

Abstract
D-3-Phosphoglycerate dehydrogenase from M. tuberculosis displays substantial substrate inhibition by its physiological substrate in the direction of NADH oxidation, hydroxypyruvic acid phosphate (HPAP). Previous investigations showed that plots of substrate concentration versus activity derived from steady state assays could be fit with the equation for complete uncompetitive inhibition and that the mechanism may be allosteric. This investigation uses a simulation of transient kinetic data to demonstrate that the mechanism is consistent with the interaction of substrate at a second site called the anion-binding site. While substrate addition at the active site is ordered, with HPAP binding before NADH, NADH can compete with the substrate for binding to the allosteric site and thereby eliminate the substrate inhibition. Fluorescence resonance energy transfer analysis of mutants with specific tryptophan residues converted to phenylalanine residues demonstrates that the main interaction of NADH with the enzyme, in the absence of substrate, is at the allosteric anion-binding site. This is further confirmed by mutations of basic residues at the anion-binding site which also demonstrates that these residues are necessary for inhibition by L-serine when it binds to the regulatory domain. This may indicate that a ligand must be bound to the anion-binding site for L-serine inhibition, providing a potential mechanism for low levels of activity in the presence of high levels of inhibitor.
D-3-phosphoglycerate dehydrogenase (PGDH1) ( EC 1.1.1.95) from Mycobacterium tuberculosis (M. tb) is a homotetramer whose subunit's are identical in their primary structure but not in their tertiary structure. The crystal structure of M. tb PGDH (1) shows that the subunits are present in two different conformations that arise from a rotation about an inter-domain connecting strand. Each subunit is made up of four distinct globular domains termed the nucleotide binding domain, the substrate binding domain, the intervening domain, and the regulatory domain. Residues 1-99 and 283-319 form the substrate binding domain, and residues 100-282 form the nucleotide binding domain. The intervening domain is made up of residues 320-454 and residues 455-529 complete the regulatory domain. The conformational rotation takes place in the strand that connects the substrate binding domain to the intervening domain.
As with E. coli PGDH (2), M. tb PGDH is inhibited by L-serine (3), which binds at the interface between adjacent regulatory domains in both enzymes. The regulatory domains are members of the ACT domain family (4) that are found in a large number of bacterial proteins mostly involved in amino acid metabolism or transcriptional regulation of amino acid biosynthesis.
The intervening domain represents an additional domain not found in E. coli PGDH that “intervenes” between the substrate binding and regulatory domains. The interface between adjacent subunits at the point where the intervening domain and the regulatory domain meet, forms an anion-binding site that is made up of residues with cationic side chains contributed by both adjacent subunits (Figure 1). Four of the five cationic residues found at each anion-binding site come from the intervening domain and an additional cationic residue is contributed by the regulatory domain (Figure 2). The site was recognized in the crystal structure (1) where it is observed to be binding a tartrate molecule, which is a component of the buffer used to crystallize the enzyme.
Figure 1. The Structure of M. tb PGDH.

The M. tb PGDH tetramer is oriented so that the anion and serine-binding sites between two adjacent subunits are clearly discernable in the upper right. Two subunits of the tetramer are shown as ribbon diagrams and the other two are shown as line drawings. Subunits A and A' are green and light gray and subunits B and B' are purple and dark gray, respectively. The domains in the two ribbon diagrams are labeled. The intervening domains are the two 4-stranded beta sheet structures located next to the substrate binding domains and below the regulatory domains. Two serine molecules bound to the regulatory domain are shown as line drawings. The cationic residues that comprise the anion-binding sites are shown in ball and stick configuration. The catalytic histidine residues are shown in red space filling depiction to highlight the location of the active sites relative to the other sites.
Figure 2. The Anion and Serine-Binding Sites of M. tb PGDH.

A close-up view of the anion and serine-binding sites depicted in Figure 1 is shown. At each interface between subunits A and B, there are two of each site related by approximate 180° symmetry. The view is down the axis connecting the two sites of the same type. One site is in front and is shown by blue ball and stick models. The second site is in back and is depicted by smaller gray ball and stick models. L-serine molecules are shown bound to the regulatory domain and the side chains of the cationic residues are shown in the anion-binding sites.
A unique dual pH optimum has been described for the M.tb enzyme (5) that appears to be related to the substrate inhibition mechanism. This is characterized by a pH dependent increase in substrate inhibition between pH 5-7 that produces a depression in activity between two peaks of increased activity in the pH profile. The mechanism of substrate inhibition was shown to be linked to this pH dependent depression in activity by the demonstration that both could be eliminated by a triple mutation to the cationic residues at the anion-binding site (5).
