

Abstract
Calcium ions have been implicated in apoptosis for many years, however the precise role of this ion in the cell death process remains incomplete. We have extensively examined the role of Ca2+ on nuclear degradation in vitro using highly purified nuclei isolated from non-apoptotic rat thymocytes. We show that these nuclei are devoid of CAD (caspase-activated DNase), and DNA degradation occurs independent of caspase activity. Serine proteases rather than caspase-3 appear necessary for this Ca2+-dependent DNA degradation in nuclei. We analyzed nuclei treated with various concentrations of Ca2+ in the presence of both a physiological (140 mM) and apoptotic (40 mM) concentration of KCl. Our results show that a 5-fold increase in Ca2+ is required to induce DNA degradation at the physiological KCl concentration compared to the lower, apoptotic concentration of the cation. Ca2+-induced internucleosomal DNA degradation was also accompanied by the release of histones, however the apoptotic-specific phosphorylation of histone H2B does not occur in these isolated nuclei. Interestingly, physiological concentrations of K+ inhibit both Ca2+-dependent DNA degradation and histone release suggesting a reduction of intracellular K+ is necessary for this apoptosis-associated nuclear degradation in cells. Together, these data define an inherent caspase-independent catabolic pathway in thymocyte nuclei that is sensitive to physiological concentrations of interacellular cations.
Keywords: Apoptosis, Nuclease, CaCl2, DNA fragmentation, Histone H2B
Introduction
Intra and extra-cellular ions play an important role in a variety of cellular functions and the concentration of intra-cellular ions is tightly regulated in cells by numerous energy-requiring cellular mechanisms [1]. For example, extra-cellular signaling pathways regulate cellular function by means of calcium ions acting through effector enzymes [2]. Alterations in cellular Ca2+ also occur during cell growth, differentiation, survival and apoptosis [3]. The regulation of cellular Ca2+ concentration is strongly associated with apoptotic states and the initiation of apoptosis [4, 5]. During the early phase of apoptosis, the Ca2+ concentration increases in the cytoplasm in various apoptotic model systems, reaching a level much higher than in normal cells [6]. Additionally, Bcl-2 and related proteins are key components of the apoptotic process that modulate endoplasmic reticulum-associated Ca2+ influx and maintain Ca2+ homeostasis [7]. However, the mechanisms regulating the increase of intracellular Ca2+ along with a role for Ca2+ during apoptosis is not completely understood [8].
An increase of Ca2+ is observed when glucocorticoids induce apoptosis in lymphocytes [5, 9] and primary thymocytes [10]. Treatment of cells in vitro with multiple apoptotic agents also induces an increase in intracellular Ca2+ (ionomycin, A23187 or thapsigargin) resulting in apoptosis morphologically and biochemically similar to glucocorticoid treatment of lymphatic cells [5, 9, 11]. This phenomenon is thought to result from the apoptotic stimulus inducing Ca2+ influx into the cytoplasm from the extra-cellular environment, as well as release of Ca2+ from the endoplasmic reticulum pools. The elevated Ca2+ concentration in the cytoplasm results in a further Ca2+ influx into both mitochondria and nuclei. In nuclei, Ca2+ has been reported to modulate gene transcription to activate apoptosis-associated enzymes that initiate DNA fragmentation and chromatin condensation [12]. Apoptotic DNA degradation is largely thought to occur by caspase-dependent endonucleases such as CAD [13]. Interestingly, Lechardeur et al. [14] has determined that ICAD/CAD exist in both the cytoplasmic and nuclear fraction of apoptotic cells, however the role of caspase-independent pathways in DNA degradation is less well defined [15].
Studies using in vitro systems have shown that DNA degradation can be induced in isolated nuclei from non-apoptotic thymocytes by the addition of Ca2+ [16], suggesting that Ca2+-dependent endonucleases may pre-exist in the nuclei in a repressed state in non-apoptotic cells. Activation of these endonucleases during apoptosis to digest chromosomal DNA and produce DNA fragmentation has been proposed as a mechanism for the propagation of apoptosis [17]. Therefore, it has been proposed that the increase in Ca2+ concentration could be one of the triggers for the initiation of apoptosis [8], as well as being a late component of the process. Thus, an open issue is how the activation of these endogenous endonucleases is repressed in the nuclei of non-apoptotic cells.
Previous work from our laboratory has suggested that inhibition of apoptosis in non-apoptotic cells is caused by repressing the activation and activity of proapoptotic enzymes including endonucleases and proteases via maintenance of a homeostatic concentration of intracellular potassium [18]. Normal cells maintain high intracellular potassium in the range of 120 to 150 mM [19]. These studies have shown that apoptosis-associated enzymes are activated concomitantly with a decrease in K+ concentration to approximately 30 to 50 mM [20, 21]. Based on these data, we have hypothesized that the physiological concentration of K+ prevents the activation of various apoptotic-associated enzymes. Consequently, these apoptotic enzymes are activated or disinhibited by ion fluxes that also result in a change in cell volume, or cell shrinkage during apoptosis [18].
We have now extensively examined the effect of Ca2+ on DNA degradation and chromatin structure in isolated nuclei obtained from non- apoptotic rat thymocytes, and evaluated the relationship between Ca2+-dependent DNA degradation and intracellular K+. We show here that thymocyte nuclei contain an endogenous calcium-activated, caspase-independent nuclease that results in apoptotic DNA fragmentation and histone release. Furthermore, both Ca2+-dependent nuclear degradation and histone release are highly sensitive to inhibition by physiological levels of intracellular cations.
Materials and Methods
Isolation of nuclei
Adrenalectomized rats (male, Sprague Dawley) around one to two months old (body weight, 60 to 75 g) were decapitated. Thymus was surgically removed and was minced with scissors in PBS solution. The minced tissues were shaken and free thymocyte cells were released. The cell suspension was filtered through a nylon mesh (200 μm Nitex mesh, Tetko Inc., New York, NY) twice and free thymocytes were recovered by centrifugation at room temperature. Animal studies were done in accordance with institutional guidelines for the care and use of laboratory animals. For the isolation of nuclei from thymocytes, methods were modified from Widlak et al [22]. The thymocytes were washed once in isolation buffer (20 mM Hepes pH 7.5, 10 mM KCl, 3 mM MgCl2, 0.25 M sucrose, 1 mM DTT and 1 mM PMSF). The cells were resuspended in isolation buffer, and treated with 0.25 % NP-40 in buffer for 10 min at 4°C. The buffer containing 1.2 M sucrose was underlayed as a cushion and centrifuged. The nuclear pellet was washed once in isolation buffer. Aliquoted nuclei were either used immediately, or stored at −80°C with 50% glycerol in the buffer and used within two weeks. We observed no comparable difference between fresh or frozen nuclei that were used interchangeable in our experiments within this two-week period.
Incubation of nuclei with CaCl2
Nuclei (2 × 106) were suspended in 200 μl of incubation buffer (20 mM Hepes pH 7.5, 3 mM MgCl2, 0.25 M sucrose, 1 mM DTT and 1 mM PMSF) and incubated with or without CaCl2 for 60 or 90 min at 37°C. In this incubation system, effect of ions (monovalent and divalent cations), their chelaters, and various chemicals (inhibitors for caspase or non-caspases, and K+ cations containing compounds) were examined. For the examination of the effect of osmolarity on the Ca2+-dependent DNA degradation, nuclei were incubated with or without 1.0 mM CaCl2 in the buffer containing additional sucrose (330 mM and 530 mM) that is equivalent to the addition of 40 mM and 140 mM KCl in the buffer, respectively.
