

Abstract
Single-stranded DNA (ssDNA) has many applications in molecular biology and biotechnology. The conventional method for the preparation of ssDNA from phagemids is laborious, costly, and inefficient. Here, we describe an integrated protocol for consistent production of phagemid ssDNA from a bacteria/phagemid/help phage complex and rapid isolation and purification of the ssDNA with a silica column followed with Duplex-Specific Nuclease digestion. The major advantages of our method are the expediency, low-cost, and consistent yield of highly pure ssDNA that is suitable for direct sequencing and other applications. This method is especially useful for large-scale preparation of high quality ssDNA.
Keywords: single-stranded DNA, isolation, purification, phagemid, Duplex-specific nuclease, DNA mismatch repair, green fluorescent protein
Whereas the double-stranded DNA (dsDNA) is the most commonly used DNA molecules, many applications and technologies in molecular biology and biotechnology require the use of single-stranded DNA (ssDNA) [1]. Such applications include, but not limited to, the preparation of a template DNA for sequencing [2], site-directed mutagenesis [3], correction of frameshift mutations [4], and strand-specific DNA synthesis. Standard approaches for the preparation of ssDNA most frequently utilize phagemids [5], which are chimeric vectors derived from the single-stranded bacteriophage M13, fd, or f1 and can replicate as plasmids in bacterial hosts. When their host bacteria are infected by a helper bacteriophage, the gene II product encoded by the helper phage introduces a strand-specific nick into the intergenic region of the phagemids initiating a rolling-circle-like replication of one strand [6]. Thereafter, single-stranded copies of the phagemid DNA are packaged into bacteriophage particles and extruded into the medium. With a polyethylene glycol (PEG) precipitation procedure, these phage particles containing phagemid ssDNA can be recovered from the supernatant, and subsequently extracted and purified by the classical phenol/chloroform and ethanol precipitation procedure [6]. Although this protocol has been widely used for sometime, there are several drawbacks. First, multiple factors can significantly affect the yield of ssDNA. They include the nature of the helper phage, the density of the culture at the time of infection, the multiplicity of infection of the helper phage, and the length of time after phage infection [7]. Because it is not easy to control these factors, the protocol can yield poor and sometimes irreproducible results. Furthermore, the use of helper phage can be costly and tedious. Second, the conventional phenol-chloroform method for the extraction of ssDNA from phagemids is not only laborious but also poses other disadvantages such as incomplete lyse of phage particles, contamination with residual protein, and hazardous concern on the chemicals. Third, many phagemid host strains such as JM109 and XL1-Blue can release their chromosomal and phagemid DNA into the medium due to cell lysis [8] resulting in the contamination of ssDNA with chromosomal DNA and double-stranded plasmid DNA. As such, ssDNA without further purification may not satisfy the requirement of various applications. Hence, the conventional protocol for the extraction of phagemid ssDNA is still tedious, laborious, costly, and inefficient.
Here, we describe an integrated method for the production, isolation and purification of ssDNA from phagemids, which provides a complete solution to the deficiencies of the conventional method. The novelty of our integrated method resides in (1) the generation of a bacterium clone stably transformed with a phagemid and a helper phage; (2) the isolation of ssDNA with a silica column; and (3) the purification of ssDNA with Duplex-Specific Nuclease. This rapid and efficient method for the preparation of highly pure ssDNA from phagemids was developed in connection with one of our ongoing projects where high quality and large quantities of ssDNA from a phagemid called pGEM5Z(+)-EGFP was needed for making a heteroduplex plasmid used for the measurement of DNA mismatch repair activity in live cells [9]. Here, we use the phagemid pGEM5Z(+)-EGFP as an example to describe the three innovative approaches in our method for the generation of ssDNA from pGEM5Z(+)-EGFP.