Recent transient kinetic studies (6) have shown that the binding of substrates to E. coli PGDH is ordered with NADH binding first. An initial transient kinetic study of substrate binding in M. tb PGDH suggests that the order is reversed, with substrate binding before coenzyme (7). This was inferred from the observation that although binding of NADH to M. tb PGDH in the absence of substrate can be demonstrated by transient kinetic analysis, the binding appears to be too slow to be able to account for the rate of the catalytic reaction, which has a kcat of approximately 2500 s-1 per tetramer. NADH binding in the presence of substrate occurs within the mixing time of the instrument. This is in contrast to a kcat of approximately 8-12 s-1 per tetramer for E. coli PGDH where the individual rate constants for both coenzyme and substrate can be determined by stopped-flow analysis and clearly demonstrated that NADH binds first in an ordered mechanism.
Steady-state kinetic analysis (2) of substrate inhibition in M. tb PGDH suggested that it conformed to a model for complete uncompetitive inhibition. On the other hand, single mutants of the cationic residues in the anion-binding site displayed substrate inhibition patterns characteristic of partial uncompetitive inhibition. The classical kinetic view of uncompetitive substrate inhibition, shown in scheme 1, is that it occurs when the second substrate in the ordered mechanism binds to the enzyme-product complex after release of the first product but before the second product is released.
scheme 1.

Scheme 1
In the case of an ordered bireactant dehydrogenase like M. tb PGDH, where HPAP binds before coenzyme, this kinetic mechanism is inconsistent with substrate inhibition by HPAP. However, substrate inhibition by the first substrate to bind can be accounted for by an allosteric mechanism where its binding to a second site has the effect of decreasing the catalytic turnover of the enzyme.
The relatively fast rates of substrate and subsequent coenzyme binding for M. tb PGDH do not allow a study of its transient binding kinetics by stopped-flow analysis. However, the stopped-flow instrument can be used to observe the full time course of the NADH turnover under conditions where substrate is depleted. In this investigation, the NADH turnover, in conjunction with the fluorescence resonance energy transfer for NADH binding to the native and mutant enzymes, provides a picture of the kinetic consequences of the interaction of anionic ligands with the anion-binding site. This work also demonstrates that while substrate inhibition in M. tb PGDH occurs through an allosteric site separate from the site that binds L-serine, the two are linked by common elements.
Experimental Procedures
NADH and the dimethylketal tricyclohexylammonium salt of hydroxypyruvic acid phosphate (HPAP) were purchased from Sigma. The latter was converted to HPAP according to directions supplied by the manufacturer. The concentration of each HPAP preparation was determined by the amount of NADH that could be converted to NAD+ when NADH is in large excess. HPAP was stored in frozen aliquots prior to use. PGDH concentration was determined using an E 1% of 5.6 (4). The concentration of the trytophan mutants was determined based on the proportion of tryptophan residues relative to the native enzyme.
Steady state PGDH activity was determined by monitoring the continuous change in absorbance at 340 nm from the conversion of NADH to NAD+ in the presence of HPAP. Assays were performed in 200 mM potassium phosphate buffer, pH 7.5, 1 mM dithiothreitol, 1 mM EDTA at 25 ° C. Plots of activity versus coenzyme concentration were fit to a form of the Hill equation,
| (Eq. 1) |
where Vm is the maximal velocity, S is the varied substrate concentration, K 0.5 is the substrate concentration at half maximal velocity, and n is the Hill coefficient.
Native and mutant PGDH were expressed and purified as previously described (2). M. tb PGDH R451A-R501A-K439A and K439A-R451A-R501A were produced previously (2). Tryptophan mutants W29F, W130F, and W327F were produced in the same manner by standard PCR based mutagenesis as previously described (7). The W130F mutant protein did not express well and could not be obtained. All other mutant proteins expressed well.
Fluorescence resonance energy transfer (FRET), between tryptophan residues in the enzyme and NADH, was monitored by measuring the amount of fluorescence at 420 nm when the enzyme is excited at 295 nm. The contribution to the 420 nm signal by NADH in solution was subtracted before plotting. There is no appreciable inner filter effect by NADH over the concentration range studied. The data were fit to a form of the Hill equation,
| (Eq. 2) |
where Fmax is the maximal fluorescence, L is the varied ligand concentration, K 0.5 is the ligand concentration at half maximal fluorescence, and n is the Hill coefficient.
The theoretical relative amount of energy transfer between tryptophan and NADH was estimated from distances obtained from the crystal structure using the equation
| (Eq. 3) |
where E is the relative energy transfer, R0 is the Förster distance (25 Å) for tryptophan and NADH (8), and r is the approximate distance between loci in the structure.
Kinetic time course studies were performed on an Applied Photophysics SX-20 stopped-flow spectrometer. The time course of the conversion of NADH to NAD+ was measured by following the absorbance at 340 nm after rapid mixing. When pre-incubation of enzyme with coenzyme was performed, it was allowed to proceed for 30 min prior to initiation of the reaction. The reaction and all reagents were thermostated at 25° C with a circulating water bath.
Kinetic simulations were performed with Global Kinetic Explorer Professional, v. 2.0, from KinTek corporation (9, 10). The program allows input of a mechanism by simple text description and then derives the differential equations needed for numerical integration. The program can be used both for fitting data to a proposed mechanism or for simulating curves to a proposed mechanism as a function of values of the rate constants input by the operator.