DNA analysis with gel electrophoresis
For the analysis of DNA degradation, nuclei were harvested after incubation and were treated with 1 mg/ml of DNase-free RNase for 30 min at 37°C, and then 400 μg/ml proteinase K overnight. DNA (1 or 2 μg) was run on 1.8 % agarose gel for 90 min at 100 V. The DNA was stained with 5 μg/ml of ethidium bromide. A size marker of DNA (100 base pairs of DNA ladder, Invitrogen) was run together with the DNA samples. DNA patterns were photographed with a Spectroline UV-transilluminator. For the fluorometric TUNEL analysis, CaCl2 treated nuclei were fixed with 4% paraformaldehyde and plated on cover slips. The nuclei were examined with a fluorometric TUNEL assay kit (Promega) and observed with an immunoflurescent microscope (Olympus IX70, AX10: Version 3.0) (Magnification, X 200).
Pulsed field gel electrophoresis
For the analysis of higher molecular weight DNA fragments, pulsed field gel electrophoresis (PFGE) was conducted by the method of Hughes and Cidlowski [23]. Nuclei (2 × 107) were incubated in isolation buffer containing 40 or 140 mM KCl with or without 1 mM CaCl2. After 90 min at 37°C, the nuclei were then treated with RNase for 1 h. The nuclei were suspended in 0.5 % low melting temperature agarose, (NuSieve GTG Agarose, CAMBREX) in PBS 37°C, transferred to a plug mold, and cooled to 4°C before extruding into 100 mM EDTA and 1 % N-sarcosyl for lysis at 37°C overnight. The plugs were then treated with 1 mg/ml proteinase K in the same buffer overnight at 50°C. The plugs were embedded in a 1% agarose gel (SeaKem GTG, Cambrex) and subjected to PFGE using a Beckmann Geneline II system. The DNA was stained with 0.5 μg/ml of ethidium bromide for 3 h. DNA size standards for PFGE were Lamda DNA ladder, MW, 48.5 to 1000 kb, and H. Wingei, MW, 1 to 3 mb (BioRad).
SDS gel electrophoresis and Western blotting
For the detection of histones, soluble chromatin in lysis buffer was directly mixed with SDS loading buffer and boiled for 5 min, then run on a 14% SDS gel (Invitrogen Inc.) at 150 V for 2 h. Proteins were stained with 0.2 % Commassie Brilliant blue. For the detection of caspase 3 and CAD, cytoplasmic or nuclear proteins (50 μg) were run on the SDS gel. Western blots were carried out using an anti-cleaved caspase 3 antibody (Cell Signaling) or anti-CAD antibody (Santa Crutz). During analysis of histone H2B phosphorylation, thymocytes were treated with 1 μM dexamethasone for 4 h and nuclei were incubated with CaCl2 and harvested. Histones were extracted [24] and western blotted against anti-phospho S14H2B (Upstate) [25].
Analysis with Phase, Confocal and Electron Microscopy
Thymocyte cells and nuclei were observed by phase contrast microscopy (Nikon eclipse; TE200-E, SPOT version 4.0.6). For confocal microscopy, cells and nuclei were stained with Hoechst 33342 (5 μg/ml final) for 30 min, washed in their appropriate buffer and examined on a LSM 510 UV mounted on an Axiovert 200M microscope (Carl Zeiss, Inc.) with a Plan-Apo 63× oil N.A.=1.4 objective lens to obtain simultaneous DIC and UV images. Hoechst was excited at 361 nm from an Enterprise ion laser (Coherent Laser, Auburn, CA), and emission was collected with a 385LP filter. For the analysis by electron microscopy, pellets of the thymocytes or nuclei were fixed with a modified Karnovsky fixate. Each sample was subsequently processed and embedded in Spurr’s resin. Thin sections (approximately 90 nm) from each epoxy block were cut, mounted on 200-mesh copper grid, stained with 10 % methanolic uranyl acetate and Reynold’s lead citrate, and then examined on a JEOL 100S transmission electron microscope.
Measurement of caspase-3 activity
Caspase-3-like protease activity was measured using a fluorometric assay according to the recommendation of the manufacturer (Kamiya Biomedical Co., Thousand Oaks, CA). For the preparation of samples, thymocytes (5 X 107 cells) were lysed by sonication in lysis buffer or, 0.4 N NaCl soluble nuclear extracts were prepared from the nuclei with or without 1 mM CaCl2. Debris was pelleted at 100,000 × g for 20 min. The supernatant was saved, and the amount of protein was measured. Reaction mixture (500 μl) consisted of 420 μl of caspase/CPP32 buffer (2 mM dithiothreitol, 0.1M HEPES pH 7.4, 1% sucrose, 0.1% CHAPS), 50 μl of 100 mM DTT, 10 μl of 2.5 mM substrate solution (Z-DEVD-AFC, Kamiya Biochemical Co.) and 20 μl of sample proteins (10, 25 and 50 μg). The mixture was incubated for 1 h at 30°C with or without 100 μM of noncompetitive inhibitor Z-DEVD-aldehyde (DEVD-al). Aliquoted samples were further incubated for 12 h at 30°C, and their fluorescence was determined at 400 nm (excitation) and 505 nm (emission). A standard curve of fluorescence versus free AFC (7-amino-4-trifluoromethyl coumarin, Sigma) was used for the positive control, and for the calculation of a specific activity of caspase-3-like enzymes in samples. FU value of each sample was subtracted with the FU value of no sample proteins.
Results
Morphology and the examination of CAD in isolated thymocyte nuclei
Following the isolation of rat thymocyte nuclei from non-apoptotic cells, we analyzed the nuclear structure by phase microscopy (Fig. 1a, left), confocal microscopy (Fig. 1a, middle) and electron microscopy (Fig. 1a, right), comparing primary thymocytes to isolated nuclei. The isolated nuclei were judged intact, and free of contamination of cytoplasm by all three techniques. These data indicate that isolated nuclei from non-apoptotic thymocytes represent a pure population containing no contamination of intact thymocytes. Additionally, western blot analysis shows that only the cytoplasm of thymocytes contains CAD, while the nuclei isolated from non-apoptotic cells incubated in the absence or presence of 1mM CaCl2 do not have detectable amounts of this nuclease (Figure 1b).
Fig. 1.

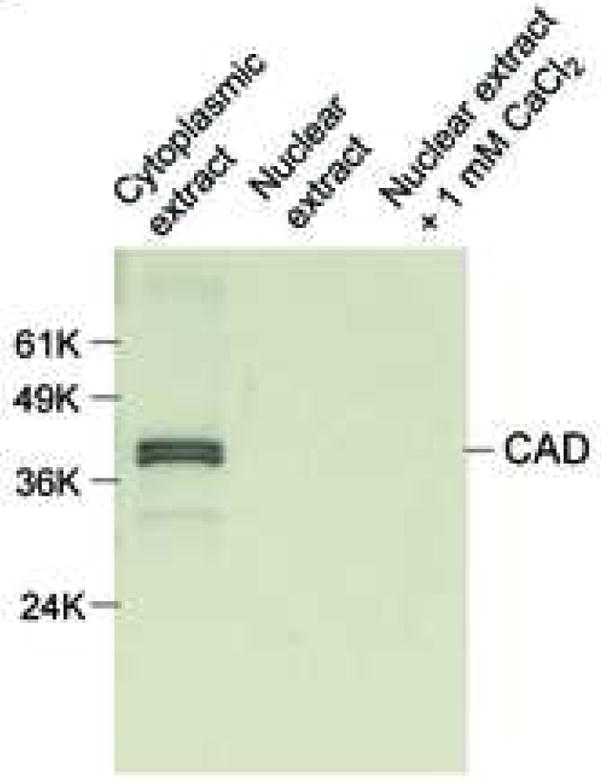
Morphological observation and the detection of CAD in nuclei isolated from viable rat thymocytes. (a) Thymocytes and isolated nuclei were obtained as described in Materials and Methods. They were observed by phase contrast microscopy (left panel; (Magnification, 200X), confocal microscopy (middle panel) or electron microscope (right panel, Magnification, 5,400X). The pictures of the nuclei indicate that cell membrane and cytoplasm are completely removed. Figures of phase contrast and electron microscopical analyses other than conforcal microscopy represent one of three independent experiments. (b) Isolated nuclei do not contain CAD. Fifty ug of cytosolic and nuclear proteins were analyzed by western blot with anti-CAD antibody. The data shown represents one of four independent experiments.
