

Since the isolation of deoxynojirimycin in 1976,1 glycosidase inhibitors have become the subject of intense scrutiny because of their profound effect on glycoprotein processing, oligosaccharide metabolism, and cell-cell and cell-virus recognition processes.2 The schulzeines (1-3), a new class of marine alkaloids isolated by Fusetani and co-workers from extracts of the marine sponge Penares schulzei, display potent (IC50 = 48-170 nM) α-glucosidase-inhibitory activity (Figure 1).3 While these compounds, which are comprised of a benzo[a]quinolizidine core with a trisulfated C28 amide sidechain, bear little apparent resemblance to the more familiar iminosugar family of inhibitors, they are structurally related to other glucosidase inhibitors, including penasulfate4 and the penarolides,5 which similarly are O-sulfated fatty acids derivatives. The structure of 1-3 was elucidated by Fusetani, through a combination of spectral analysis and chemical degradation, while their absolute configuration was determined by modified Mosher analysis of the fragments obtained upon acidic hydrolysis of the lipid side chains. Herein we describe the enantioselective total synthesis of schulzeine A, B and C, together with a revision of the previously proposed stereochemistry at the C20' position of schulzeine A. 6
Figure 1.
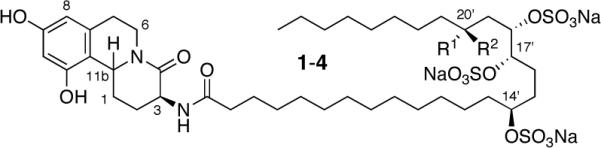
(+)-Schulzeine A (revised structure) (1; C11b-Hβ; R1 = Me R2 = H); (-)-schulzeine B (2; C11b-Hα; R1, R2 = H); (+)-schulzeine C (3; C11b-Hβ; R1, R2 = H); (+)-schulzeine A (proposed structure) (4; C11b-Hβ; R1 = H; R2 = Me).
From a strategic standpoint, we envisioned that both the trans- and cis-3-aminobenzo[a]quinolizidine subunits present in schulzeines A and C, and B, respectively, could be constructed from L-glutamate derivative 8, through a Pictet-Spengler-type cyclocondensation (Scheme 1). Although Lee7 and others8 have demonstrated that γ- and β-substituents in hydroxy lactams related to 9 can affect varying degrees of stereocontrol at the C11b ring junction, it was unclear at the outset of this study whether efficient 1,4-asymmetric induction could be achieved during the cyclization of the N-acyliminium ion generated from 8.6,9
Scheme 1. Synthesis of the Benzo[a]quinolizidine Subunita.
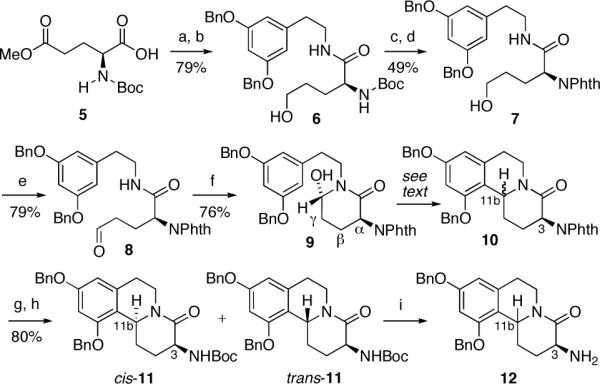
aConditions: (a) i-BuOCOCl, NMM, CH2Cl2, -35 °C, 15 min, then 2-[3,5-bis(benzyloxy)phenyl]ethylamine, DMF, CH2Cl2, rt, 12 h; (b) NaBH4, LiCl, THF, MeOH, 0 °C, 5.5 h; (c) HCl-Et2O, CH2Cl2, rt, 2 h; (d) PhthCO2Et, Na2CO3, THF, rt, 16 h; (e) Swern oxidation; (f) AcOH, CH2Cl2, rt, 2 h; (g) H2NNH2, EtOH, rt, 24 h; (h) Boc2(O), CH2Cl2, rt, 1 h, cis-11 (57%), trans-11 (26%); (i) TFA, CH2Cl2, 0 °C, 1 h cis-12 (C11b-Ha, 98%), trans-12 (C11b-Hb, 85%).