Results
Time Course of the Reaction Catalyzed by M. tb PGDH Under Conditions Where Substrate is Rapidly Depleted
After rapid mixing of enzyme with coenzyme and substrate, the full time course of the reaction was remarkable in that an initial rapid decrease in absorbance at 340 nm was followed by an abrupt transition to a very slow decrease in signal followed by an increase in rate until the NADH concentration became limiting (Figure 3). On the other hand, when the enzyme is pre-incubated with NADH prior to rapid mixing with HPAP, the conversion from rapid to slower rate is not observed. Rather, the absorbance rapidly decreases in a continuous fashion.
Figure 3. Time Course of M. tb Catalysis.

The conversion of NADH to NAD+ by native M. tb PGDH is monitored under conditions where substrate is depleted. Two mixing protocols where performed on a stopped flow instrument. The data are represented by symbols and the simulation to Scheme 2 is represented by a solid line. For clarity, not every data point is shown. 1) Enzyme (2.5 μM) was rapidly mixed with HPAP (1000 μM) and NADH (350 μM), Ο; 2) Enzyme (2.5 μM) and NADH (175 μM) were rapidly mixed with HPAP (1000 μM) and NADH (175 μM), □.
Based on previous observations that substrate inhibition occurred when substrate interacted with the anion-binding site (5), the transient change in catalytic rate was thought to be a function of this interaction. The preliminary interpretation was that the initial fast drop in absorbance resulted from catalysis at the active site before appreciable interaction of substrate at the anion-binding site occurred. The conversion to a slower rate of catalysis represented subsequent binding of substrate to the anion-binding site resulting in a slower turnover of catalysis. The observation that pre-incubation of enzyme with NADH could eliminate the abrupt transition from fast to slower rate suggested that NADH could bind to the enzyme in such a manner that it prevented productive interaction of substrate at the anion-binding site.
Based on this initial interpretation, a simulation was set up according to the following general model: where E, S, and N, represent enzyme, HPAP, and NADH, respectively. P and Q represent the products, phosphoglycerate and NAD+. When S or N appear to the right of E, it designates binding to the active site. When they appear to the left of E, it designates binding to a second separate site, which was presumed to be the anion-binding site. In setting up the model, it is assumed that there is no product inhibition, and that HPAP and NADH will be depleted. It was also assumed that all of the NADH containing complexes possessed the same extinction coefficient for NADH. The previously determined on and off rates for NADH binding to the enzyme in the absence of substrate ( E + N = NE) were fixed. The simulations shown in Figure 3 were performed by first attempting to fit the data to the model to derive starting parameters for the individual rate constants to be used in subsequent simulation of the model. This fitting procedure demonstrated that while the data could be approximated pretty well with respect to the general shapes of the curves, the model was not well constrained by the data and that the values for many of the rate constants exhibited relatively large errors. The estimated rate constants obtained from the fitting procedure were used as starting points for further simulation using the scrolling feature of KinTek global explorer. This produced bounds which represent the range over which the rate constants could be changed without appreciably affecting the shapes of the curves. It was assumed that pre-incubation with saturating levels of NADH would drive the equilibrium in favor of the formation of the NE complex. Thus, the initial concentration of NE in the simulation was set equal to the enzyme concentration. Figure 3 show the results of the simulation to the model in Scheme 2. The data from the stopped-flow experiment in shown with symbols and the simulation is shown with a solid line. The simulated rate constants are presented in Table 1 along with the bounds for each determined from the simulation. Although the simulation is in relatively good agreement with the data, the model is not well constrained. Nonetheless, if the results are viewed more qualitatively, general trends can be deduced from and a reasonable assessment of the contribution of the various steps in the model can be made. This is evaluated further in the Discussion section.
scheme 2.
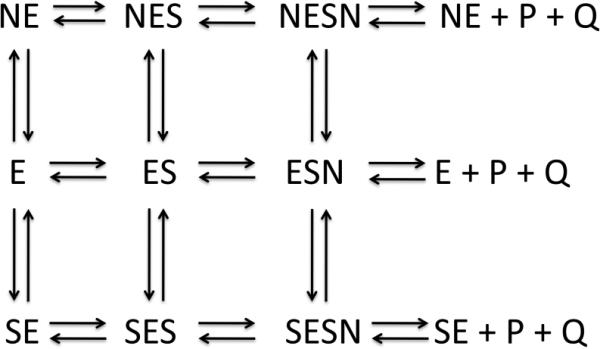
Scheme 2
Table 1.