Ca2+ induces DNA degradation and histone release in isolated nuclei
Isolated nuclei were further examined by a fluorometric terminal deoxynucleotidyl transferase dUTP-biotin nick-end-labeling (TUNEL) assay to determine the extent of degraded DNA in the presence of 0.5 mM CaCl2. All nuclei treated with CaCl2 were TUNEL positive (Fig. 2a, top), whereas control nuclei showed only a few TUNEL positive nuclei (Fig. 2a, bottom). Apoptotic cells are known to shrink condensed chromatin during this cell death process, however we observed that nuclei treated with CaCl2 were not significantly compacted as judged by DAPI staining (Fig. 2a). We next examined the effect of CaCl2 treatment on DNA degradation in the concentration range of 0.1 μM to 1 mM final. Internucleosomal DNA ladders characteristic for apoptosis were observed in nuclei isolated from non-apoptotic thymocytes in buffer containing 50 μM to 1.0 mM CaCl2 (Fig. 2b). Treatment of nuclei from non-apoptotic thymocytes with CaCl2 concentration below 50 μM did not result in internucleosomal DNA degradation within the time frame of our experiments. During the incubation, internucleosomal DNA degradation was observed as early as 15 min after treatment, and reached a maximum level around 60 to 90 min post treatment (data not shown). These data strongly suggests that the Ca2+-dependent DNA degradation machinery pre-exists in isolated nuclei obtained from non-apoptotic thymocytes. To examine the specificity of this Ca2+-dependent DNA degradation, we examined the effects of EDTA, a chelater of divalent cations, or EGTA, which has a high specificity for Ca2+ than EDTA, on DNA degradation. Both EDTA and EGTA markedly inhibited Ca2+-dependent DNA degradation (Fig. 2c). The inhibitory effect of EGTA (Fig. 2c, lane 8) appeared greater than EDTA (Fig. 2c, lane 7) as predicted based on their relative affinities for Ca2+.
Fig. 2.
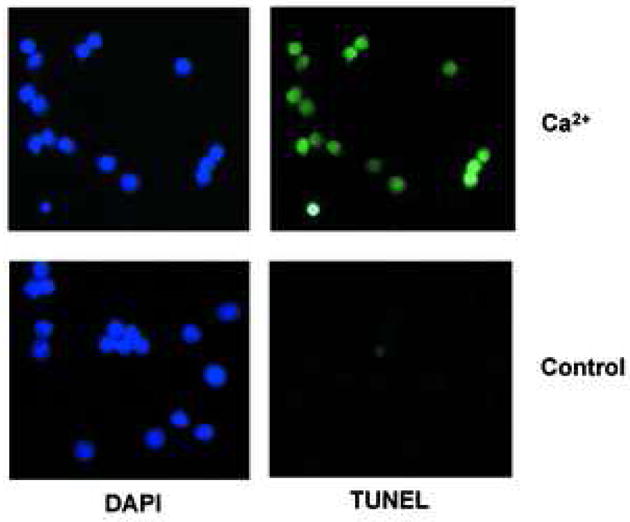
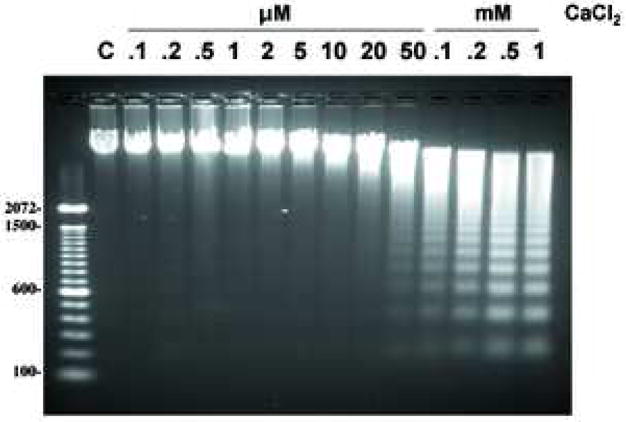

Ca2+ induces DNA cleavage and histone release in isolated nuclei from non-apoptotic thymocytes. (a) Induction of DNA cleavage in nuclei with 0.5 mM CaCl2 treatment. 1× 104 nuclei were treated with a fluorometric TUNEL assay (right) and counter stained with DAPI (left). The data shown represents one of three independent experiments. (b) Induction of DNA degradation with various concentrations of CaCl2 ranging from 0.1 μM to 1 mM. C is a control. Nuclei (2 × 106) were incubated in vitro at 37°C for 90 min with or without CaCl2 as described in Materials and Methods. Nuclei were harvested and treated with protease K and RNase. DNA (2 μg) from the nuclei was run on an agarose gel and stained with ethidium bromide. Typical internucleosomal DNA ladders were observed starting around 50 μM CaCl2. The data shown represents one of at lease three independent experiments. (c) Divalent cations and Ca2+ specific DNA degradation in isolated nuclei. EDTA and EGTA block Ca2+ dependent DNA degradation in nuclei obtained from thymocytes. The data shown represents one of two independent experiments.
We also evaluated the effect of CaCl2 treatment on histone release and observed the release of histones from CaCl2 treated nuclei, specifically at the higher concentration of CaCl2 (over 0.2 mM) that accompanies internucleosomal DNA fragmentation (Fig. 3a). Previous studies have shown that histone H2B is specifically phosphorylated during apoptosis in intact cells [24, 25], however it is unknown if Ca2+ treatment of isolated nuclei can also promote this apoptotic-specific phosphorylation of H2B. Figure 3b shows that H2B is phosphorylated in thymocytes induced to undergo apoptosis by dexamethasone, whereas H2B in isolated nuclei treated with 1 mM Ca2+ was not phosphorylated, despite the occurrence of degraded DNA. Thus our data suggest that while Ca2+ alone can induce the degradation of nucleosomes, as well as DNA, H2B phosphorylation only occurs in nuclei in intact cells and is not necessary for the occurrence of DNA degradation in isolated nuclei in the presence of elevated Ca2+.
Fig. 3.