Our route to the schulzeines commenced from L-glutamic acid γ-methyl ester 5,10 which was coupled with 2-[3,5-bis(benzyloxy)phenyl]ethylamine,11 via the mixed anhydride (Scheme 1). Chemoselective reduction of the methyl ester, using lithium chloride-sodium borohydride12 then proceeded smoothly to provide primary alcohol 6 as a single enantiomer.13 Unanticipated interference of the N-Boc group during the subsequent Pictet-Spengler reaction now mandated the exchange of this carbamate for a more robust N-phthalimide group. This was accomplished by treatment of 6 with ethereal HCl and N-phthaloylation of the resulting primary amine with N-(ethoxycarbonyl)phthalimide.14 Oxidation of alcohol 7, under Swern conditions,15 provided unstable aldehyde 8, which upon exposure to acetic acid, underwent cyclization to form hydroxy lactam 9.7b Gratifyingly, exposure of either 8 or 9 to a range of Pictet-Spengler promoters, including BF3•Et2O, acetic or trifluoroacetic acid, lead to the selective formation of cis-10 with cis/trans selectivity ranging from 2:1 (8, TFA-CH2Cl2, -78 °C, 93%) to 9:1 (9, AcOH-CHCl3, reflux, 72%). While cis and trans-10 proved to be inseparable by flash chromatography, diastereomer separation could be achieved after hydrazinolysis of the phthalimide group and formation of carbamates cis-11 and trans-11.16 Treatment of each diastereomer with TFA then afforded trans- and cis-12, the benzo[a]quinolizidine subunits required for the synthesis of schulzeines A and C, and schulzeine B, respectively.17
Construction of the proposed C28 trisulfate sidechain of 1 began from hydroxy ester 13,18 which was converted to the corresponding aldehyde via Swern oxidation.15 The C14 stereocenter was then introduced by asymmetric allylation under Keck's conditions,19 which afforded 14 in high enantiomeric excess (>98% e.e.).13 After silylation of the hydroxyl group, a sequence of hydroboration-oxidation, Mitsunobu reaction of the primary alcohol with 1-phenyl-1H-tetrazole-5-thiol and S-atom oxidation provided sulfone 15 in excellent overall yield. Assembly of the complete lipid subunit was now accomplished using the Julia-Kocienski protocol.20 Thus, selective deprotonation of 15 with KHMDS followed by treatment of the sulfone anion with (S)-3-methylundecanal furnished E-alkene 16 with high diastereoselectivity, albeit in moderate yield. Treatment of 16 with HF-TBAF then aqueous NaOH resulted in the sequential deprotection of the O-TBS ether and tert-butyl ester. Carboxylic acid 17 was then coupled with trans-12 using EDC to afford compound 18. Sharpless asymmetric dihydroxylation in the presence of the (DHQ)2PHAL ligand21 then provided 1,4,5-triol 19 as a 4:1 mixture of diastereomers.
Persulfation of 19 was accomplished by treatment with excess sulfur trioxide-pyridine complex in DMF for 2 h.22 Purification of the resulting trisulfate by reverse phase chromatography and hydrogenolysis of the benzyl ethers now provided epi-C20'-schulzeine A (4). Unfortunately, the 1H and 13C NMR spectra of this material, although similar to the natural product, were not identical.3 That these discrepancies were restricted to those resonances from and near the C20' stereocenter of the lipid side chain, prompted us to the postulate that the original stereochemical assignment at this position should be reevaluated.
In light of these findings, we undertook the synthesis of compound 1, the C20' diastereomer of 4 (Scheme 2). Returning to the Julia-Kocienski olefination, the six-step sequence to the target was now repeated using sulphone 15 and (R)-3-methyl-undecanal, in place of the (S)-enantiomer, to provide schulzeine A (1). Gratifyingly, the 1H and 13C NMR spectra of 1 were found to be consistent with those reported by Fusetani for the natural product,3 thus supporting our earlier premise that the stereochemistry at C20' should be reassigned as the R-configuration.
Scheme 2. Total Synthesis of Schulzeine A (1).a.
