

Abstract
Transgenic mice lacking renal aquaporins (AQPs), or containing mutated AQPs, have been useful in confirming anticipated AQP functions in renal physiology and in discovering new functions. Mice lacking AQPs 1–4 manifest defects in urinary concentrating ability to different extents. Mechanistic studies have confirmed the involvement of AQP1 in near-isosmolar fluid absorption in proximal tubule, and in countercurrent multiplication and exchange mechanisms that produce medullary hypertonicity in the antidiuretic kidney. Deletion of AQPs 2–4 impairs urinary concentrating ability by reduction of transcellular water permeability in collecting duct. Recently created transgenic mouse models of nephrogenic diabetes insipidus produced by AQP2 gene mutation offer exciting possibilities to test new drug therapies. Several unanticipated AQP functions in kidney have been discovered recently that are unrelated to their role in transcellular water transport. There is evidence for involvement of AQP1 in kidney cell migration following renal injury, of AQP7 in renal glycerol clearance, of AQP11 in prevention of renal cystic disease, and possibly of AQP3 in regulation of collecting duct cell proliferation. Future work in renal AQPs will focus on mechanisms responsible for these non-fluid-transporting functions, and on the development of small-molecule AQP inhibitors for use as aquaretic-type diuretics.
Keywords: Water transport, AQP, urinary concentrating mechanism
Introduction
Multiple aquaporin (AQP) subtypes are expressed in kidney: AQP1 in plasma membranes of proximal tubule, thin descending limb of Henle and outer medullary descending vasa recta, AQP2 in the apical membrane and intracellular vesicles of principal cells in collecting duct, AQP3 and AQP4 in the basolateral membrane of the same cells, AQP6 in intracellular vesicles of collecting duct intercalated cells, and AQP7 in the S3 segment of proximal tubule (Fig. 1A) (reviewed in ref. 1). This distribution suggests the involvement of renal AQPs primarily in osmotically driven water transport across kidney tubules and vasa recta. Here, we review data from AQP knockout mice confirming the role of AQPs in the urinary concentrating mechanism, as well as recent findings suggesting novel functions of AQPs in kidney that are not related to transcellular water transport.
Figure 1. Impaired urinary concentrating ability in AQP null mice.
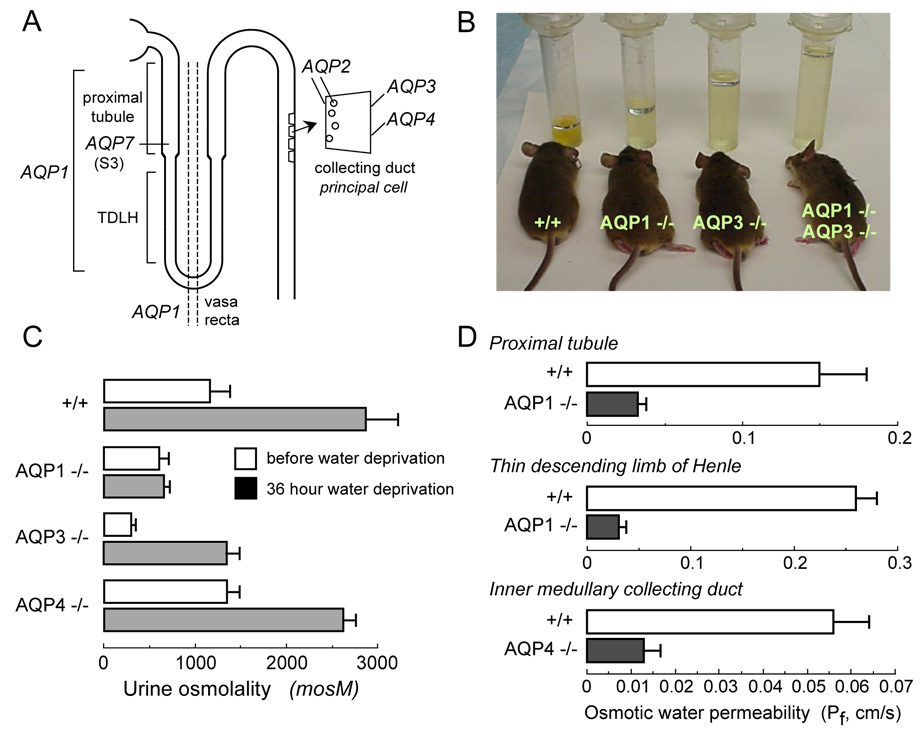
A. Sites of AQP expression in kidney. B. Twenty-four hour urine collections showing polyuria in mice lacking AQP1 and AQP3, individually and together. C. Urine osmolalities before and after 36 hour water deprivation (S.E.). D. Transepithelial osmotic water permeability (Pf) in microperfused proximal tubule, thin descending limb of Henle, and inner medullary collecting from mice of indicated genotype (S.E.). Data summarized from refs. 15–18, 20, 21.
Analysis of extrarenal phenotypes has provided exciting data suggesting new roles of AQPs, some with direct relevance to kidney function (reviewed in ref. 2). One paradigm that has emerged is a role for AQPs in near-isosmolar transepithelial fluid transport, as occurs in kidney proximal tubule. Mice lacking AQP1 manifest reduced production of aqueous fluid by the ciliary body (3) and of cerebrospinal fluid by the choroid plexus (4), and mice lacking AQP5 have impaired secretion of saliva (5) and airway submucosal gland fluid (6). AQP4, which is expressed in brain glial cells, plays an important role in brain water accumulation and removal in various brain pathologies such as stroke, tumor, infection and hydrocephalus (7–9). AQP4 also appears to play a role in neuroexcitation, including seizure activity and cortical spreading depression, by a mechanism that may involve facilitated reuptake of K+ from the extracellular space by glial cells (10, 11). AQP3-facilitated glycerol transport by epidermal keratinocytes is involved in maintaining high glycerol in epidermis and stratum corneum to support skin hydration and biosynthetic functions (12). AQP7-facilitated glycerol transport in adipocytes appears to be important in regulating fat cell glycerol content to prevent adipocyte hypertrophy and progressive fat accumulation (13, 14). Other new roles of AQPs in cell migration and proliferation are discussed further below.