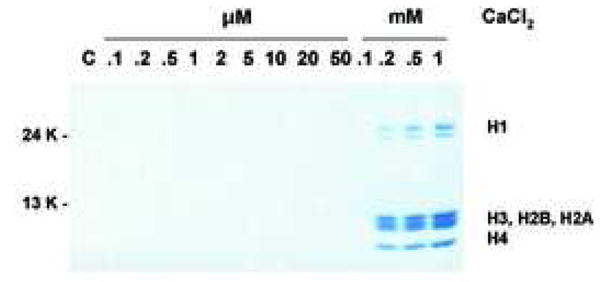
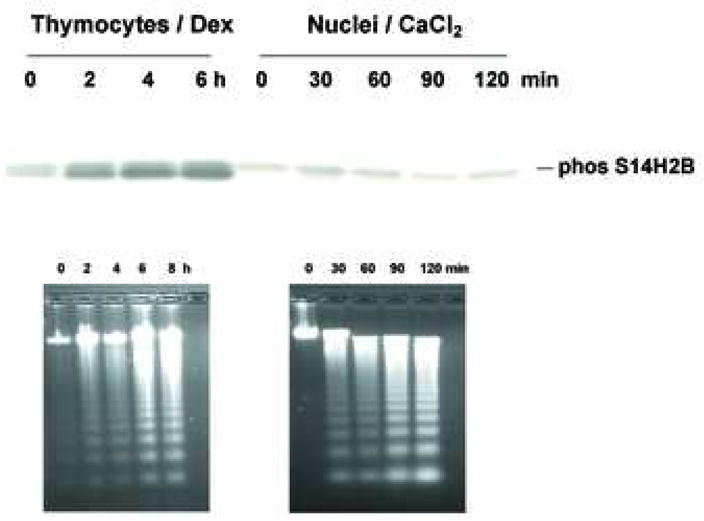
Histone release without H2B phosphorylation. (a) Histone release accompanying DNA fragmentation during incubation with CaCl2. After incubation of nuclei at each CaCl2 concentration as described for figure 2b, aliquots of lysis buffer soluble fractions were analyzed by SDS PAGE. Proteins were stained with Commasie blue. The data shown represents one of three independent experiments. (b) Ca2+ does not induce apoptosis-specific H2B phosphorylation in isolated nuclei. Western blot analysis with anti-phospho Ser14H2B for thymocytes cultured with dexamethasone or nuclei incubated with 1 mM of CaCl2 at 37°C for the indicated times. Histones were extracted were extracted from these samples and 10 μg were blotted against anti-phospho S14H2B. DNA was extracted and analyzed by agarose gel electrophoris. The data shown represents one of three independent experiments.
Examination of protease involvement during Ca2+-dependent DNA degradation
It is well known that numerous proteases are activated in apoptotic cells. We therefore analyzed whether proteases exist or are activated in isolated nuclei during DNA degradation observed in response to CaCl2. Figure 4a shows that none of the three classical caspase inhibitors (Z-VAD for pan caspases, DEVD for caspase-3, and IETD for casapase-8) altered the Ca2+-dependent internucleosomal DNA degradation in isolated nuclei. Additionally, western blot analysis for caspase-3 showed that isolated non-apoptotic nuclei have a negligible amount of either intact or cleaved caspase-3 (Fig. 4a, ii).
Fig. 4.

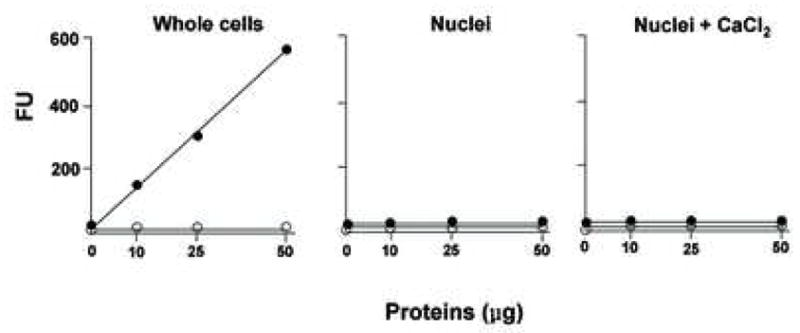
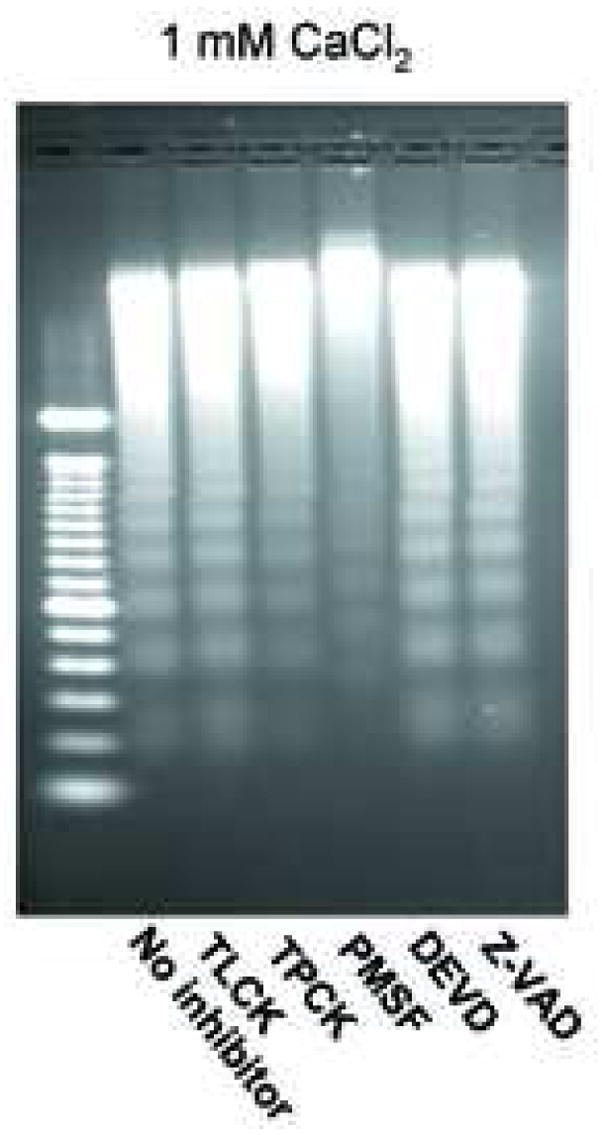
Effect of protease inhibitors on Ca2+-dependent DNA degradation. (a) i; Analysis of caspase inhibitors on Ca2+-dependent DNA degradation. Nuclei were incubated with 0.5 mM CaCl2 and 100 μM of caspase inhibitors in buffer containing 40 mM KCl prior to DNA was analyzed. C and Ø are control and calcium treatment without inhibitor, respectively. Z, D and I are: Z-VAD, DEVD and IETD with calcium, respectively. ii; Western blotting of caspase 3. Anti-caspase 3 antibody was used against protein extracts (100 μg of protein) from cytoplasm and nuclear extracts from non-apoptotic thymocytes. The data shown represents one of three independent experiments. (b) Measurement of caspase-3 activities in the nuclear proteins. Cell lysate were prepared from thymocytes and 0.4 N NaCl soluble nuclear extracts were obtained from nuclei or nuclei incubated with 1 mM CaCl2 for 90 min at 37°C. Capase-3 activity was measured based on the ability to cleave specific fluorogenic substrates (Z-DEVD-AFC-cleavable activity) in the extracts. FU: Fluorescent units. The data shown represents two experiments. (c) Effect of protease inhibitors on the Ca2+-dependent DNA degradation. Nuclei were incubated with 1 mM Ca2+ and a protease inhibitor (100 μM each) in buffer containing 40 mM KCl. The data shown represents one of two independent experiments.