Relative Kinetic Constants Determined from Simulation
| Step | k+ia | Bounds b | k-ia | Bounds b |
|---|---|---|---|---|
| E + S = ES | 1270 | 900 - > 1270 | 111 | 0 - <111 |
| ES + N = ESN | 409 | 408 - 410 | 70 | 40 - 95 |
| ESN = E + P + Q | 3410 | 3400 - 3450 | 0 | 0 - >280 |
| E + S = SE | 0.1 | 0 - 0.15 | 0.8 | 0.79 - 0.81 |
| ES + S = SES | 1.55 | 1.54 - 1.56 | 1.0 | 0.95 - 1.05 |
| SE + S = SES | 0.001 | 0.0009 - 0.0011 | 1.18 | 1.17 - 1.19 |
| SES + N = SESN | 0.19 | 0.18 - 0.20 | 3 | 2.9 - 3.1 |
| SESN = SE + P + Q | 0.32 | 0.31 - 0.33 | 0 | 0 - 0.001 |
| E + N = EN | 0.37 c | 0.22 c | ||
| NE + S = NES | 1.13 | 1.12 - 1.14 | 40100 | 40000 - 41000 |
| NES + N = NESN | 759 | 750-765 | 12800 | 12700 - 12900 |
| NESN = NE + P + Q | 4800 | 4790 - 4810 | 0 | 0 - 0.01 |
| ES + N = NES | 0 | 0 - 0.05 | 0 | 0 - 1.5 |
| ESN + S = SESN | 0 | 0 - 0.001 | 0 | 0 - 0.01 |
| ESN + N = NESN | 0 | 0 - 0.001 | 0 | 0 - 1 |
Kinetic constants for substrate or coenzyme binding are in units of μM-1s-1 and all others are in units of s-1.
Bounds indicate the approximate interval over which a value can be changed without affecting the shape of the simulated curve.
These constants are determined from transient binding measurements and are fixed during the simulation.
Time Course of the Reaction Catalyzed by M. tb PGDH Mutated at the Anion-Binding Site Under Conditions Where Substrate is Rapidly Depleted
The time course of the reaction with the triple mutant R501A-R451A-K439A M. tb PGDH shows a remarkably different pattern (Figure 4). The mutations to the anion-binding site have completely eliminated the transition from fast to slow conversion of NADH as seen for the native enzyme. Now, both mixing protocols produce very similar time courses. It appears that the mutations at the anion-binding site eliminate the productive interaction of substrate at this site and that, as predicted, this interaction was responsible for the multiphasic kinetics of NADH turnover observed for the native enzyme.
Figure 4. Time Course of R451A-R501-K439A M. tb Catalysis.

The conversion of NADH to NAD+ by R451A-R501A-K439A M. tb PGDH is monitored under conditions where substrate is depleted. two mixing protocols are the same as in Figure 1. The data are represented with the symbols and, for clarity, not every data point is shown. 1) Enzyme (2.5 μM) was rapidly mixed with HPAP (1000 μM) and NADH (350 μM), 0 ; 2) Enzyme (2.5 μM) and NADH (175 μM) were rapidly mixed with HPAP (1000 μM) and NADH (175 μM),□.
Titration of M. tb PGDH and Mutant M. tb PGDH with NADH
Both E. coli and M. tb PGDH possess a tryptophan residue within 10-12 Å of the NADH binding site. In the case of E. coli PGDH, there is a relatively strong fluorescence resonance energy transfer (FRET) from the single tryptophan residue in the enzyme to NADH when it binds at the active site (11). However, M. tb PGDH produces a FRET signal that is only approximately 25% of that of E. coli PGDH. The relatively lower FRET presumably results from the inability of NADH to bind to the active site in the absence of substrate. M. tb PGDH contains two additional tryptophan residues, one of which is within 16-20 Å of the anion-binding site (Trp-327) and the other (Trp-29) is relatively distant from both sites but is closer to the active site than the anion-binding site. Thus, the tryptophan residues appear to be well placed for assessing the relative degree of NADH binding to each site based on their relative FRET. The relative contributions for each tryptophan residue for binding at the active site, calculated using equation 3, are 24% for Trp-29, 57% for Trp-130 and 19% for Trp-327. On the other hand, the relative contributions for each tryptophan residue for binding at the anion-binding site are 3% for Trp-29, 2% for Trp-130 and 95% for Trp-327. The NADH titration for the native and the tryptophan mutants are shown in Figure 5. If NADH is binding to the active site, a relative loss in FRET would be incurred with the W29F and W130F mutants. Unfortunately, the W130F protein could not be isolated but the FRET with the W29A mutant is very comparable to the wild type in magnitude and affinity. No appreciable change in FRET would be expected from the W327F mutant but that is not what is observed. On the other hand, if NADH were binding to the anion-binding site, the only significant reduction in FRET would be that from the W327F mutant. This is essentially what is observed in Figure 5.
Figure 5. Fluorescence Resonance Energy Transfer in Native and Mutant M. tb PGDH.

Equimolar amounts of enzyme (2.5 μM subunit) were titrated with NADH in 200 mM potassium phosphate buffer, pH 7.5. The samples were excited at 295 nm and the fluorescence at 420 nm was monitored as a function of NADH concentration. Native, M. tb PGDH, • W29F, ▲; W327F, ■; and R451A-R501A-K439A,♦. The solid lines are fits to the Hill equation (Eq. 2).