AQPs and urinary concentrating function
A major and anticipated role of AQPs in kidney is in water transport across kidney tubules and vasa recta for the formation of a concentrated urine in the antidiuretic kidney. Deletion of AQP1 and/or AQP3 in mice results in marked polyuria, as seen in 24 hour urine collections (Fig. 1B) (15, 16). Fig. 1C summarizes urine osmolalities before and after a 36 hour water deprivation. Urinary osmolality in AQP1 null mice is low and does not increase with water deprivation, resulting in severe dehydration. AQP3 null mice are able to generate a partially concentrated urine in response to water deprivation. AQP4 null mice manifest only a mild defect in maximum urinary concentrating ability. Urinary diluting ability in these AQP null mice is unimpaired, as the mice are able to generate urine of <120 mOsm after infusion of half-normal saline.
AQP1 deletion produces polyuria and unresponsiveness to vasopressin or water deprivation by two distinct mechanisms – impaired near-isosmolar water reabsorption by the proximal tubule, and reduced medullary hypertonicity resulting from impaired countercurrent multiple and exchange. Transepithelial osmotic water permeability (Pf) in the isolated microperfused S2 segment of proximal tubule was reduced by ~5-fold in AQP1 knockout mice (Fig. 1D) (17), indicating that the major pathway for osmotically-driven water transport in this segment is transcellular and AQP1-dependent. Free-flow micropuncture with end-proximal tubule fluid sampling showed ~50% reduced fluid absorption, with little effect on single nephron glomerular filtration rate (18). The data support a 3-compartment model in which mild luminal hypotonicity drives osmotic water movement through highly water permeable cell membranes. As predicted by this model, a substantial ~40 mOsm transepithelial osmotic gradient was found in end proximal tubule fluid of AQP1 null mice, much greater than that of ~10 mOsm in wildtype mice (19). AQP1 also provides the major route for transepithelial water permeability in thin descending limb of Henle (TDLH) and outer medullary descending vasa recta (OMDVR) (20). These results indicate that AQP1 is the principal water channel in TDLH, and support the conclusion that osmotic equilibration in TDLH plays a key role in the renal countercurrent concentrating mechanism. Analysis of AQP1-dependent water transport in OMDVR in response to gradients of NaCl vs. raffinose indicated that solutes larger than NaCl are able to drive water movement through both AQP1 and an AQP1-independent, mercurial-insensitive pathway that may involve paracellular transport (20). The primary interpretation of microperfusion and micropuncture data is that AQP1 deletion impairs urinary concentrating ability by impairing near-isosmolar fluid absorption by proximal tubule and by interfering with the normally hypertonic medullary interstitium generated by countercurrent multiplication and exchange. The precise degree of impairment in urinary concentrating ability is also influenced by secondary renal mechanisms, such as tubuloglomerular feedback, as well as altered expression of other genes in AQP1 null mice.
AQP3 and AQP4 are expressed at the basolateral membrane of collecting duct epithelium, with relatively greater expression of AQP3 in cortical and outer medullary collecting duct and AQP4 in inner medullary collecting duct. In contrast to AQP1 null mice, countercurrent multiplication and exchange mechanisms in AQP3/AQP4 null mice are basically intact. The polyuria in AQP3 null mice results in large part from the more than three-fold reduction in Pf of cortical collecting duct basolateral membrane (16). In addition, AQP2 expression is reduced in AQP3 null mice, which appears to be a maladaptive renal response seen in various forms of acquired polyuria. AQP4 null mice manifest only a mild impairment in maximal urinary concentrating ability (21), despite a four-fold reduced water permeability in microperfused inner medullary collecting duct (Fig. 1D) (22). These results support the conclusion that the majority of water is reabsorbed in the cortical and outer medullary segments of the collecting duct.
Mouse models of AQP2 gene mutation/deletion
Mutations in the AQP2 water channel produce the rare genetic disorder nephrogenic diabetes insipidus (NDI). Some AQP2 mutations produce non-X-linked NDI by a recessive mechanism, which involves defective mutant AQP2 protein folding/function; a few AQP2 mutations produce a dominant form of NDI resulting from ER/Golgi interactions between wildtype and mutant AQP2 that prevent plasma membrane targeting of wildtype AQP2 (reviewed in ref. 23). We have been interested in ‘protein folding’ diseases, such as cystic fibrosis caused by the ΔF508 mutation, which produces defective cell function by retention of the misfolded mutant protein at the endoplasmic reticulum and consequent prevention of plasma membrane expression. An emerging paradigm is the therapy of protein folding diseases by chemical or molecular chaperones, which may facilitate folding of the mutant protein by direct binding and/or modulation of components of the molecular quality control machinery. T126M is one of several AQP2 mutations that cause autosomal recessive NDI (24). Analysis of AQP2-T126M folding at the endoplasmic reticulum in mammalian cell culture models indicated ER retention, with remarkable resistance to membrane extraction by non-ionic detergents (25). Interestingly, despite it is presumed misfolding, the endoplasmic reticulum-retained AQP2-T126M functions as a water channel, suggesting that maneuvers which correct AQP2 trafficking would restore water permeability in collecting duct cells. One effective strategy to correct the trafficking defect involved the use of 'chemical chaperones' (26). Growth of AQP2-T126M-expressing cells in medium containing glycerol or trimethyamine-N-oxide for 48 hours corrected AQP2-T126M mistrafficking as assessed by localization studies and restoration of cell membrane water permeability.
We have focused attention on mouse models of NDI caused by the AQP2-T126M mutation in order to test drugs that might correct its cellular misprocessing. Initially, an AQP2-T126M knock-in mouse model was generated by targeted gene replacement using a Cre-loxP strategy in which the targeted gene locus contained an engineered T126M mutation (Fig. 2A) (27). Unfortunately, though the homozygous mutant mice appeared normal just after birth, they failed to thrive (mice at age 5 days shown in Fig. 2B, left) and generally died in the first week of life. As expected from cell culture studies, immunoblot analysis of kidneys of the homozygous mutant mice confirmed complex glycosylation of wildtype AQP2, but mainly endoglycosidase H-sensitive core glycosylation of AQP2-T126M, indicating ER-retention (Fig. 2B, right). Urine/serum analysis showed a urinary concentrating defect with serum hyperosmolality and low urine osmolality. Kidneys from mutant mice showed collecting duct dilatation and papillary atrophy, probably resulting from the marked polyuria. Though these mice provided in vivo verification of the expected defect in AQP2-T126M processing, their early mortality precluded analysis of protein folding ‘corrector’ therapy.