Caspase-3-like protease activity was also directly analyzed in cytosolic and nuclear extracts by a fluorometric assay using a peptide of caspase-3 conjugated with a fluorescent dye, AFC (Figure 4b). In the normal cytosolic thymocyte lysate, caspase-3 activity is evident in a protein-dependent manner (filled circles in Fig 4b, left). This caspase-3 activity was completely inhibited by a caspase-3 inhibitor, DEVD-FMK (open circles in Fig 4b, left). In contrast, no significant caspase-3 activity was observed in the soluble nuclear proteins from isolated nuclei (Fig 4b, middle). Additionally, caspase-3 activity was also not detected in nuclei incubated with 1 mM CaCl2 (Fig 4b, right), despite the extensive occurrence of nuclear DNA degradation (as shown in Fig. 2b). Together these data in conjunction with the data on the incubation of nuclei with caspase inhibitors and western blot analysis suggest that Ca2+-dependent DNA degradation in nuclei is independent of caspase-3-like activity.
We further evaluated other protease inhibitors, such as the serine protease inhibitors TLCK, TPCK and PMSF, for their ability to inhibit Ca2+-dependent DNA degradation. Among the three serine protease inhibitors examined, TLCK had not effect, whereas both TPCK and PMSF showed a partial inhibition of degraded DNA, with PMSF appearing to be more effective (Fig. 4c). Again, the caspase inhibitors DEVD and Z-VAD had no effect on preventing DNA degradation (Fig. 4c). Our experiments with protease inhibitors suggest that isolated nuclei do not have caspases, but rather caspase-independent protease activities, which may be involved in nuclear DNA degradation triggered by calcium.
Physiological concentrations of K+ inhibit Ca2+-dependent DNA degradation
Since the intracellular concentration of K+ rapidly decreases in the early phase of apoptosis, the effect of K+ ion concentration was extensively examined during Ca2+-dependent DNA degradation in vitro. Forty mM of KCl was used to reflect the KCl concentration found in apoptotic cells [20, 21], whereas 140 mM of KCl was used as a physiological concentration of KCl found in normal healthy cells [19]. In the presence of 0.5 mM CaCl2, a fluorometric TUNEL assay was performed on isolated nuclei treated in the presence of either 40 or 140 mM KCl (Fig. 5a). TUNEL positive nuclei were observed for a majority of the nuclei incubated in the 40 mM KCl buffer in the presence of CaCl2, whereas the occurrence of TUNEL-positive staining was significantly reduced at 140 mM KCl. These data suggest that Ca2+-dependent DNA cleavage in nuclei can occur at low (apoptotic) concentrations of KCl, but is largely inhibited at the physiological concentrations of KCl. We also examined the effect of various KCl concentrations ranging from 5 to 150 mM in the presence of CaCl2 to determine the extent of Ca2+-dependent DNA degradation in isolated nulei. A significant inhibitory effect of KCl was observed at 90 mM KCl (Fig. 5b). Over 110 mM, KCl has a profound inhibitory effect on internucleosomal DNA degradation. Interestingly, at the lower concentration of K+ previously reported to reflect the intracellular concentration of K+ in apoptotic cells (30 to 50 mM K+) [20, 21], a distinct pattern of internucleasomal DNA degradation was observed (Fig. 5b).
Fig. 5.
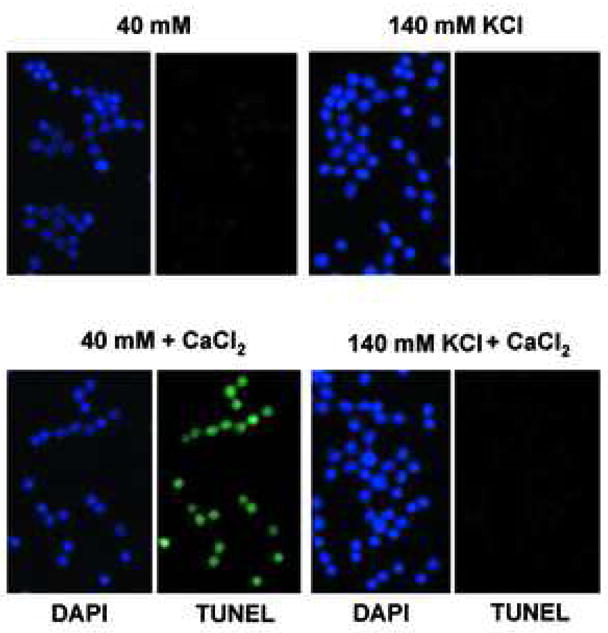

Physiological concentration of K+ blocks Ca2+-dependent DNA degradation in nuclei. (a) TUNEL assay, Nuclei (1 × 104) were incubated under “apoptotic” (40 mM) and “physiological” concentration (140 mM) of K+ with 1 mM CaCl2. Right, fluorescent TUNEL assay. Left, counter staining with DAP I. The data shown represents at least three experiments. (b) Analysis of Ca2+-dependent DNA degradation under various concentration of K+. DNA was extracted and run on agarose gel electrophoresis. Nuclei (2 × 106) were incubated with 1 mM CaCl2 together with an increasing concentration of KCl. For each sample, 1 μg of DNA was analyzed. “Ø” denotes control nuclei maintained on ice. The data shown represents one of three independent experiments.
Previous studies examining the effect of high KCl on DNA degradation has focused mainly on increasing concentrations of KCl, thus we wished to determine how various concentrations of CaCl2 at either an apoptotic or physiologic concentration of KCl would effect Ca2+-dependent DNA degradation. At a physiological concentration of K+ (140 mM), internucleosomal DNA ladders were observed at approximately 1 mM CaCl2 (Fig. 6a, as indicated by an arrow), with DNA degradation being significantly reduced at the lower concentrations of Ca2+. In contrast, in the presence of 40 mM K+, a buffer reflecting an “apoptotic concentration” of K+, the effective concentration of Ca2+ necessary for DNA degradation was significantly lower than observed in the 140 mM K+ buffer (Fig. 6b). Under this apoptotic K+ concentration, DNA ladders were observed at concentrations of CaCl2 as low as 0.2 mM (Fig. 6b, indicated by an arrow). Together, these data suggest a 5-fold lower concentration of the stimulus is required in the presence of an apoptotic concentration of KCl, compared to the physiological level of KCl. Interestingly, under both KCl conditions, CaCl2 in the range of 8 to 10 mM had a slight inhibitory effect on the extent of DNA degradation. Since it is known that other cations can have a similar inhibitory effect on DNA degradation [18], our data suggests that high concentrations of CaCl2 may prevent the occurrence of DNA fragmentation, even under conditions where CaCl2 is used as the stimulatory agent.
Fig. 6.