In order to consistently produce large amount of ssDNA and save the cost for the helper phage, we attempted to produce a clone of E. coli JM109, which would be stably transformed with pGEM5Z(+)-EGFP and infected with the helper phage M13KO7. A similar approach was reported by Jupin and Gronenborn [7], in which an E. coli strain was first infected with M13KO7 and then made competent for stable transformation with phagemids. While this approach was reported to be easy and yielded abundant and reproducible amount of phagemid ssDNA, we found that the infection with the helper phage often led to a low survival rate when the JM109 bacteria were made competent and transformed with a phagemid resulting in extremely low transformation efficiency. Hence, it was not easy to obtain individual clones of the bacteria/phage/phagemid complex with this procedure. We adopted the concept and generated a bacteria/phagemid/phage complex. We first transformed competent JM109 cells (Takara Co., Ltd., Tokyo, Japan) with pGEM5Z(+)-EGFP and selected transformed clones on an ampicillin-containing LB plate. A single colony of JM109/pGEM5Z(+)-EGFP cells was then picked from the LB plate, resuspended in 5 ml of LB broth containing 50 mg/L ampicillin and incubated at 37°C and 200 rpm. M13KO7 helper phage (New England Biolabs Inc., Beverly, Massachusetts, USA) was added to the culture at a multiplicity of infection of 10 when the A600 of the culture reached 0.05. Incubation at 37 °C was continued for another 60 min and kanamycin was added to a final concentration of 75 mg/L. After an additional incubation for several hours when the culture reached logarithmic growth phase, twenty microliters of the culture solution were spread onto a selective LB plate containing 50 mg/L ampicillin and 75 mg/L kanamycin. A parallel control plate was prepared by spreading 20 ul of JM109/pGEM5Z(+)-EGFP bacterial culture onto the two antibiotic-containing LB plate. Because M13KO7 confers kanamycin resistance and pGEM5Z(+)-EGFP confers ampicillin resistance, numerous colonies grew out in the plate spread with JM109/pGEM5Z(+)-EGFP/M13KO7 culture, but none in the control plate spread with JM109/pGEM5Z(+)-EGFP culture (Figure 1). The individual colonies of JM109/pGEM5Z(+)-EGFP/M13KO7 complex can be directly inoculated into liquid medium and grown overnight at 37 °C with strong agitation for ssDNA production or for storage at −80°C for future use. Using these individual clones of JM109/pGEM5Z(+)-EGFP/M13KO7 complex, we were able to prepare large quantities of ssDNA of pGEM5Z(+)-EGFP with reduced cost and time, and increased consistency due to the elimination of helper phage infection step.
Figure 1. Generation of individual colonies of JM109/pGEM5Z(+)-EGFP/M13KO7 complex.
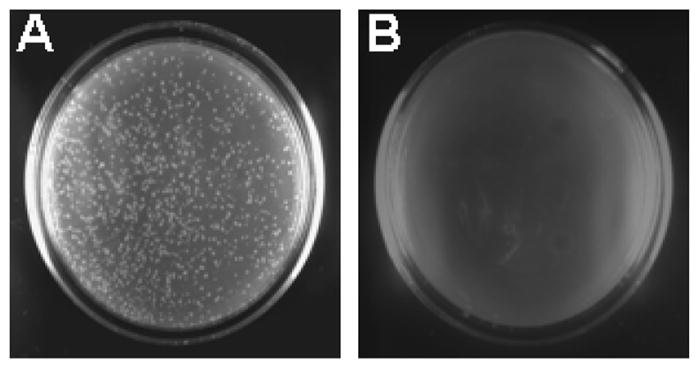
Twenty microliters of the culture of JM109/pGEM5Z(+)-EGFP/M13KO7 complex (A) or JM109/pGEM5Z(+)-EGFP (B) described in the text were spread on a LB plate containing 50 mg/L ampicillin and 75 mg/L kanamycin. After an overnight incubation at 37 °C, numerous colonies were formed in Plate A, but not in Plate B
To avoid the use of conventional phenol and chloroform extraction for the isolation of ssDNA from phage particles, we devised a procedure for isolating phagemid ssDNA using silica columns. The phage particles produced in a 50 ml overnight culture of JM109/pGEM5Z(+)-EGFP/M13KO7 complex were recovered from the supernatant by the classical PEG precipitation procedure [6] and lysed in 2 ml of a disruption buffer (5 M Guanidinium Hydrochloride, 1% Triton X-100, and 10 mM MOPS, pH 6.5) with a short incubation of 5 min at room temperature. A silica column (Tiangen Biotech Co., LTD, Beijing, China) with a maximum DNA adsorption quantity of 500 μg was placed inside a 50-ml Falcon tube. The cell lysate was poured into the column, which was then centrifuge at 10,000g for 2 min. The flow-through solution was discarded and the column was washed twice with 5 ml of 80% isopropanol and centrifugation at 10,000g for 2 min. The column was then transferred to a new Falcon tube and incubated with 1 ml of sterile TE buffer for 2–3 min to elute the bound DNA. The eluted DNA in the TE buffer was collected by centrifugation at 10,000g for 2 min. The 50 ml culture usually yielded a total 150–200 μg ssDNA. On the other hand, a parallel preparation of ssDNA from 50 ml of M13KO7-infected JM109/pGEM5Z(+)-EGFP culture following the conventional procedure [8] usually yielded no more than 50 μg ssDNA. Furthermore, the ssDNA isolated with the silica column method often appeared purer with less contaminants than that isolated with the conventional method after electrophoresis in agarose gel (Lanes 1 and 2 in Figure 2A), and the 260/280 absorbance ratio of the isolated ssDNA was usually below 1.6 with the conventional method and was always between 1.8 and 2.0 with the silica column method. In addition, the conventional phenol/chloroform extraction method can take 2 hr whereas the silica column method only takes about 15 min with a two to four-fold higher yield. Thus, this silica column-based method offers the advantages of speed and yield as well as safety and convenience.