Figure 2. AQP2 transgenic mouse models of nephrogenic diabetes insipidus.
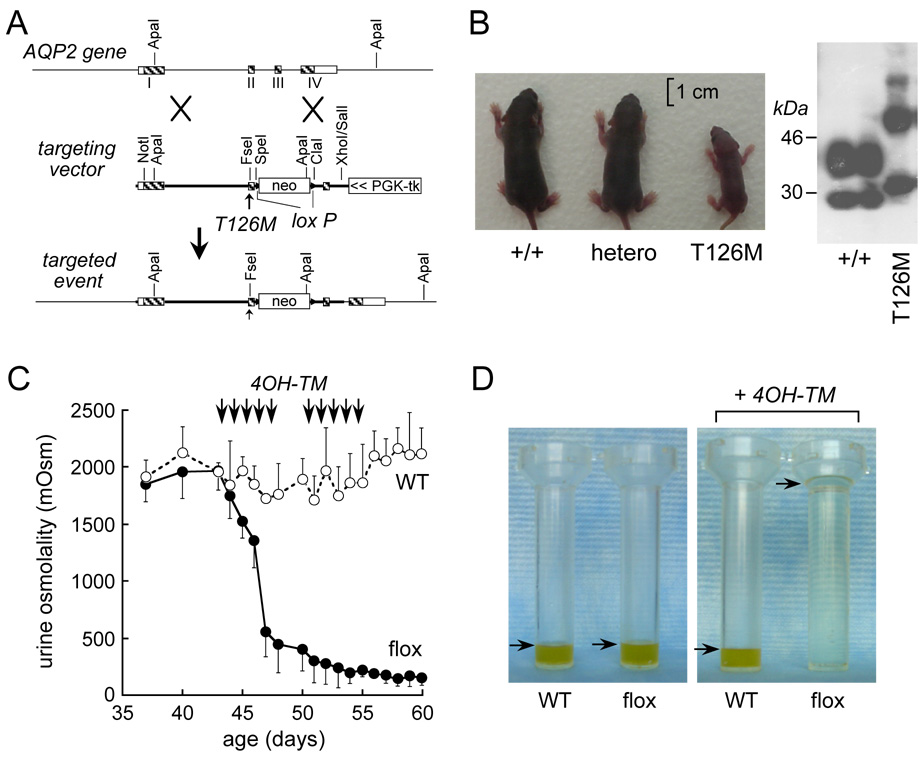
A. Targeting strategy for introduction of the T126M mutation into AQP2 gene replacement. Homologous recombination results in replacement of the indicated segment (thick line) of the AQP2 gene by a 1.8-kb neomycin selection cassette flanked by loxP sites. B. (left) Photograph of mice of indicated genotypes at 5 days after birth. (right) Immunoblot of kidney homogenates. C. Urine osmolality in tamoxifen-treated wildtype mice (open circles) and AQP2flox mice (filled circles) given free access to food and water (S.E.). Arrows indicate tamoxifen injections. D. Twenty-four hour urine output in untreated (left) and tamoxifen-treated (right) WT and AQP2flox mice (arrows indicate urine level). Adapted from refs. 27, 28.
As a first step in developing an AQP2-T126M ‘conditional knock-in’ model of NDI, we generated an inducible mouse model of AQP2 gene deletion (‘conditional knock-out’ mouse) manifesting severe polyuria in adult mice (28). LoxP sequences were inserted into introns 1 and 2 in the mouse AQP2 gene by homologous recombination in embryonic stem cells. Mating of germ-line AQP2-loxP mice with tamoxifen-inducible Cre-expressing mice produced offspring with inducible homozygous Cre-AQP2-loxP, which had normal phenotype. Tamoxifen administration resulted in Cre recombinase expression and AQP2 gene excision, resulting in undetectable full-length AQP2 transcript in kidney and >95 % reduction in immunoreactive AQP2 protein. The tamoxifen-treated mice had severe polyuria and inability to concentrate their urine in response to water deprivation (Fig. 2C, 2D). Importantly, unlike the neonatal polyuric mice the adult polyuric mice survived well. Recently, we used a novel ‘conditional gene knock-in’ strategy to generate adult, AQP2-T126M mutant mice (unpublished data). Mice heterozygous separately for floxed wild-type AQP2 and AQP2-T126M were bred to produce hemizygous mice containing a floxed wild-type AQP2 allele and a mutant AQP2-T126M allele. Conditional deletion of the wild-type AQP2 gene in adult mice by tamoxifen administration produced mice expressing only the mutant AQP2-T126M protein. The conditional knock-in adult mice showed polyuria, urinary hypoosmolality and ER-retention of AQP2-T126M in collecting duct, as expected, and so should be useful for testing of protein folding ‘corrector’ therapies.
Several other mouse models of NDI caused by AQP2 deletion/mutation have been generated recently. Collecting duct-specific AQP2 gene knockout produced a curious phenotype of severe NDI, as anticipated, through the mice were able to survive until adulthood (29). It is unclear whether the ability of mice to survive in this model is related to incomplete excision of the AQP2 gene in collecting duct, to strain or environment differences, or to some unique function of AQP2 in connecting tubule. As another model of autosomal recessive NDI, AQP2-F204V mutant mice were identified by forward genetic screening of ethylnitrosourea-mutagenized mice (30). AQP2-F204V is relatively mild mutation compared with AQP2-T126M (or the null mutant), permitting these mice to survive beyond the neonatal period with a milder form of NDI. Last, a model of autosomal dominant NDI was produced by knock-in of an AQP2 with a C-terminus frame shift mutation (31). The heterozygous mice were polyuric, as anticipated, and showed AQP2 mislocalization in kidney collecting duct with basolateral membrane targeting. Though these various mouse models are unlikely to yield new insights into the renal physiology of NDI caused by AQP2 impairment, since other animal models of NDI as well as humans with NDI have been studied extensive, they should have application in testing of new therapies.