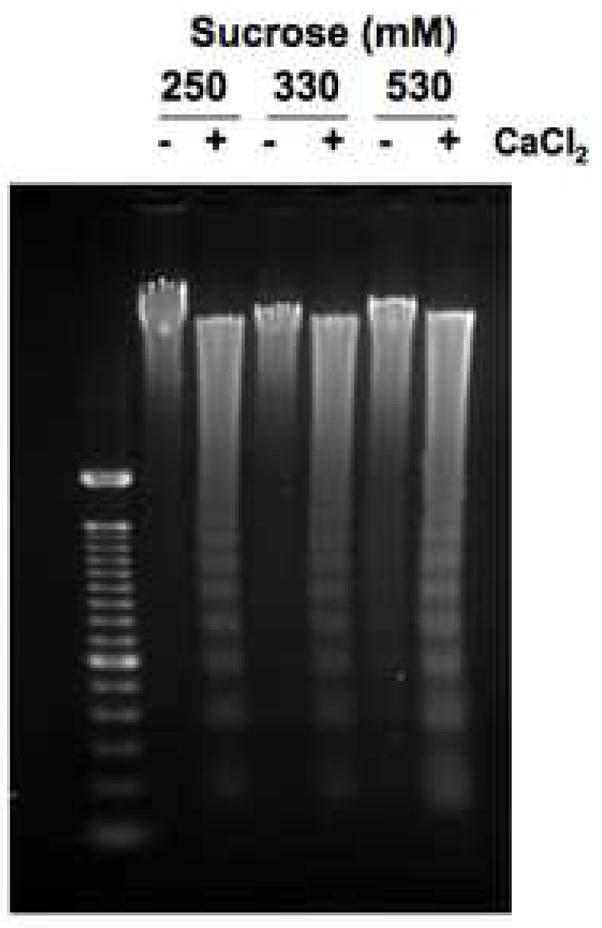
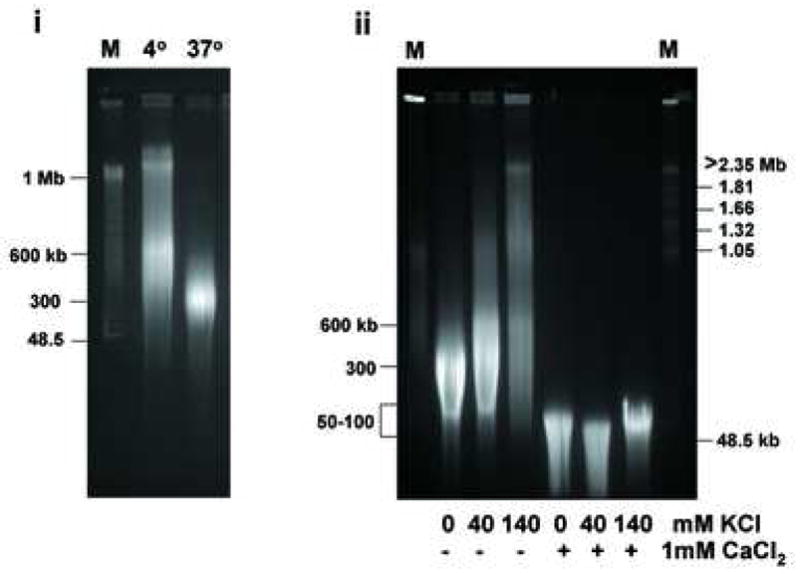
Extensive analysis of KCl effect on DNA degradation. (a) The effect of Ca2+ under the physiological concentration of K+ at 140 mM on Ca2+-dependent DNA degradation. Nuclei were incubated in isolation buffer containing 140 mM KCl with an increasing concentration of CaCl2. The arrow indicates the initial concentration of CaCl2 with considerable internucleosomal DNA degradation. “Ø” denotes control nuclei maintained on ice that was not exposed to either 40 mM or 140 mM KCl buffer. “0” denotes nuclei resuspended in the respective KCl buffer however in the absence of CaCl2. The data shown represents one of three independent experiments. (b) The effect of Ca2+ in the “apoptotic concentration” of K+ at 40 mM on Ca2+-dependent DNA degradation. Nuclei were incubated in the same condition as above except with the buffer containing 40 mM KCl. In a buffer containing 40 mM KCl, a 5 times smaller Ca2+ concentration is sufficient to trigger the degradation of internucleosomal DNA. The data shown represents one of three independent experiments. (c) Effect of osmolarity of sucrose in buffer on the Ca2+-dependent DNA degradation. Nuclei (2 × 106) were incubated in buffer containing sucrose (330 and 530 mM equivalent concentration to the addition of 40 and 140 mM KCl, respectively), with or without 1.0 mM CaCl2. The data shown represents one of two independent experiments. (d) Pulsed field gel electrophoresis (PFGE) of DNA degradation products in the presence of K+. Nuclei (2 × 106) were incubated in buffer containing 40 or 140 mM KCl with or without 1 mM CaCl2. i, DNA degradation during the incubation of nuclei. ii, Inhibition of the Ca2+-dependent and independent DNA degradation by K+. M; size marker of DNA, C; Control. The data shown represents two experiments.
To evaluate whether the change in osmolarity resulting from adding KCl to the buffer was responsible for this inhibitory effect, DNA degradation of nuclei in sucrose containing buffer with an equivalent osmolarity to KCl was examined. Nuclei in buffer prepared using either the normal concentration of sucrose (250 mM) in the buffer; 330 mM sucrose, which is equivalent to the 40 mM KCl buffer; or 530 mM sucrose, which is equivalent to the 140 mM KCl buffer were incubated with or without CaCl2 and their DNA was analyzed by agarose gel (Fig. 6c). Increasing the osmolarity using sucrose did not prevent DNA degradation upon CaCl2 treatment, suggesting that a change of osmolarity is not responsible for the inhibition of DNA fragmentation, thus demonstrating specificity based on either 40 or 140 mM KCl in the buffer.
We also examined degradation of large DNA molecules, in the mega-base pair range, in nuclei incubated in the absence and presence of CaCl2 by pulse field gel electrophoresis (PFGE). First, DNA from nuclei maintained at 4°C was compared with DNA from nuclei incubated at 37°C (Fig. 6d, i). When nuclei were maintained at 4°C, DNA molecules between 600 kb to 2 mega base pairs (mb) were observed, whereas virtually no DNA of this size existed in nuclei at 37 °C. In contrast at 37 °C, the DNA was fragmented to an average size of approximately 300 kb, even in the absence of CaCl2. However, addition of either 40 or 140 mM KCl consistently suppressed this degradation (Fig. 6d, ii, lanes 2 and 3), indicating that the physiological concentration of K+ also inhibits fragmentation of high molecular weight DNA. The addition of 1 mM CaCl2 resulted in the large DNA being degraded into fragments smaller than 100 kb even in the presence of 40 or 140 mM KCl (Fig. 6d, ii). Thus physiological K+ can inhibit both mega base and internucleosomal DNA degradation in the absence of exogenous Ca2+. In contrast, in the presence of Ca2+, physiological K+ can only inhibit DNA fragmentation at the level of internuclesomal ladder formation, and does not prevent the degradation of large DNAs of more than 100 kb in size.
Prevention of histone release at a physiological concentration of K+
SDS gel electrophoresis was conducted to further evaluate the release of histones from nuclei treated with CaCl2 as shown earlier (see Figure 3a), in the presence of either the apoptotic or physiologic concentration of KCl. In 40 mM KCl with 0.5 mM CaCl2, core histones (H2A, H2B, H3 and H4) were clearly observed around 13 Kd, and H1 was observed around 32 K (Fig. 7, right), along with a robust amount of degraded DNA (Fig. 7, left). In contrast, the amount of histone released in the presence of 140 mM KCl was negligible (Fig. 7, right), coordinate with the inhibition of internucleosomal DNA (Fig. 7, left). Together these data indicate that a physiological concentration of K+ prevents both histone release along with internucleosomal DNA degradation during Ca2+-treatment of isolated nuclei.
Fig. 7.