Figure 2. Generation of highly pure ssDNA.
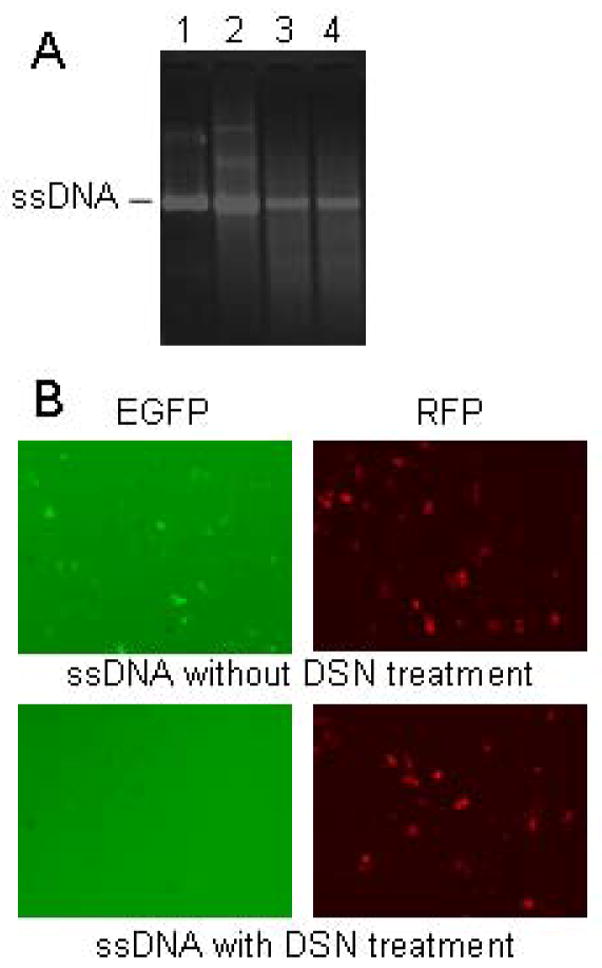
(A) Electrophoretic analysis of the ssDNA products. Lane1 is ssDNA isolated with silica column from the culture of JM109/pGEM5Z(+)-EGFP/M13KO7 complex. Lane2 is ssDNA isolated from M13KO7-infected JM109/pGEM5Z(+)-EGFP culture with the conventional method. Lanes 3 and 4 are the same ssDNA samples as Lanes 1 and 2, respectively, after treatment with DSN. (B) Fluorescent images of HCT116 cells co-transfected with ssDNA and pDsRed1-N1. ssDNA (3 μg) isolated from JM109/pGEM5Z(+)-EGFP/M13KO7 complex was treated with or without DSN, and then co-transfected with pDsRed1-N1 (1 μg) into HCT116 cells with Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The representative green and red fluorescent images were taken 24 hr later. Co-transfection with pDsRed1-N1 was used to ascertain that the transfection efficiencies were comparable.