AQP7 and renal glycerol clearance
AQP7 is expressed selectively in the apical membrane of epithelial cells in the relatively short S3 straight segment of the proximal tubule. AQP7, like AQP3, is an ‘aquaglyceroporin’ that transports both water and glycerol, whereas most of the other AQPs primarily transport water. A curious phenotype was found for mice lacking AQP7 (32). While urinary concentrating function was intact, there was increased urinary glycerol clearance. Urinary glycerol concentration in AQP7 null mice was remarkably greater than that in wildtype mice (1.7 vs. 0.005 mg/ml), suggesting that AQP7 in proximal straight tubule is required for reabsorption of filtered glycerol. However, the large magnitude of this difference seems hard to reconcile with the very limited AQP7 expression, and the significance of this observations is unclear because serum glycerol concentration was not reduced in the AQP7 null mice. Evidence was reported that AQP7 may play a small, supportive role in the urinary concentrating mechanism, as osmotic water permeability in apical membrane vesicles from proximal tubule of AQP7 deficient kidneys was slightly reduced by ~10%, and urinary concentrating ability of AQP1/AQP7 double knockout mice was reduced compared to that of AQP1 null mice. The absence of significantly impaired urinary concentrating function in AQP7 deficiency in mice may be related to the co-expression of AQP4 in mice in the S3 segment of proximal tubule, as well as to expression of AQP1 throughout the proximal tubule. Direct measurements of transepithelial water and glycerol permeability in proximal straight tubule are needed to further understand the renal phenotype of AQP7 null mice, as well as rigorous glycerol clearance measurements.
AQP11 and renal cystic disease
AQP11 has a somewhat different amino acid sequence from other AQPs in that it lacks one of the two conserved asparagine-proline-alanine (NPA) motifs. Whether AQP11 functions as a water channel has been debated, though a recent report showed a small increase in water permeability in liposomes reconstituted with recombinant AQP11 (33). In kidney, AQP11 appears to be expressed in intracellular vesicles in proximal tubule (34), though available antibodies are limited in their specificity. AQP11 null mice manifest an interesting phenotype of vacuolization and cyst formation in proximal tubule, similar in some respects to polycystic kidney disease. Over the first few weeks of life the kidneys in AQP11 null mice become enlarged, with cysts occupying the whole cortex (Fig. 3), leading to renal failure and death. Prior to the appearance of cysts, the proximal tubule epithelium contains vacuoles. The mechanisms responsible for the association of AQP11 deficiency with renal cystogenesis remain unknown, though their elucidation might offer new insights on how and why renal cysts form in polycystic kidney disease. We found that primary cultures of proximal tubule cells from neonatal kidneys of AQP11 null mice manifest a mild defect in endosomal acidification (34). However, it is unclear whether defective endosomal acidification is involved in cyst pathogenesis or constitutes an epiphenomenon. Further analysis is needed of the cellular functions of AQP11 and the mechanisms by which AQP11 deletion in mice produces renal cystic disease.
Figure 3. Renal cyst formation and defective endosomal acidification in proximal tubule cells in AQP11 deficiency.
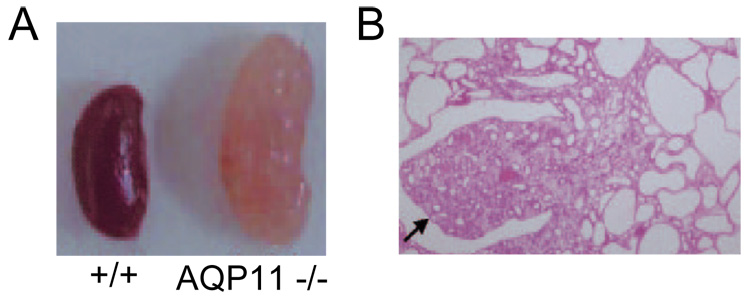
A. Kidney of 30 day old AQP11 null mouse showing numerous large cysts. B. Kidney histology (arrow indicates papilla). Adapted from ref. 34.
AQPs and cell migration
As reviewed recently (35), we discovered a novel cellular role for AQPs in cell migration, which was initially demonstrated in endothelial cells and various transfected cells (36), and subsequently found in brain astroglial cells (37), tumor cells (38), corneal epithelium cells (39) and epidermal cells (40). The general finding, in multiple cell types, is that AQP expression increases cell water permeability and migration toward a chemotactic stimulus, with little or no effect on cell size, adherence or other properties. Demonstrated consequences of AQP-facilitated cell migration include AQP1-facilitated tumor angiogenesis, AQP4-facilitated glial scar formation, AQP-facilitated tumor invasion and metastasis, and AQP-facilitated wound healing. In multiple cell types AQP expression is polarized to the leading edge of migrating cells and associated with increased number and dynamics of membrane protrusions, or lamellipodia. We proposed that AQP-dependent water transport at the leading edge of migrating cells facilitates water entry in lamellipodia, as driven by actin depolymerization and ion transport, resulting in more rapid lamellipodial extension and consequent cell migration (Fig. 4A). Additionally, increased cell membrane water permeability could facilitate cell migration through narrow spaces, such as in the extracellular space in brain or across vascular endothelia, where marked changes in cell shape and volume occur.
Figure 4. AQP1-facilitated migration of proximal tubule epithelial cells.