The time course data presented earlier showed that the apparent substrate inhibition could be eliminated by pre-incubation with NADH. If both ligands competed for the same site, it would be expected that the FRET due to NADH binding in the R501A-R451A-K439A mutant would be significantly reduced because the energy transfer from Trp-327 would be eliminated. Figure 5 demonstrates that the FRET in the triple mutant is significantly reduced compared to the native enzyme.
The Interaction of NADH with M. tb PGDH in the Presence of Substrate
Since the presence of both substrates produces a very fast catalytic reaction and NADH binds last in the ordered reaction, the binding of NADH to the catalytic site cannot be followed by FRET or stopped flow analysis. However, the relative interaction of NADH with the active site can be assessed by following the dependence of the steady state catalysis as a function of NADH concentration. Figure 6 demonstrates that the K 0.5 values for native enzyme and the mutants are comparable. This suggests that none of the mutations has an appreciable effect on NADH binding at the active site when the enzyme is turning over. Also note that the K 0.5 for NADH for the FRET interactions are in the range of 2-4 mM while those for activity are 80-100 mM (Table 2). This reinforces the conclusion that the NADH interaction for each is occurring at different sites.
Figure 6. Activity of Native and Mutant M. tb PGDH as a function of the NADH Concentration.

The activity of the enzymes was determined with the standard assay at varying concentrations of NADH. HPAP is present at 400 μM, which corresponds to the concentration at which optimal activity is observed. Native PGDH, •; W29F,▲; W327F, ■; and R451A-R501A-K439A, ♦. The solid lines are fits to the Hill equation (Eq. 1).
Table 2.
NADH Dependence of Activity and FRET for Native and Mutant M. tb PGDH a
| Mutation | Activityb | FRETc | ||
|---|---|---|---|---|
| n | K0.5 μM | n | K0.5 μM | |
| Native | 2.5 ± 0.2 | 106 ± 5 | 1.6 ± 0.1 | 3.2 ± 0.1 |
| W29F | 2.2 ± 0.1 | 99 ± 2 | 1.2 ± 0.1 | 1.9 ± 0.1 |
| W327F | 2.0 ± 0.1 | 89 ± 4 | 1.7 ± 0.3 | 3.0 ± 0.3 |
| W130F | NDd | NDd | ||
| R451A, | ||||
| R501A, | ||||
| K439A | 1.5 ± 0.2 | 83 ± 13 | 2.3 ± 0.2 | 3.6 ± 0.1 |
Performed in 200 mM potassium phosphate, pH 7.5.
The HPAP concentration is 400 μM and the NADH concentration is varied
The fluorescence at 420 nm is measured with excitation at 295 nm as a function of NADH concentration.
ND, Not determined. Protein could not be isolated.
Inhibition of catalytic activity by L-serine in native and mutant M. tb PGDH.
Since the anion-binding site lies between the regulatory site, where serine binds, and the catalytic site, the effect of mutations of the anion-binding site residues on serine inhibition was investigated. Each of the five protonatable cationic side chain residues in the anion-binding site were changed to alanine residues. In addition, the triple mutant, K439A-R451A-R501A was also tested. The serine inhibition patterns of these mutants are presented in Table 3. Serine inhibition was measured using two HPAP levels, a high level where substrate inhibition is significant, and a lower level, approximately at the peak of activity and before obvious substrate inhibition is observable by steady state analysis.
Table 3.
Serine Inhibition of Native and Mutant M. tb PGDH a
| Mutation | Peak Substrate | High Substrate | Ki, HPAPb μM | ||
|---|---|---|---|---|---|
| n | K0.5 μM | n | K0.5 μM | ||
| Native | 1.6 ± 0.1 | 19 ± 1 | 1.5 ± 0.1 | 42 ± 2 | 950 ± 120 |
| R451A | 1.6 ± 0.1 | 93 ± 3 | 1.7 ± 0.3 | 290 ± 34 | 950 ± 150 |
| R501A | 1.5 ± 0.1 | 421 ± 21 | 1.7 ± 0.1 | 1276 ± 69 | 1020 ± 30 |
| K439A | 1.2 ± 0.1 | 8.6 ± 0.6 | 1.2 ± 0.1 | 6.8 ± 0.3 | 54 ± 4 |
| H447A | 1.7 ± 0.1 | 90 ± 6 | 1.5 ± 0.1 | 129 ± 9 | 870 ± 50 |
| R446A | 1.3 ± 0.1 | 117 ± 6 | 0.9 ± 0.1 | 120 ± 19 | 289 ± 15 |
| R446A, | |||||
| R501A, | |||||
| K439A | 1.0 ± 0.1 | 26 ± 3 | NDc | NDc | 66 ± 18 |
| R451A, | |||||
| R501A, | |||||
| K439A | NId | NId | |||
Performed in 200 mM potassium phosphate, pH 7.5.
From reference 5.
ND, not determined
NI, no inhibition observed at 5000 μM L-serine.
