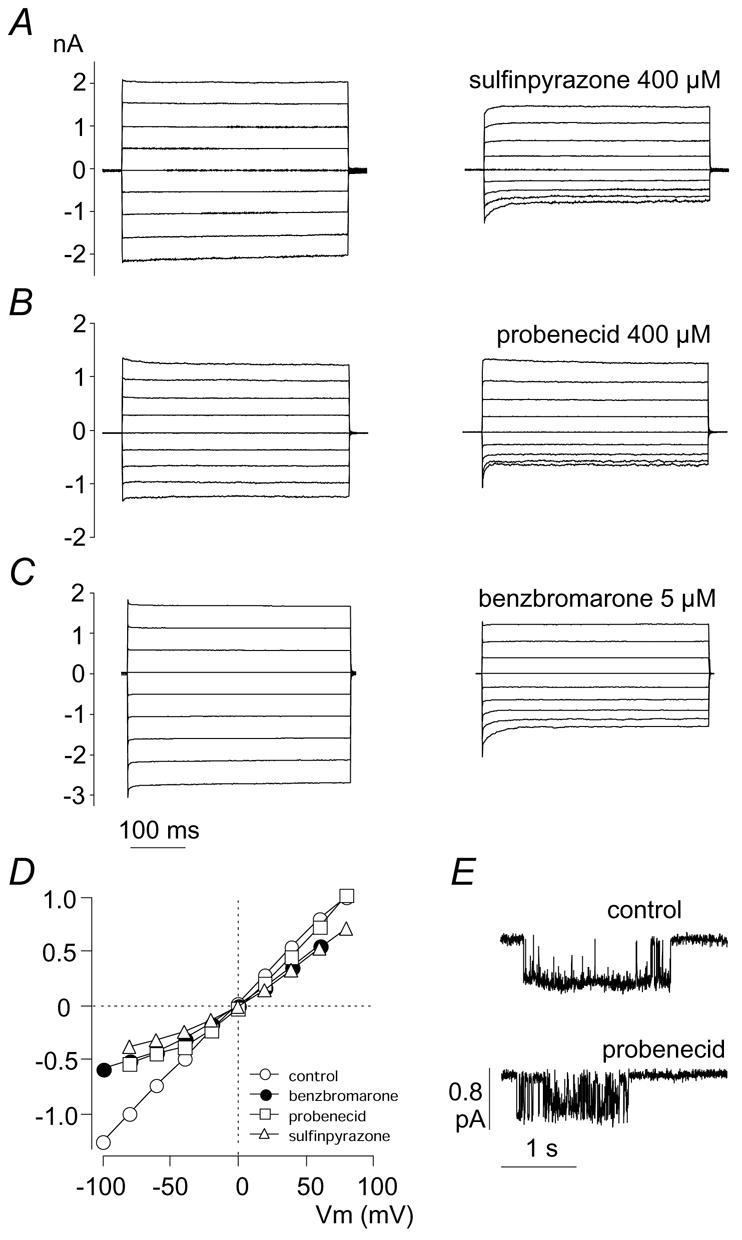
Patch-clamp analysis of mechanism of CFTR block by multidrug resistance-associated protein inhibitors. (A–C) Representative whole-cell recordings of CFTR Cl− currents. CFTR was activated with forskolin (0.5 μM). Each panel shows superimposed membrane currents elicited at different membrane potentials (in 20 mV steps) in the absence (left) and in the presence (right) of sulfinpyrazone, probenecid, or benzbromarone at the indicated concentrations. The range of applied membrane potentials was–80 to +80 mV for sulfinpyrazone and probenecid and–100 to +60 mV for benzbromarone. (D) Voltage-dependence of CFTR currents in the absence and presence of inhibitors. Membrane currents are normalized for the value measured at–80 mV in the absence of inhibitors. (E) Cell-attached recording of a single CFTR channel. Traces show a single burst of channel opening in absence and presence of probenecid (400 μM). Openings are represented by downward deflections from the baseline.
The mechanism of CFTR block was also investigated at the single channel level (Fig. 2E). In cell-attached recordings, the effect of probenecid mainly consisted in a large increase in the frequency of brief closures occurring within a burst of activity with a consequent decrease of the mean open time (from 256 ± 70 in control conditions to 78 ± 20 ms with 400 μM probenecid at −60 mV; p < 0.05).