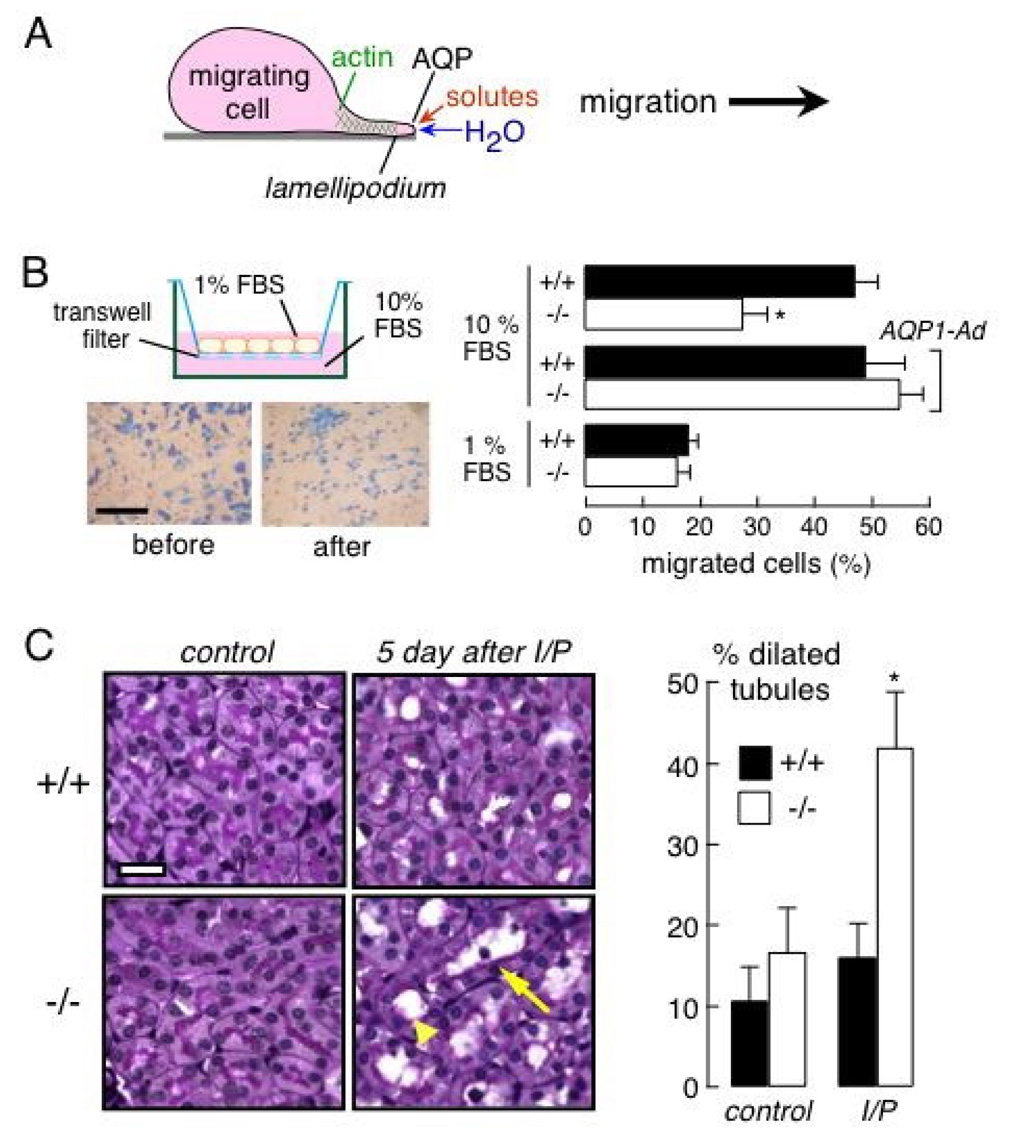
A. Proposed mechanism for AQP-facilitated cell migration showing water transport into lamellipodia at the leading edge of migrating cells. See text for further explanations. B. (left) In vitro Boyden chamber assay of proximal tubule cell migration. (top) Schematic of modified Boyden chamber containing a transwell porous membrane filter. Cells migrate from top to bottom in response to a chemotactic stimulus. (bottom) Photographs of adherent proximal tubule cells at the completion of an assay, shown before scraping (left), and after scraping the upper surface (right) to visualize migrated cells. Bar, 100 µm. (right) Percentage of migrated cells at 6 h after cell plating (S.E., * p < 0.01). The bottom chamber contained 10% or 1% FBS. ‘AQP1-Ad’ indicates adenoviral-mediated AQP1 gene delivery. C. Ischemia-reperfusion model of acute renal tubular injury. (left) Periodic acid-Schiff and hematoxylin staining of kidney sections from control wildtype and AQP1 null mice (left), and at 5 days after ischemia/reperfusion (I/P) (right). Bar, 20 µm. Arrowhead, dilated tubule; arrow, degenerated tubule. (right) Percentage of proximal tubules with inner diameter greater than 5 µm (S.E., * p < 0.05). Adapted from ref. 41.
AQP-dependent cell migration may be important in the renal response to various types of injury and in the spread of renal tumors. In a proof-of-concept study, migration was compared in primary cultures of proximal tubule cells from wildtype and AQP1 null mice (41). The cultures were indistinguishable in their appearance, growth/proliferation and substrate adherence, though as expected the AQP1-deficient cells had reduced plasma membrane water permeability. Migration of AQP1-deficient cells was reduced by more than 50% compared to wild-type cells, as measured in a Boyden chamber in the presence of a chemotactic stimulus (Fig. 4B) as well as in a scratch assay of wound healing. Adenoviral-mediated introduction of AQP1 in the AQP1-deficient cells corrected their defects in water permeability and migration. The potential relevance of these cell culture findings to the intact kidney was tested in an in vivo model of acute tubular injury caused by 30 min renal artery occlusion-reperfusion. At 5 days following ischemia-reperfusion, kidneys in AQP1 null mice showed remarkably greater tubular injury than in wildtype mice (Fig. 4C), with F-actin disorganization. These findings suggest the involvement of AQP1 in the response of the proximal tubule to injury. Whether other AQPs are involved in the renal response to various acute and chronic insults remained to be determined.