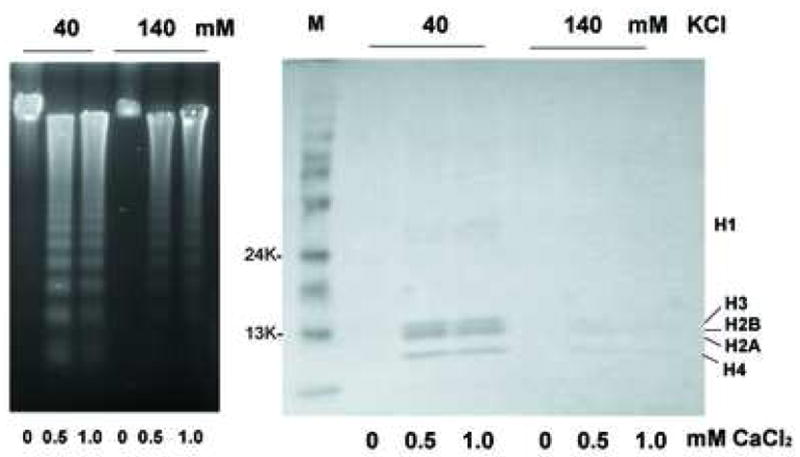
The prevention of histone release by physiological concentration of KCl. Nuclei were treated with 0.5 or 1 mM CaCl2 under the KCl at either 40 or 140 mM KCl. Soluble chromatin was run on SDS gel electrophoresis and stained with Coommacie Briliant Blue (right). The DNA was analyzed from the identical chromatin fractions (left). The data shown represents one or three independent experiments.
Discussion
Mechanism of DNA degradation in isolated nuclei by Ca2+
We have examined apoptotic DNA degradation that occurs in the presence of Ca2+ during incubation with highly purified nuclei from non-apoptotic cells. Our studies showed that Ca2+ induces internucleosomal DNA degradation in isolated nuclei from non-apoptotic thymocytes in the absence of CAD. We also analyzed in detail the effects of Ca2+ on DNA degradation in isolated purified nuclei in regards to the effects of various concentrations of KCl known to prevent apoptotic nuclease activity. Our data indicate that greater than 1 mM Ca2+ is required for DNA fragmentation in the presence of a physiological concentration of KCl. In contrast, at an apoptotic concentration of KCl, a 5-fold lower Ca2+ concentration results in a similar extent of DNA fragmentation. We show that the inhibition of DNA cleavage is not due to an overall increase in osmolality of the buffer, as comparable concentrations of sucrose did not prevent nuclease activity. The presence of physiological concentrations of KCl also prevents histone release, while the release of histones is observed at an apoptotic concentration of KCl. Finally, the phosphorylation of histone H2B, a characteristic often implicated with apoptotic cells, does not occur in isolated nuclei upon Ca2+ treatment, suggesting that this phosphorylation is not required for Ca2+-dependent DNA degradation. Thus, our studies indicate that the DNA degradation machinery that exists in nuclei from primary thymocytes and is activated upon CaCl2 treatment is similar to that reported in typical apoptotic cells.
Interestingly, Ca2+ can induce apoptotic DNA degradation without influence of cytoplasmic molecules, suggesting an additional mechanism to induce apoptosis. Calcium ions can easily penetrate from cytoplasm to nuclei through the nuclear envelope. Therefore, small changes of cytosolic Ca2+ cause equally rapid changes in nuclear Ca2+, consistent with the free diffusion of Ca2+ through nuclear pores [26]. It is also known that the Ca2+ concentration is approximately 100 nM in the cytoplasm and nuclei, but 100 to 1000 μM in the endoplasmic reticulum [19] and the intracellular concentration of Ca2+ can increase to approximately 300 to 500 nM upon treatment with Vitamin D compounds [27,28]. The concentration of CaCl2 necessary for extensive degradation such as internucleosomal DNA degradation in our study was 0.2 to 1.0 mM, in contrast to approximately 5 mM in a previous report [29], which is much higher than a physiological concentration of Ca2+. Our data suggest that in apoptotic cells, other factors or unique mechanisms likely exists to facilitate DNA degradation. One possible regulatory mechanism we identified is the lowering the intracellular K+ as observed during the loss of cell volume during apoptosis to facilitate Ca2+-dependent DNA degradation.
A caspase-associated endonuclease (CAD) has been identified which is Mg2+-dependent, activated by caspases, and has often been implicated in the generation of internucleosomal DNA degradation [13, 30]. We did not detect the presence of CAD in isolated nuclei from non-apoptotic thymocytes. Additionally, we found isolated nuclei to be devoid of caspase-3 protein and caspase-3-like activity in the presence and absence of Ca2+. Therefore, this model system does not appear to have either caspase activity or CAD, suggesting other endonucleases are involved in the internucleosomal DNA degradation response to CaCl2. While Ca2+-induced DNA degradation mimics the internucleosomal, ladder-like pattern observed in apoptotic cells, we can not rule out if single or multiple nucleases exists in cells that may be responsible for DNA fragmentation in vivo. In whole cells, cytoplasmic factors may influence the resulting activation of apoptotic nucleases. Additionally, this activation may depend on the specific cell type or stimulus employed to induce cell death. However, our data shows that enzymes exist in isolated nuclei that are exquisitely sensitive to the level of Ca2+ present under various cationic conditions.
Our laboratory initially described a calcium-dependent endonuclease (NUC18) from apoptotic rat thymocytes [17]. NUC18 activity was detected in nuclear extracts of thymocytes of both control and glucocorticoid-treated thymocytes, and was associated with apoptotic DNA degradation. Furthermore, we have shown that partial protein sequencing of pure NUC18 generated two peptide sequences that have a remarkable homolog to rat cyclophilin A, and other cyclophilin family members such as cyclophilin B and C [31]. These cyclophilins have a calcium/magnesium-dependent nuclease activity with biochemical and pharmacological properties similar to those of NUC18, and result in high molecular weight DNA cleavage [31]. These data in conjunction with recent genetic analysis in C. elegans raise the intriguing possibility that cyclophilin-related proteins may play a role in DNA degradation during apoptosis [32]. Another possible endonuclease among the potential apoptosis-associated endonucleases [33] is DNase-gamma as this enzyme is also Ca2+- and Mg2+-dependent, and was originally isolated from the nuclei of rat thymocytes [34].
Caspase independent DNA degradation
During the incubation of nuclei with CaCl2, we observed that isolated nuclei from non-apoptotic cells do not have caspase activity. Thus, our data also suggest that non-caspase proteases are activated in nuclei, and DNA degradation can be activated with Ca2+ in the absence of caspase-3. Previous studies indicate that endonucleases other than CAD or serine proteases are involved in DNA degradation [33]. TLCK and TPCK are known irreversible serine protease inhibitors of trypsin or trypsin-like proteases and chymotrypsin or chymotrypsin-like proteases, respectively. PMSF is known to be a more general serine protease inhibitor, along with inhibiting various cysteine proteases. However, these inhibitors have also been shown exhibit other biochemical effects on cells. Specifically, TLCK and TPCK have been shown to block activation of NFκB [35], prevent the activation of pp70(s6k), a mitogen-related kinase [36], and to suppress the processing of caspases in some model systems [37, 38]. In contrast, these two proteases have been shown to both induce and enhance cell death upon treatment with various cytotoxic agents [39–41].
McConkey [29] reported that TPCK, a serine protease inhibitor, prevented Ca2+-dependent DNA degradation in isolated thymocyte nuclei, and suggested the existence of an alternative pathway of DNA degradation mediated by a Ca2+ stimulated nuclear protease. Hughes et al. [42] has also reported that non-caspase proteases are required for chromatin degradation during apoptosis. In a study of apoptosis in human melanoma cells induced by etoposide, Bruin et al. [43] recently found that a serine protease is involved in the initiation of DNA damage-induced apoptosis. Our data showed that the broad-spectrum protease inhibitor PMSF was most effective in preventing DNA degradation upon Ca2+ treatment. Thus, it is possible that there are at least two different signaling pathways in isolated nuclei to activate endonucleases: one where by Ca2+ activates endonucleases directly; and another where by endonucleases are activated indirectly by a proteolysis resulting from Ca2+-dependent proteases.