All of the mutations had a significant effect on the K 0.5 for serine while R446A and K439A had significant effects on the Hill coefficient. R501A significantly decreased the sensitivity of the enzyme to serine while K439A had the opposite effect, increasing sensitivity to serine substantially. In contrast, R501A by itself has little effect on substrate inhibition whereas K439A and R446A greatly increase it. The catalytic activity of the triple mutant, K439A-R451A-R501A, was unable to be inhibited by serine even at very high concentrations. The significantly reduced sensitivity to serine of the two enzymes containing the R501A mutation appeared to implicate this residue as being crucial for serine inhibition. However, another triple mutation, R446A-R501A-K439A, containing R501A, demonstrates that the situation is more complex than this simple interpretation. Serine inhibition at high HPAP concentrations generally followed the trend for low HPAP concentrations but with the K0.5 being somewhat increased for native, R451A, R501A, and H447A.
Discussion
The simulation of the model shown in scheme 2 to the data for the two mixing strategies require that the model contain steps for both substrate and coenzyme binding at a second site and that complexes with one or both sites occupied can turnover to product. Furthermore, substrate binding for catalysis is ordered, with HPAP (S) binding first to the active site. In the absence of HPAP, NADH (N) can only bind to the second site while HPAP can bind to both sites. If these conditions are not met in the model, the simulation fails to produce a reasonable approximation to the data. However, the values produced for the rate constants (listed in Table 1) are not well constrained by the model. Therefore, these values were used as starting approximations to further simulate the data curves. While the absolute values of many of the rate constants are not constrained well enough to be reliable in an absolute sense, the relative magnitudes of the rate constants, and the ranges over which they can change without affecting the simulations, can be considered to provide a qualitative assessment of the contributions of each step to the mechanism. The simulation to the model allows the following conclusions: 1) certain steps do not occur or are very slow since they are not necessary in satisfying the simulation. 2) This represents a model for partial substrate inhibition by HPAP since there must be turnover of the SESN complex. Originally it was thought that substrate inhibition was of the complete type (5) where the SESN complex would not turn over. 3) Turnover of the SESN complex is slow relative to that for the ESN and NESN complexes. 4) Substrate inhibition occurs through binding of HPAP to a second site on the enzyme. 5) In the absence of HPAP, the enzyme can also bind NADH at a second site converting the enzyme to an NE complex that can still bind substrate and coenzyme at the active site and undergo catalysis. This appears to block substrate binding at the second site, suggesting that the binding of the two is competitive. This effect can also be observed in the steadystate assay where the initial velocity is measurably faster if the enzyme is pre-incubated with NADH than if it is pre-incubated with HPAP. 6) Even though NADH can bind at the second site, it is not as productive in the sense that it is not greatly inhibitory to the rate of turnover. 7) The SE complex must dissociate to form free enzyme in order for the rate of substrate depletion to match the data. 8) The second order rate constants for binding of ligands to the second site are small compared to those for binding to the active site. The Kd of NADH at this site can be calculated to be approximately 0.6 μM from the rates of NADH binding determined in the absence of substrate. The Kd for HPAP can be estimated to be approximately 8 μM from the simulation. If this were not the case, substantial inhibition would be seen at low substrate concentrations.
In order to test the conclusions about binding loci reached from the simulation, the energy transfer to NADH was measured using mutations to the tryptophan residues. The triple mutation at the anion-binding site that was shown to eliminate substrate inhibition previously was also tested. If NADH were binding to the free enzyme at the anion-binding site, the major contribution to the FRET would come from Trp-327. This is supported by the demonstration that when this residue is mutated to a phenylalanine, there is a significant decrease in the FRET that does not occur when Trp-29 is mutated. Conversely, if NADH bound to the active site of the free enzyme, a drop in FRET would be expected for the W29F mutant. This is not observed. The FRET is reduced significantly when the cationic residues at the anion-binding site are replaced by nonprotonatable side chains, indicating again that NADH is binding to this site in the native enzyme. However, the FRET is not completely eliminated by the mutations, suggesting that some non-productive binding of NADH is still occurring. The triple mutation does not have a significant effect on NADH interaction with the active site during catalysis. Taken together, these findings support the conclusion that both HPAP and NADH can bind at the anion-binding site in a competitive manner, but only the HPAP binding is functional in causing significant substrate inhibition. This is consistent with the observation that tartrate binding to the anion-binding site also does not effect the rate of catalysis (5, 7). From this, one would infer that productive binding, in the sense of producing significant substrate inhibition, involves specific interaction related to the structure of the substrate.