A role for AQP3 in epithelial cell proliferation
New evidence from extrarenal tissues supports a role for AQP3 in cell proliferation that may be of relevance in kidney collecting duct epithelium, where AQP3 is strongly expressed in the basolateral membrane. AQP3 null mice manifest defective cell proliferation in multiple tissues where AQP3 is normally expressed. At the ocular surface, where AQP3 is expressed in corneal and conjunctival surface epithelium, AQP3 deletion results in delayed restitution of the corneal surface after denudation, and impaired proliferation of corneal epithelial cell (39). In colon, where AQP3 is expressed in enterocytes, AQP3 deficiency results in increased mortality and colonic destruction in chemical models of colitis, with impaired proliferation of colonic epithelial cells (42). A similar result was found for healing of skin wounds, where AQP3 deficiency impairs wound healing and epidermal cell proliferation (40). In each case glycerol administration restored, at least partially, the impairment in cell proliferation. Recently, we discovered a remarkable phenotype using a skin tumor model of epidermal cell proliferation in which cutaneous papillomas were produced by repeated inducer-promoter exposure (43). Mice lacking AQP3 failed to produce papillomas following the same treatment in which nearly all wildtype mice produced multiple papillomas. Biochemical studies suggested that AQP3-mediated glycerol transport was responsible for the impaired proliferation phenotype by a mechanism involving reduced glycerol content in AQP3-deficient epidermis resulting in reduced cellular glycerol metabolites and ATP, with consequent impaired MAP kinase signaling and lipid biosynthesis. Whether this mechanism established in epidermis applies to other cell types in which AQP3 is expressed remains to be determined, as does the relevance to the kidney of the defective cell proliferation phenotype in AQP3-deficient skin, eye and colon.
Role of aquaporins in peritoneal fluid transport
The peritoneal cavity is lined by a membranous barrier that provides a large surface for fluid movement between peritoneal capillaries and the peritoneal cavity. Although the peritoneal cavity normally contains little fluid, marked ascites can accumulate in conditions associated with reduced serum oncotic pressure, increased portal venous pressure, or peritoneal cavity inflammation/infection. The large peritoneal surface is exploited in peritoneal dialysis, where water and solutes are extracted from blood by repeated infusion and removal of dialyzate solutions into the peritoneal cavity. Kinetic models of peritoneal dialysis postulate distinct classes of 'pores' that transport water and solutes to various extents, including an ultrasmall, 'water-only' pore. Early expression studies and measurements of mercurial inhibition of peritoneal water transport suggested that the water-only pore is AQP1 (44), which has been localized to capillary endothelia and mesangium near the peritoneal luminal surface.
Measurements in AQP1 null mice provided direct evidence for involvement of AQP1 in peritoneal water transport (45). Hyperosmolar saline (saline + 300 mM sucrose) was infused rapidly into the peritoneal cavity via a catheter and serial fluid samples were withdrawn to quantify the time course of osmotic equilibration, using albumin as a volume marker. The albumin dilution data showed a ~2.5-fold reduced water flux in the AQP1 null mice (Fig. 5). As expected, because AQP1 is a water-only pathway, the transport of radiolabeled urea was not affected by AQP1 deletion. A more recent study on clinically relevant aspects of AQP1 in peritoneal dialysis focused on sodium sieving and peritoneal water transport in wildtype vs. AQP1 null mice during peritoneal dialysis (46). The results supported the conclusion that AQP1 is the ultrasmall pore pathway that accounts for approximately 50% of ultrafiltration in peritoneal dialysis in mice.
Figure 5. AQP1-facilitated peritoneal fluid absorption in peritoneal dialysis.
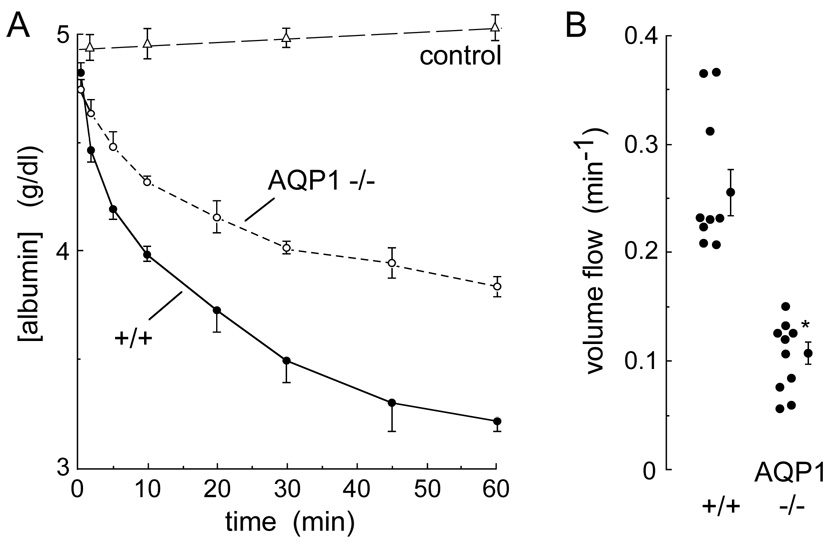
Osmotically induced water transport. The peritoneal cavity was infused with 2 ml of a physiological solution containing 300 mM sucrose and 5 g/dl albumin (600 mosM), and serial fluid samples were obtained. Control measurements were done with isosmolar instillate (‘control’), omitting sucrose. A. Time course of albumin concentration in peritoneal fluid (S.E.) from wildtype and AQP1 null mice. B. Summary of initial volume flow rates in each mouse studied. * p < 0.001. Adapted from ref. 45.
Summary and directions
Renal phenotype studies of AQP knockout mice have confirmed the predicted roles of AQPs in the urinary concentrating mechanism, and have suggested potential new roles of AQPs in renal cell migration and proliferation, glycerol clearance, and cystogenesis. Several transgenic mouse models of AQP2 mutations are now available for testing of potential drug therapies. Although NDI is an extremely rare disorder and so unlikely to be a candidate for development of ‘new chemical entity’ drugs, existing or new approved drugs developed for other indications, such as other diseases of protein folding, may be of benefit in NDI. Further work in renal AQPs is needed to elucidate the cellular mechanisms of their new roles that are unrelated to transcellular water transport. Last, and potentially of greatest significance, is the development of small-molecule inhibitors and inducers of renal AQPs for therapy of a variety of disorders such as dysnatremias and total body water overload. Selective inhibitors of renal AQPs will also be useful in confirming phenotype data from knockout mice, without the potentially confounding effects of chronic AQP deletion on expression of other genes.