4. Discussion
CFTR is unique within the large family of ABC transporters because it is the only protein with ion channel function. Despite the differences in function, ABCC drug transporters and CFTR share some structural features. First of all, they have a high level of amino acid conservation at some critical residues in the NBDs. Furthermore, they have a similarity in the overall organization of the transmembrane domains. Our aim was to verify whether inhibitors of ABCC transporters have the ability to affect CFTR function. We selected three compounds which have been reported to block various types of ABCC multidrug resistance proteins (Schinkel and Jonker, 2003).
We tested the inhibitors on FRT cells with stable expression of human CFTR. The three compounds were all able to inhibit the CFTR-dependent Cl− currents although with quite different potencies. Block by sulfinpyrazone and probenecid required high micromolar concentrations, with the latter compound being the least potent. Their affinity was comparable to that of CFTR blockers like glibenclamide or diphenylamine-2-carboxylate (Schultz et al., 1999). Conversely, benzbromarone was much more potent with a significant block obtained at low micromolar levels, a potency comparable to that of CFTR inhibitors discovered by high-throughput screening of large chemical libraries (Muanprasat et al., 2004). We investigated the mechanism of CFTR block by using the whole-cell configuration of the patch-clamp technique. We found that the effect of benzbromarone, sulfinpyrazone, and probenecid is sensitive to membrane potential, the block being progressively stronger as the membrane potential is more negative (i.e. when Cl− net flux is directed outward). Correspondingly, the shape of the current-voltage relationship changes from linear to outwardly rectifying. Since these compounds are negatively charged in solution at physiological pH, the voltage dependence and the outward rectification may be explained by postulating that the binding site lies on the cytosolic side of CFTR pore in a region sensing the transmembrane electrical field. According to this model, positive and negative membrane potentials decrease and increase, respectively, CFTR block by altering the residency time of the inhibitor within the channel pore. Therefore, benzbromarone, sulfinpyrazone, and probenecid behave similarly to other CFTR blockers like diphenylamine-2-carboxylate and glibenclamide which act from the intracellular side (Linsdell and Hanrahan, 1999; Hwang and Sheppard, 1999; Schultz et al., 1999). The type of block of single channel currents, i.e. a decrease of mean open time due to the appearance of brief intraburst closures, is also consistent with a mechanism involving occlusion of the channel pore. Occlusion of the pore was proposed by Linsdell and Hanrahan (1999) to explain the block of CFTR channels by multidrug resistance protein inhibitors. Actually, these authors observed a decrease in single channel conductance whereas, in our study, we mainly found a reduction in channel open time. The difference may be due to the intrinsic characteristics of compounds used in the two studies. However, we may also consider the higher frequency of data filtering in our study (500 vs 50 Hz) which probably allowed us to better resolve brief closure events.
It has been proposed that the CFTR pore has a large cytosolic vestibule which can accomodate several types of molecules (Hwang and Sheppard, 1999) and this may explain its sensitivity to many different inhibitors having a net negative charge in common. The shared sensitivity of CFTR and ABCC drug transporters to inhibitors could imply a similarity in the organization of the transmembrane domains and in the interaction with anionic molecules. Indeed, it has been shown that charged amino acids at equivalent positions in the transmembrane helices of ABCC1 and CFTR are important in their corresponding transport function (Haimeur et al., 2004; Dawson et al., 1999).
Acknowledgments
This work was supported by Telethon-ITALY (GGP05103), CIPE-Regione Liguria (Biofarma 2) and the NIH (P30 DK072517). We also thank Dr. Kerry Rhoden for kindly reading and correcting the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson MP, Gregory RJ, Thompson S, Souza DW, Paul S, Mulligan RC, Smith AE, Welsh MJ. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991;253:202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- Bell CL, Quinton PM. T84 cells: anion selectivity demonstrates expression of Cl− conductance affected in cystic fibrosis. Am J Physiol. 1992;262:C555–C562. doi: 10.1152/ajpcell.1992.262.3.C555. [DOI] [PubMed] [Google Scholar]