In systems where substrate binding is ordered, substrate inhibition can occur through either a kinetic or an allosteric mechanism. The classical view of kinetic uncompetitive substrate inhibition is that the second substrate to bind productively can also form a dead-end complex at the active site before the product of the first substrate dissociates (Scheme 1). This mechanism is not consistent with uncompetitive substrate inhibition by the first substrate to bind and can thus be ruled out as a mechanism for M. tb PGDH. Rather, the evidence suggests that the substrate inhibition in this case has an allosteric mechanism. A mechanism of allosteric substrate inhibition in E. coli phosphofructokinase has also been demonstrated using a hybrid tetramer approach (12) as well as crystallographic analysis (13). However, in this enzyme, the allosteric communication came about through binding of MgATP in one active site of the tetramer and fructose-6-phosphate in another active site of the tetramer. In other words, the allosteric interaction did not involve a “new” site, other than the active site, as is the case for M. tb PGDH. Allosteric substrate inhibition mechanisms have also been proposed for other enzymes such as tyrosine hydroxylase (14), ornithine carbamoyl transferase (15), and aspartate transcarbamylase (16). However, in these cases the identity of the allosteric site where substrate binds has not been determined. Non-substrate inhibitors have also been reported to bind at both the active site and an allosteric site in some enzymes such as L-threonine dehydratase (17).
Interestingly, the triple mutation at the anion-binding site eliminated both substrate inhibition and inhibition by serine, which binds at the regulatory site. Based on the single site mutations, the reduction of serine inhibition would appear to be largely due to mutation of R501. This single mutation has little effect on the substrate inhibition mechanism. Conversely, the presence of substrate at levels that produce significant substrate inhibition decrease further the sensitivity of the R501A mutant enzyme to serine. However, another triple mutation that also removes the R501 side chain, R446AR501A-K439A, displays very good sensitivity to serine and significantly decreases the Ki for substrate inhibition. Thus, the interaction of substrate at this site is complex.
It is noted that the R451A-R501A-K439A triple mutation removes all of the inter-domain contacts from one of the subunits. Thus, hydrogen bond or ionic interaction across the domain interface through the binding of a ligand would be eliminated. The complete elimination of substrate and serine inhibition by this particular triple mutation may come through the elimination of the interaction between the adjacent intervening domains.
This raises the question of whether or not the binding of a ligand at the anionbinding site is required for serine inhibition to occur. If so, it could provide a mechanism where serine inhibition of the enzyme is not significant at low substrate levels but increases as substrate concentration increases, providing an additional level of control where basal levels of enzymatic activity could occur even at high serine levels. This notion does not appear to be consistent with the data in Table 3 where higher levels of substrate generally appear to decrease sensitivity to serine. However, these data are for two widely different substrate concentrations, with the low concentration being relatively high at approximately 400 μM. Furthermore, the difference for the native enzyme is not particularly large. In order to test this idea more rigorously, serine inhibition at low levels of substrate would need to be accurately measured. This presents a significant technical challenge since further decreases of already low levels of activity would have to be measured accurately. Alternatively, the specific residues at the anion-binding site may be necessary for efficient transmission of the signal elicited by serine binding to reach the active site without the requirement for it to be occupied by a ligand. However, the crystal structure does not show any other obvious interactions of these side chains with other elements of the structure.
This investigation has clearly demonstrated the interaction of substrate and coenzyme at a second, non-catalytic site in M. tb PGDH. The data confirm that the substrate inhibition that is observed has an allosteric mechanism and that the anion-binding site is the allosteric site. Binding of coenzyme at this site is competitive with substrate but does not produce appreciable inhibition, indicating that precise interactions with the substrate are required for inhibition. This is the second allosteric site that has been identified in M.tb PGDH, the first being the serine-binding site. These two sites, the serine-binding site and the anion-binding site, are in proximity to each other and the data suggest that they do not act independently. This interaction may be the basis for a complex control mechanism for modulating catalytic activity of this enzyme in the tuberculosis organism brought about by the addition of the intervening domain in the structure of the protein. While other PGDH enzymes also possess an intervening domain, very little is known about them so it is not possible to say whether this mechanism may be unique to the M. tb enzyme. Human PGDH, which contains both a regulatory or ACT domain and an intervening domain, displays substrate inhibition by HPAP but is not inhibited by L-serine (unpublished observation). Therefore, for this one example at least, the overall control mechanism will not be similar.
The potential involvement of the anion-binding site in the mechanism of serine inhibition in M. tb PGDH also has interesting implications. For instance, the development of a very tight binding inhibitor for the anion-binding site may act by itself or synergistically with serine or other substances to effectively irreversibly inhibit the enzymatic activity. Since PGDH has been shown to be an essential enzyme for Mycobacterium tuberculosis (18), the anion-binding site provides a potential new target for drug development.
Acknowledgments
Supported by grant # GM 56676 (G. A. G.) from the National Institutes of Health
Footnotes
The abbreviations used are: PGDH, D-3-phosphoglycerate dehydrogenase;M. tb, Mycobacterium tuberculosis; HPAP, hydroxypyruvic acid phosphate (also called phosphohydroxypyruvate); FRET, fluorescence resonance energy transfer.