Acknowledgments
Supported by grants DK35124, DK72517, EY13574, EB00415, HL59198 and HL73856 from the National Institutes of Health, and Research Development Program and Drug Discovery grants from the Cystic Fibrosis Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Nielsen S, Frokiaer J, Marples D, et al. Aquaporins in the kidney: from molecules to medicine. Physiol Rev. 2002;82:205–244. doi: 10.1152/physrev.00024.2001. [DOI] [PubMed] [Google Scholar]
- 2.Verkman AS. More than just water channels: unexpected cellular roles of aquaporins. J Cell Sci. 2005;118:3225–3232. doi: 10.1242/jcs.02519. [DOI] [PubMed] [Google Scholar]
- 3.Zhang D, Vetrivel L, Verkman AS. Aquaporin deletion in mice reduces intraocular pressure and aqueous fluid production. J Gen Physiol. 2002;119:561–569. doi: 10.1085/jgp.20028597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oshio K, Song Y, Verkman AS, et al. Reduced intraventricular pressure and cerebrospinal fluid production in mice lacking aquaporin-1 water channels. Faseb J. 2005;18:76–78. doi: 10.1096/fj.04-1711fje. [DOI] [PubMed] [Google Scholar]
- 5.Ma T, Song Y, Gillespie A, Carlson EJ, et al. Defective secretion of saliva in transgenic mice lacking aquaporin-5 water channels. J Biol Chem. 1999;274:20071–20074. doi: 10.1074/jbc.274.29.20071. [DOI] [PubMed] [Google Scholar]
- 6.Song Y, Verkman AS. Aquaporin-5 dependent fluid secretion in airway submucosal glands. J Biol Chem. 2001;276:41288–41292. doi: 10.1074/jbc.M107257200. [DOI] [PubMed] [Google Scholar]
- 7.Verkman AS, Binder DK, Block O, et al. Three distinct roles of aquaporin-4 in brain function revealed by knockout mice. Biochem Biophys Acta. 2006;1758:1085–1093. doi: 10.1016/j.bbamem.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 8.Manley GT, Fujimura M, Ma T, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nature Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- 9.Papadopoulos MC, Manley GT, Krishna S, Verkman AS. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. Faseb J. 2004;18:1291–1293. doi: 10.1096/fj.04-1723fje. [DOI] [PubMed] [Google Scholar]
- 10.Binder DK, Yao X, Sick TJ, et al. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia. 2006;53:631–636. doi: 10.1002/glia.20318. [DOI] [PubMed] [Google Scholar]
- 11.Padmawar P, Yao X, Bloch O, et al. K+ waves in brain cortex visualized using a long-wavelength K+-sensing fluorescent indicator. Nature Meth. 2005;2:825–827. doi: 10.1038/nmeth801. [DOI] [PubMed] [Google Scholar]
- 12.Hara M, Verkman AS. Glycerol replacement corrects defective skin hydration, elasticity, and barrier function in aquaporin-3-deficient mice. Proc Natl Acad Sci U S A. 2003;100:7360–7365. doi: 10.1073/pnas.1230416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hara-Chikuma M, Sohara E, Rai T, et al. Progressive adipocyte hypertrophy in aquaporin-7 deficient mice: Adipocyte glycerol permeability as a novel regulator of fat accumulation. J Biol Chem. 2005;280:15493–15496. doi: 10.1074/jbc.C500028200. [DOI] [PubMed] [Google Scholar]
- 14.Hibuse T, Maeda N, Funahashi T, et al. Aquaporin 7 deficiency is associated with development of obesity through activation of adipose glycerol kinase. Proc Natl Acad Sci U S A. 2005;102:10993–10998. doi: 10.1073/pnas.0503291102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma T, Yang B, Gillespie A, et al. Severely impaired urinary concentrating ability in transgenic mice lacking aquaporin-1 water channels. J Biol Chem. 1998;273:4296–4299. doi: 10.1074/jbc.273.8.4296. [DOI] [PubMed] [Google Scholar]
- 16.Ma T, Song Y, Yang B, et al. Nephrogenic diabetes insipidus in mice deficient in aquaporin-3 water channels. Proc Natl Acad Sci USA. 2000;97:4386–4391. doi: 10.1073/pnas.080499597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chou CL, Knepper MA, Van Hoek AN, et al. Reduced water permeability and altered ultrastructure in thin descending limb of Henle in aquaporin-1 null mice. J Clin Invest. 1999;103:491–496. doi: 10.1172/JCI5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schnermann J, Chou CL, Ma T, et al. Defective proximal tubular fluid reabsorption in transgenic aquaporin-1 null mice. Proc Natl Acad Sci USA. 1998;95:9660–9664. doi: 10.1073/pnas.95.16.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vallon V, Verkman AS, Schnermann J. Luminal hypotonicity in proximal tubules of aquaporin-1 knockout mice. Am J Physiol. 2000;278:F1030–F1033. doi: 10.1152/ajprenal.2000.278.6.F1030. [DOI] [PubMed] [Google Scholar]
- 20.Pallone TL, Edwards A, Ma T, et al. Requirement of aquaporin-1 for NaCl driven water transport across descending vasa recta. J Clin Invest. 2000;105:215–222. doi: 10.1172/JCI8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma T, Yang B, Gillespie A, et al. Generation and phenotype of a transgenic knock-out mouse lacking the mercurial-insensitive water channel aquaporin-4. J Clin Invest. 1997;100:957–962. doi: 10.1172/JCI231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou CL, Ma T, Yang B, et al. Four-fold reduction in water permeability in inner medullary collecting duct of aquaporin-4 knockout mice. Am J Physiol. 1998;274:C549–C554. doi: 10.1152/ajpcell.1998.274.2.C549. [DOI] [PubMed] [Google Scholar]
- 23.Robben JH, Knoers NV, Deen PM. Cell biological aspects of the vasopressin type-2 receptor and aquaporin-2 water channel in nephrogenic diabetes insipidus. Am J Physiol. 2006;291:F257–F270. doi: 10.1152/ajprenal.00491.2005. [DOI] [PubMed] [Google Scholar]