- Dawson DC, Smith SS, Mansoura MK. CFTR: mechanism of anion conduction. Physiol Rev. 1999;79:S47–S75. doi: 10.1152/physrev.1999.79.1.S47. [DOI] [PubMed] [Google Scholar]
- Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–1166. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- Grant CE, Valdimarsson G, Hipfner DR, Almquist KC, Cole SP, Deeley RG. Overexpression of multidrug resistance-associated protein (MRP) increases resistance to natural product drugs. Cancer Res. 1994;54:357–361. [PubMed] [Google Scholar]
- Haimeur A, Conseil G, Deeley RG, Cole SP. Mutations of charged amino acids in or near the transmembrane helices of the second membrane spanning domain differentially affect the substrate specificity and transport activity of the multidrug resistance protein MRP1 (ABCC1) Mol Pharmacol. 2004;65:1375–1385. doi: 10.1124/mol.65.6.1375. [DOI] [PubMed] [Google Scholar]
- Hipfner DR, Deeley RG, Cole SP. Structural, mechanistic and clinical aspects of MRP1. Biochim Biophys Acta. 1999;1461:359–376. doi: 10.1016/s0005-2736(99)00168-6. [DOI] [PubMed] [Google Scholar]
- Hwang TC, Sheppard DN. Molecular pharmacology of the CFTR Cl− channel. Trends Pharmacol Sci. 1999;20:448–453. doi: 10.1016/s0165-6147(99)01386-3. [DOI] [PubMed] [Google Scholar]
- Ishikawa T. The ATP-dependent glutathione S-conjugate export pump. Trends Biochem Sci. 1992;17:463–468. doi: 10.1016/0968-0004(92)90489-v. [DOI] [PubMed] [Google Scholar]
- Jedlitschky G, Leier I, Buchholz U, Center M, Keppler D. ATP-dependent transport of glutathione S-conjugates by the multidrug resistance-associated protein. Cancer Res. 1994;54:4833–4836. [PubMed] [Google Scholar]
- Jedlitschky G, Leier I, Buchholz U, Barnouin K, Kurz G, Keppler D. Transport of glutathione, glucuronate, and sulfate conjugates by the MRP gene-encoded conjugate export pump. Cancer Res. 1996;56:988–994. [PubMed] [Google Scholar]
- Kruh GD, Zeng H, Rea PA, Liu G, Chen ZS, Lee K, Belinsky MG. MRP subfamily transporters and resistance to anticancer agents. J Bioenerg Biomembr. 2001;33:493–501. doi: 10.1023/a:1012827221844. [DOI] [PubMed] [Google Scholar]
- Kruh GD, Belinsky MG. The MRP family of drug efflux pumps. Oncogene. 2003;22:7537–7552. doi: 10.1038/sj.onc.1206953. [DOI] [PubMed] [Google Scholar]
- Leier I, Jedlitschky G, Buchholz U, Cole SP, Deeley RG, Keppler D. The MRP gene encodes an ATP-dependent export pump for leukotriene C4 and structurally related conjugates. J Biol Chem. 1994;269:27807–27810. [PubMed] [Google Scholar]
- Leslie EM, Deeley RG, Cole SP. Toxicological relevance of the multidrug resistance protein 1, MRP1 (ABCC1) and related transporters. Toxicology. 2001;167:3–23. doi: 10.1016/s0300-483x(01)00454-1. [DOI] [PubMed] [Google Scholar]
- Linsdell P, Hanrahan JW. Glutathione permeability of CFTR. Am J Physiol. 1998;275:C323–C326. doi: 10.1152/ajpcell.1998.275.1.C323. [DOI] [PubMed] [Google Scholar]
- Linsdell P, Hanrahan JW. Substrates of multidrug resistance-associated proteins block the cystic fibrosis transmembrane conductance regulator chloride channel. Br J Pharmacol. 1999;126:1471–1477. doi: 10.1038/sj.bjp.0702458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJ, Verkman AS. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol. 2004;124:125–137. doi: 10.1085/jgp.200409059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicka M, Kalinowska M, Skierski J, Lewandowski W. A review of selected anti-tumour therapeutic agents and reasons for multidrug resistance occurrence. J Pharm Pharmacol. 2004;56:1067–1081. doi: 10.1211/0022357044265. [DOI] [PubMed] [Google Scholar]
- Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55:3–29. doi: 10.1016/s0169-409x(02)00169-2. [DOI] [PubMed] [Google Scholar]
- Schmitt L, Tampe R. Structure and mechanism of ABC transporters. Curr Opin Struct Biol. 2002;12:754–760. doi: 10.1016/s0959-440x(02)00399-8. [DOI] [PubMed] [Google Scholar]
- Schultz BD, Singh AK, Devor DC, Bridges RJ. Pharmacology of CFTR chloride channel activity. Physiol Rev. 1999;79:S109–S144. doi: 10.1152/physrev.1999.79.1.S109. [DOI] [PubMed] [Google Scholar]
- Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev. 1999;79:S23–S45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- Verkman AS, Lukacs GL, Galietta LJV. CFTR chloride channel drug discovery–inhibitors as antidiarrheals and activators for therapy of cystic fibrosis. Curr Pharm Des. 2006;12:2235–2247. doi: 10.2174/138161206777585148. [DOI] [PubMed] [Google Scholar]