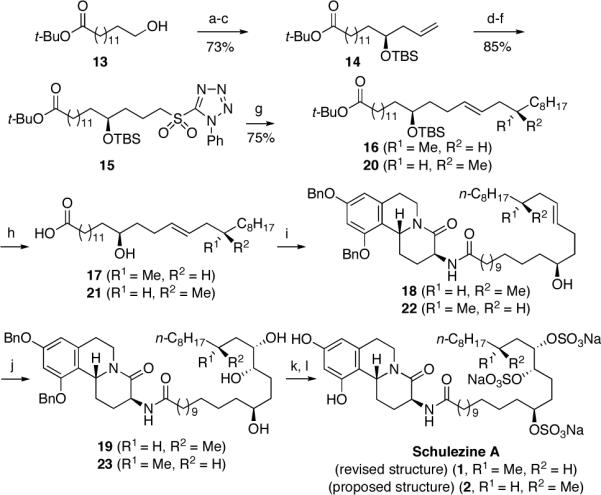
aConditions: (a) Moffat-Swern oxidation; (b) (R)-(+)-1,1'-bi-2-napthol (10 mol%), Ti(O-i-Pr)4, 4 Å m.s., CH2Cl2, reflux 1 h; then -78 °C, allyltri-n-butylstannane, -20 °C, 5 d; (c) TBSCl, imd, CH2Cl2, 0 °C®rt, 18 h; (d) 9-BBN, THF, 0 °C®rt, 14 h, then H2O2, NaOH, B(OH)3, rt °C, 1 h; (e) 1-phenyl-1H-tetrazole-5-thiol, DIAD, Ph3P, THF, 0 °C®rt, 12 h; (f) (NH4)6Mo7O24•4H2O (20 mol%), H2O2, EtOH, 0 °C®rt, 14 h; (g) KHMDS, DME, -60 °C, 30 min, then (S)-3-methylundecanal, -60 °C®rt, 16 h, 16, 45%; KHMDS, DME, -60 °C, 30 min, then (R)-3-methylundecanal, -60 °C®rt, 16 h, 20, 75%, d.r. > 20:1; (h) HF, TBAF, THF, rt, 48 h; NaOH (1 M), EtOH, THF, reflux, 48 h, 17, 70%; 21, 84%; (i) EDC, CH2Cl2, rt, 16 h, 18, 73%; 22, 80%; (j) AD-mix a, MeSO2NH2, t-BuOH-t-BuOMe-H2O-THF, 0 °C (1:1:1:1), 18 h, 19, 83%, d.r. = 4:1; AD-mix a, MeSO2NH2, t-BuOH-t-BuOMe-H2O (1:1:1), 0 °C, 48 h, 23, 77%, dr = 91:9; (k) SO3-pyr, DMF, rt, 2 h; (l) H2, Pd/C, EtOH, rt, 8 h, 4, 88% (two steps); 1, 75% (two steps).
Following the same strategy employed in the assembly of 1, the total synthesis of schulzeines B (2) and C (3) was also completed.23 In the case of target 2, the use of cis-12, the 3-aminobenzo[a]quinolizidine derived from compound cis-11, was mandated. Notably, the 1H and 13C NMR spectra of synthetic 2 and 3, closely matched the data reported for the natural products.
In summary, the total asymmetric synthesis of the α-glucosidase inhibitors schulzeine A, B and C has been completed and the absolute configuration of the C20' stereocenter of schulzeine A has been reassigned. In addition to providing material for further biological evaluation, our findings serve to reaffirm the key role that total synthesis plays in establishing the actual structure of promising natural products.
Supplementary Material
Acknowledgment
We thank UIC and the National Institutes of Health (GM59157) for financial support and Professor Yoichi Nakao for helpful discussions.
Footnotes
Supporting Information Available: Experimental details and spectroscopic data (PDF). This material is free of charge via the Internet at http://pubs.acs.org.
References
- (1).Stütz AE, editor. Iminosugars as glycosidase inhibitors: Nojirimycin and beyond. Wiley-VCH; Weinheim: 1999. [Google Scholar]
- (2).(a) Pearson MSM, Mathé-Allainmat M, Fargeas V, LeBreton J. Eur. J. Org. Chem. 2005:2159. [Google Scholar]; (b) Afarinkia K, Bahar A. Tetrahedron Asym. 2005;16:1239. [Google Scholar]; (c) Lillelund VH, Jensen HH, Liang X, Bols M. Chem. Rev. 2002;102:515. doi: 10.1021/cr000433k. [DOI] [PubMed] [Google Scholar]
- (3).Takada K, Uehara T, Nakao Y, Matsunaga S, van Soest RW, Fusetani N. J. Am. Chem. Soc. 2004;126:187. doi: 10.1021/ja037368r. [DOI] [PubMed] [Google Scholar]
- (4).Nakao Y, Maki T, Matsunaga S, van Soest RWM, Fusetani N. J. Nat. Prod. 2004;67:1346. doi: 10.1021/np049939e. [DOI] [PubMed] [Google Scholar]
- (5).Nakao Y, Maki T, Matsunaga S, van Soest RWM, Fusetani N. Tetrahedron. 2000;56:8977. [Google Scholar]
- (6).While this work was in progress, the synthesis of 2 and 3 were reported: Gurjar MK, Pramanik C, Bhattasali D, Ramana CV, Mohapatra DK. J. Org. Chem. 2007;72:6591. doi: 10.1021/jo070560h. Also, Kuntiyong reported a related route to the schulzeine benzo[a]quinolizidine system: Kuntiyong P, Akkarasamiyo S, Eksinitkun G. Chem. Lett. 2006;35:1008..