- 24.Deen PM, Croes H, van Aubel RA, et al. Water channels encoded by mutant aquaporin-2 genes in nephrogenic diabetes insipidus are impaired in their cellular routing. J Clin Invest. 1995;95:2291–2296. doi: 10.1172/JCI117920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamarappoo BK, Verkman AS. Misfolding of mutant aquaporin-2 water channels in nephrogenic diabetes insipidus. J Biol Chem. 1999;274:34825–34831. doi: 10.1074/jbc.274.49.34825. [DOI] [PubMed] [Google Scholar]
- 26.Tamarappoo BK, Verkman AS. Defective trafficking of AQP2 water channels in nephrogenic diabetes insipidus and correction by chemical chaperones. J Clin Invest. 1998;101:2257–2263. doi: 10.1172/JCI2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang B, Gillespie A, Carlson EJ, et al. Early neonatal mortality in a transgenic AQP2 knock-in model of nephrogenic diabetes insipidus. J Biol Chem. 2001;276:2775–2779. doi: 10.1074/jbc.M008216200. [DOI] [PubMed] [Google Scholar]
- 28.Yang B, Zhou D, Qain L, et al. Mouse model of inducible nephrogenic diabetes insipidus produced by floxed aquaporin-2 gene deletion. Am J Physiol. 2006;291:F465–F472. doi: 10.1152/ajprenal.00494.2005. [DOI] [PubMed] [Google Scholar]
- 29.Rojek A, Füchtbauer EM, Kwon TH, et al. Severe urinary concentrating defect in renal collecting duct-selective AQP2 conditional-knockout mice. Proc Natl Acad Sci U S A. 2006;103:6037–6042. doi: 10.1073/pnas.0511324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lloyd DJ, Hall FW, Tarantino LM, et al. Diabetes insipidus in mice with a mutation in aquaporin-2. PLoS Genet. 2005;19(1):e20. doi: 10.1371/journal.pgen.0010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sohara E, Rai T, Yang SS, et al. Pathogenesis and treatment of autosomal-dominant nephrogenic diabetes insipidus caused by an aquaporin 2 mutation. Proc Natl Acad Sci U S A. 2006;103:14217–14222. doi: 10.1073/pnas.0602331103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sohara E, Rai T, Miyazaki J, et al. Defective water and glycerol transport in the proximal tubules of AQP7 knockout mice. Am J Physiol. 2005;289:F1195–F1200. doi: 10.1152/ajprenal.00133.2005. [DOI] [PubMed] [Google Scholar]
- 33.Yakata K, Hiroaki Y, Ishibashi K, et al. Aquaporin-11 containing a divergent NPA motif has normal water channel activity. Biochim Biophys Acta. 2007;1768:688–693. doi: 10.1016/j.bbamem.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 34.Morishita Y, Matsuzaki T, Hara-Chikuma M, et al. Disruption of aquaporin-11 produces polycystic kidneys following vacuolization of the proximal tubule. Mol Cell Biol. 2005;25:7770–7779. doi: 10.1128/MCB.25.17.7770-7779.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papadopoulos MC, Saadoun S, Verkman AS. Aquaporins and cell migration. Pflugers Arch. 2008 doi: 10.1007/s00424-007-0357-5. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saadoun S, Papadopoulos MC, Hara-Chikuma M, Verkman AS. Impairment of angiogenesis and cell migration by targeted of aquaporin-1 gene disruption. Nature. 2005;434:786–792. doi: 10.1038/nature03460. [DOI] [PubMed] [Google Scholar]
- 37.Auguste K, Jin J, Uchida K, et al. Greatly impaired migration of implanted aquaporin-4 deficient astroglial cells in mouse brain toward a site of injury. Faseb J. 2007;21:108–116. doi: 10.1096/fj.06-6848com. [DOI] [PubMed] [Google Scholar]
- 38.Hu J, Verkman AS. Increased migration and metastatic potential of tumor cells expressing aquaporin water channels. Faseb J. 2006;20:1892–1894. doi: 10.1096/fj.06-5930fje. [DOI] [PubMed] [Google Scholar]
- 39.Levin MH, Verkman AS. Aquaporin-3-dependent cell migration and proliferation during corneal re-epithelialization. Invest Opthalmol Vis Sci. 2006;47:4365–4372. doi: 10.1167/iovs.06-0335. [DOI] [PubMed] [Google Scholar]
- 40.Hara-Chikuma M, Verkman AS. Aquaporin-3 facilitates epidermal wound healing. J Mol Med. 2008 doi: 10.1007/s00109-007-0272-4. In press. [DOI] [PubMed] [Google Scholar]
- 41.Hara-Chikuma M, Verkman AS. Aquaporin-1 facilitates migration of epithelial cells in kidney proximal tubule. J Am Soc Nephrol. 2006;17:39–45. doi: 10.1681/ASN.2005080846. [DOI] [PubMed] [Google Scholar]
- 42.Thiagarajah JR, Zhao D, Verkman AS. Impaired enterocyte proliferation in aquaporin-3 deficiency in a mouse model of colitis. Gut. 2008 doi: 10.1136/gut.2006.104620. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hara-Chikuma M, Verkman AS. Prevention of skin carcinogenesis by targeted aquaporin-3 gene deletion. Mol Cell Biol. 2008 doi: 10.1128/MCB.01482-07. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carlsson O, Nielsen S, Zakaria R, et al. In vivo inhibition of transcellular water channels (aquaporin-1) during acute peritoneal dialysis in rats. Am J Physiol. 1996;271:H2254–H2262. doi: 10.1152/ajpheart.1996.271.6.H2254. [DOI] [PubMed] [Google Scholar]
- 45.Yang B, Folkesson HG, Yang J, et al. Reduced water permeability of the peritoneal barrier in aquaporin-1 knockout mice. Am J Physiol. 1999;276:C76–C81. doi: 10.1152/ajpcell.1999.276.1.C76. [DOI] [PubMed] [Google Scholar]
- 46.Ni J, Verbavatz JM, Rippe A, et al. Aquaporin-1 plays an essential role in water permeability and ultrafiltration during peritoneal dialysis. Kidney Int. 2006;69:1518–1525. doi: 10.1038/sj.ki.5000285. [DOI] [PubMed] [Google Scholar]