Because ssDNA prepared from phagemids is often contaminated with certain amounts of chromosomal and double-stranded phagemid DNA due to the lysis of bacteria during the culture, when the ssDNA from pGEM5Z(+)-EGFP was used for the preparation of the heteroduplex EGFP plasmid in the DNA mismatch repair assay as we reported previously [9], the contamination with double-stranded pGEM5Z(+)-EGFP causes overestimation of MMR activity due to the expression of the enhanced green fluorescence protein (EGFP) from the double-stranded plasmid rather than from the repaired heteroduplex plasmid. Thus, the contamination with dsDNA can significantly confound experimental outcomes from the use of ssDNA prepared from phagemids. To remove the dsDNA (mainly bacteria chromosomal DNA and plasmid) from the ssDNA samples from pGEM5Z(+)-EGFP, we incubated 0.25 U of a duplex-specific nuclease (DSN) with 100 μg of ssDNA product at 70°C for 5 min. DSN (Evrogen, Moscow, Russia) is an enzyme purified from hepatopancreas of the kamchatka crab. It exhibits strong cleavage preference for dsDNA substrates [10]. The DSN-treated ssDNA was compared to that without DSN treatment by gel electrophoresis (Figure 2A). It is clear that high molecular weight dsDNA contaminants shown above the ssDNA band in Lanes 1 and 2 were no longer visible after DSN treatment as shown in Lanes 3 and 4 indicating efficient elimination of dsDNA. These results have been repeated for more than 20 times in our laboratory thus far. Because the phagemid pGEM5Z(+)-EGFP is a mammalian EGFP expression plasmid, its contamination in the ssDNA preparation will generate EGFP expression when the ssDNA samples are transfected into mammalian cells. Therefore, we also compared the purity of ssDNA of pGEM5Z(+)-EGFP before and after DSN treatment by transfecting it into the human colon carcinoma HCT116 cells. As shown in Figure 2B, with a similar transfection efficiency as reflected by a similar number of red fluorescent cells due to co-transfection of the red fluorescence protein (RFP) expression vector pdsRed1-N1, the ssDNA sample without DSN treatment produced numerous green fluorescent cells whereas the ssDNA sample treated with DSN produced virtually no green fluorescent cells. These results again confirm the efficient elimination of dsDNA with the DSN treatment.
In summary, we have developed an integrated method for the preparation of large amounts of high quality ssDNA from the phagemid. The major advantages of this method are the expediency, low-cost, and a high yield of highly pure single-stranded DNA. This method is especially useful for large-scale, frequent preparation of high quality ssDNA.
Acknowledgments
This work was supported by the major project of Science and Technology Department of Zhejiang Province (NO.2008C03001-2), key project of Natural Science Foundation of Zhejiang Province (NO.Z2080266), key project of Science and Technology Department of Wenzhou (NO.S20060023), 5010 key project of Wenzhou Medical College (NO.XNK06009), and NIH grant R01CA079683.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Swords WE. Preparation of single-stranded DNA from phagemid vectors. Methods Mol Biol. 2003;235:103–106. doi: 10.1385/1-59259-409-3:103. [DOI] [PubMed] [Google Scholar]
- 2.Goszczynski B, McGhee JD. Resolution of sequencing ambiguities: a universal FokI adapter permits Maxam-Gilbert re-sequencing of single-stranded phagemid DNA. Gene. 1991;104:71–74. doi: 10.1016/0378-1119(91)90466-o. [DOI] [PubMed] [Google Scholar]
- 3.Trower MK. A protocol for site-directed mutagenesis employing a uracil-containing phagemid template. Methods Mol Biol. 1996;58:469–476. doi: 10.1385/0-89603-402-X:469. [DOI] [PubMed] [Google Scholar]
- 4.Tsuchiya H, Sawamura T, Harashima H, Kamiya H. Correction of frameshift mutations with single-stranded and double-stranded DNA fragments prepared from phagemid/plasmid DNAs. Biol Pharm Bull. 2005;28:1958–1962. doi: 10.1248/bpb.28.1958. [DOI] [PubMed] [Google Scholar]
- 5.Blondel A, Thillet J. A fast and convenient way to produce single stranded DNA from a phagemid. Nucleic Acids Res. 1991;19:181. doi: 10.1093/nar/19.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 2001. Working with bacteriophage M13 vector; pp. 3.1–3.52. [Google Scholar]
- 7.Jupin I, Gronenborn B. Abundant, easy and reproducible production of single-stranded DNA from phagemids using helper phage-infected competent cells. Nucleic Acids Res. 1995;23:535–536. doi: 10.1093/nar/23.3.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin JJ, Smith M, Jessee J, Bloom F. DH11S: an Escherichia coli strain for preparation of single-stranded DNA from phagemid vectors. Biotechniques. 1992;12:718–721. [PubMed] [Google Scholar]
- 9.Lei X, Zhu Y, Tomkinson A, Sun L. Measurement of DNA mismatch repair activity in live cells. Nucleic Acids Research. 2004;32:e100. doi: 10.1093/nar/gnh098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anisimova VE, Barsova EV, Bogdanova EA, Lukyanov SA, Shcheglov AS. Thermolabile duplex-specific nuclease. Biotechnol Lett. 2009;31:251–257. doi: 10.1007/s10529-008-9850-y. [DOI] [PubMed] [Google Scholar]