- (7).(a) Lee YS, Soo SK, Jung HC, Park H. Tetrahedron. 1997;53:3045. [Google Scholar]; (b) Lee YS, Cho DJ, Kim SN, Choi JH, Park H. J. Org. Chem. 1999;64:9727. [Google Scholar]
- (8).(a) Bassas O, Llor N, Santos MMM, Griera R, Molins E, Amat M, Bosch J. Org. Lett. 2005;7:2817. doi: 10.1021/ol0505609. [DOI] [PubMed] [Google Scholar]; (b) Takayama H, Arai M, Kitajima M, Aimi N. Chem. Pharm. Bull. 2002;50:1141. doi: 10.1248/cpb.50.1141. [DOI] [PubMed] [Google Scholar]
- (9).Efficient 1,4-asymmetric induction has been observed with less conformationally flexible 2,5-diketopiperazine systems: Zawadzka A, Leniewski A, Maurin JK, Wojtasiewicz K, Siwicka A, Blachut D, Czarnocki Z. Eur. J. Org. Chem. 2003:2443.
- (10).Shimamoto K, Ishida M, Shinozaki H, Ohfune Y. J. Org. Chem. 1991;56:4167. [Google Scholar]
- (11).Zhao H, Neamati N, Mazumder A, Sunder S, Pommier Y, Burke TR. J. Med. Chem. 1997;40:1186. doi: 10.1021/jm960449w. [DOI] [PubMed] [Google Scholar]
- (12).Hamada Y, Shibata M, Sugiura T, Kato S, Shioiri T. J. Org. Chem. 1987;52:1252. [Google Scholar]
- (13).As determined by NMR analysis of the (R)-(+)-MTPA derivative.
- (14).Worster PM, Leznoff CC, McArthur CR. J. Org. Chem. 1980;45:174. [Google Scholar]
- (15).Omura K, Swern D. Tetrahedron. 1978;34:1651. [Google Scholar]
- (16).Since epimerization at the C(3) center of cis-11, under basic conditions (MeONa, MeOH, reflux) generates, a 1.5:1 mixture of cis and trans isomers, which can be separated and recycled, trans-11 is available with greater selectivity from the D-enantiomer of 5, by way of ent-cis-10.
- (17).Assignment of relative stereochemistry to cis and trans-10 was achieved through NOESY experiments; a clear cross-peak between C11b-H and C3-H was observed in cis-10, but not in the trans isomer. Further confirmation of this assignment was secured by conversion of cis and trans-11 to their respective (R) and (S)-MTPA amide derivatives, which were prepared by Fusetani as part of a degradation study of 1-3: see ref 3. For a tabular comparison of the spectral properties of these Mosher amides, see Supporting Information.
- (18).Emde U, Koert U. Eur. J. Org. Chem. 2000:1889. [Google Scholar]
- (19).Keck GE, Tarbet KH, Geraci LS. J. Am. Chem. Soc. 1993;115:8467. [Google Scholar]
- (20).Blakemore PR. J. Chem. Soc., Perkin Trans 1. 2002:2563. [Google Scholar]
- (21).Sharpless KB, Amberg W, Bennani YL, Crispino GA, Hartung J, Jeong KS, Kwong HL, Morikawa K, Wang ZM, Xu DQ, Zhang XL. J. Org. Chem. 1992;57:2768. [Google Scholar]
- (22).Gilbert EE. Chem. Rev. 1962;62:549. [Google Scholar]
- (23).For full details concerning of the total synthesis of schulzeine B (2) and C (3), see Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.