References
- 1.Dey S, Grant GA, Sacchettini JC. Crystal structure of Mycobacterium tuberculosis D-3-phosphoglycerate dehydrogenase: Extreme asymmetry in a tetramer of identical subunits. J. Biol. Chem. 2005;280:14892–14899. doi: 10.1074/jbc.M414489200. [DOI] [PubMed] [Google Scholar]
- 2.Al-Rabiee R, Zhang Y, Grant GA. The mechanism of velocity modulated allosteric regulation in D-3-phosphoglycerate dehydrogenase: Site-directed mutagenesis of effector binding site residues. J. Biol. Chem. 1996;271:23235–23238. doi: 10.1074/jbc.271.38.23235. [DOI] [PubMed] [Google Scholar]
- 3.Dey S, Hu Z, Xu XL, Sacchettini JC, Grant GA. D-3-Phosphoglycerate dehydrogenase from Mycobacterium tuberculosis is a link between the Escherichia coli and mammalian enzymes. J. Biol. Chem. 2005;280:14884–14891. doi: 10.1074/jbc.M414488200. [DOI] [PubMed] [Google Scholar]
- 4.Grant GA. The ACT domain: A small molecule binding domain and its role as a common regulatory element. J. Biol. Chem. 2006;281:33825–33829. doi: 10.1074/jbc.R600024200. [DOI] [PubMed] [Google Scholar]
- 5.Burton RL, Chen S, Xu XL, Grant GA. A novel mechanism for substrate inhibition in Mycobacterium tuberculosis D-3-phosphoglycerate dehydrogenase. J. Biol. Chem. 2007;282:31517–31524. doi: 10.1074/jbc.M704032200. [DOI] [PubMed] [Google Scholar]
- 6.Burton RL, Hanes JW, Grant GA. A stopped flow kinetic analysis of substrate binding and catalysis on Escherichia coli D-3-phosphoglycerate dehydrogenase. J. Biol. Chem. 2008;283:29706–29714. doi: 10.1074/jbc.M805180200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dey S, Burton RL, Grant GA, Sacchettini JC. Structural analysis of substrate and effector binding in Mycobacterium tuberculosis D-3-phosphoglycerate dehydrogenase. Biochemistry. 2008;47:8271–8282. doi: 10.1021/bi800212b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steinberg IZ. Long-range nonradiative transfer of electronic excitation energy in proteins and polypeptides. Ann. Rev. Biochem. 1971;40:83–114. doi: 10.1146/annurev.bi.40.070171.000503. [DOI] [PubMed] [Google Scholar]
- 9.Johnson KA, Simpson ZB, Blom T. Global Kinetic Explorer: A new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 2009;387:20–29. doi: 10.1016/j.ab.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 10.Johnson KA, Simpson ZB, Blom T. FitSpace Explorer: An algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 2009;387:30–41. doi: 10.1016/j.ab.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 11.Grant GA, Hu Z, Xu XL. Cofactor binding to Escherichia coli D-3-phosphoglycerate dehydrogenase induces multiple conformations which alter effector binding. J. Biol. Chem. 2002;42:39548–39553. doi: 10.1074/jbc.M208019200. [DOI] [PubMed] [Google Scholar]
- 12.Fenton AW, Reinhart GD. Mechanism of substrate inhibition in Escherichia coli phosphofructokinase. Biochemistry. 2003;42:12676–12681. doi: 10.1021/bi0349221. [DOI] [PubMed] [Google Scholar]
- 13.Cabrera R, Ambrosio AL, Garratt RC, Guixé V, Babul J. Crystallographic structure of phosphofructokinase-2 from Escherichia coli in complex with two ATP molecules. Implications for substrate inhibition. J. Mol. Biol. 2008;383:588–602. doi: 10.1016/j.jmb.2008.08.029. [DOI] [PubMed] [Google Scholar]
- 14.Quinsey NS, Luong AQ, Dickson PW. Mutational analysis of substrate inhibition in tyrosine hydroxylase. J. Neurochem. 1998;71:2132–2138. doi: 10.1046/j.1471-4159.1998.71052132.x. [DOI] [PubMed] [Google Scholar]
- 15.Tricot C, Villeret V, Sainz G, Dideberg O, Stalon V. Allosteric regulation in Pseudomonas aeruginosa catabolic ornithine carbamoyltransferase revisited: association of concerted homotropic cooperative interactions and local heterotropic effects. J. Mol. Biol. 1998;283:695–704. doi: 10.1006/jmbi.1998.2133. [DOI] [PubMed] [Google Scholar]
- 16.LiCata VJ, Allewell NM. Is substrate inhibition a consequence of allostery in aspartate transcarbamylase. Biophys. Chem. 1997;64:225–234. doi: 10.1016/s0301-4622(96)02204-1. [DOI] [PubMed] [Google Scholar]
- 17.Leoncini R, Pagani R, Marinello E, Keleti T. Double inhibition of L-threonine dehydratase by aminothiols. Biochim. Biophys. Acta. 1989;994:52–58. doi: 10.1016/0167-4838(89)90061-7. [DOI] [PubMed] [Google Scholar]
- 18.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]