Regulatory mechanism of apoptosis by K+ ions during DNA degradation
It has been previously reported that K+ prevents Ca2+-dependent DNA degradation [18, 44]. In the present study, we also found a suppressive effect of KCl on Ca2+ dependent DNA degradation occurring at a KCl concentration of 90 to 100 mM, with a maximum effect around the physiological range of 140 to 150 mM KCl. However, we further examined the effect of both a physiological and apoptotic concentration of KCl on Ca2+-dependent DNA degradation in response to increasing concentrations of CaCl2. In the presence of a physiological concentration of KCl (140 mM), a 5-fold high concentration of Ca2+ was required to induce internucleosomal DNA cleavage, compared to an apoptotic concentration of KCl (40 mM). Interestingly, at higher concentrations of CaCl2, a repressive effect was also observed, suggesting that while Ca2+ can induced DNA degradation, this cation may also inhibit nuclease activity in a similar manner of K+. Similarly, KCl at 140 mM in media also prevented apoptotic DNA degradation in thymocytes, as well as cultured HL-60 cells upon apoptotic stimulation (data not shown). The mechanism of preventing apoptosis by altering ionic strength is unknown, however possibilities include K+ or cationic inactivation of the endonuclease activity, or by blocking the interaction between Ca2+ and endonuclease, thus preventing the activation of the enzyme itself. Overall, these findings support a hypothesis that K+ homeostasis or maintaining a high concentration of intracellular K+ in the normal physiological range protects cells from apoptosis. Additionally, there appears to be direct relationship between the concentration of KCl and the amount of stimulus required to induce DNA degradation. Consequently, a rapid reduction of K+ in the early phase of apoptosis triggers the activation of apoptotic machinery.
Chromatin structure in isolated nuclei treated with Ca2+
In this study, we found little evidence for chromatin condensation in nuclei treated with CaCl2 at all KCl concentrations examined (0, 40 and 140 mM), supporting the notion that chromatin condensation may not be necessarily linked to or is independent of DNA degradation [45]. Previous studies have shown that histones are released in various apoptotic cells [46, 47]. Our findings indicate that 1) histones are also released in isolated nuclei treated with Ca2+ and 2) the amount of histones released corresponds approximately to the amount of internucleosomal DNA. The release of histones is prevented under physiological concentrations of KCl. Together, these findings aid our understanding of the basic mechanism of chromatin degradation in apoptotic cells.
Acknowledgments
We thank John Horton for electron microscopic analysis and for members of our laboratory for useful discussion. This research was supported by the Intramural Research Program of the NIEHS, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yu SP, Canzoniero LM, Choi DW. Ion homeostasis and apoptosis. Curr Opin Cell Biol. 2001;13:405–411. doi: 10.1016/s0955-0674(00)00228-3. [DOI] [PubMed] [Google Scholar]
- 2.Murdoch GH, Waterman M, Evans RM, Rosenfeld MG. Molecular mechanisms of phorbol ester, thyrotropin-releasing hormone, and growth factor stimulation of prolactin gene transcription. J Bio Chem. 1985;260:11852–11858. [PubMed] [Google Scholar]
- 3.Yano S, Tokumitsu H, Soderling TR. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature. 1998;396:584–587. doi: 10.1038/25147. [DOI] [PubMed] [Google Scholar]
- 4.Berridge MJ, Bootman MD, Lipp P. Calcium- a life and death signal. Nature. 1998;395:645–648. doi: 10.1038/27094. [DOI] [PubMed] [Google Scholar]
- 5.Scoltock AB, Bortner CD, St Bird GJ, Putney JW, Jr, Cidlowski JA. A selective requirement for elevated calcium in DNA degradation, but not early events in anti-Fas-induced apoptosis. J Biol Chem. 2000;275:30586–30596. doi: 10.1074/jbc.M004058200. [DOI] [PubMed] [Google Scholar]
- 6.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 7.He H, Lam M, McCormick TS, Distelhorst CW. Maintenance of calcium homeostasis in the endoplasmic reticulum by Bcl-2. J Cell Biol. 1997;138:1219–1228. doi: 10.1083/jcb.138.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rizzuto R, Pinton P, Ferrari D, Chami M, Szabadkai G, Magalhaes PJ, Di Virgilio F, Pozzan T. Calcium and apoptosis: facts and hypotheses. Oncogene. 2003;22:8619–8627. doi: 10.1038/sj.onc.1207105. [DOI] [PubMed] [Google Scholar]
- 9.Caron-Leslie LA, Cidlowski JA. Similar actions of glucocorticoids and calcium on the regulation of apoptosis in S49 cells. Mol Endocrinol. 1991;5:1169–1179. doi: 10.1210/mend-5-8-1169. [DOI] [PubMed] [Google Scholar]
- 10.Zheng LM, Zychlinsky A, Liu CC, Ojcius DM, Young JD. Extracellular ATP as a trigger for apoptosis or programmed cell death. J Cell Biol. 1991;112:279–288. doi: 10.1083/jcb.112.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Azmi S, Dhawan D, Singh N. Calcium ionophore A23187 induces apoptotic cell death in rat thymocytes. Cancer Lett. 1996;107:97–103. doi: 10.1016/0304-3835(96)04349-2. [DOI] [PubMed] [Google Scholar]
- 12.Santella L, Carafol E. Calcium signaling in the cell nucleus. FASEB J. 1997;11:1091–1109. [PubMed] [Google Scholar]
- 13.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;39:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 14.Lechardeur D, Drzymala L, Sharma M, Zylka D, Kinach R, Pacia J, Hicks C, Usmani N, Rommens JM, Lukacs GL. Determinants of the nuclear localization of the heterodimeric DNA fragmentation factor (ICAD/CAD) J Cell Biol. 2000;150:321–34. doi: 10.1083/jcb.150.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cande C, Cohen I, Daugas E, Ravagnan L, Larochette N, Zamzami N, Kroemer G. Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie. 2002;84:215–222. doi: 10.1016/s0300-9084(02)01374-3. [DOI] [PubMed] [Google Scholar]
- 16.Cohen JJ, Duke RC. Glucocorticoid activation of a calcium- dependent endonuclease in thymocyte nuclei to cell death. J Immuno. 1984;132:38–42. [PubMed] [Google Scholar]
- 17.Gaido ML, Cidlowski JA. Identification, purification, and characterization of a calcium-dependent endonuclease (NUC18) from apoptotic rat thymocytes. NUC18 is not histone H2B. J Biol Chem. 1991;266:18580–18585. [PubMed] [Google Scholar]
- 18.Hughes FM, Jr, Bortner CD, Purdy GD, Cidlowski JA. Intracellular K+ suppresses the activation of apoptosis in lymphocytes. J Biol Chem. 1997;272:30567–30576. doi: 10.1074/jbc.272.48.30567. [DOI] [PubMed] [Google Scholar]
- 19.Hajnoczky G, Davies E, Madesh M. Calcium signaling and apoptosis. Biochem Biophys Res Commun. 2003;304:445–454. doi: 10.1016/s0006-291x(03)00616-8. [DOI] [PubMed] [Google Scholar]
- 20.Barbiero G, Duranti F, Bonelli G, Amenta JS, Baccino FM. Intracellular ionic variations in the apoptotic death of L cells by inhibitors of cell cycle progression. Exp Cell Res. 1995;217:410–418. doi: 10.1006/excr.1995.1104. [DOI] [PubMed] [Google Scholar]
- 21.Hughes FM, Jr, Cidlowski JA. Glucocorticoid-induced thymocyte apoptosis: protease-dependent activation of cell shrinkage and DNA degradation. J Steroid Biochem Mol Biol. 1998;65:207–217. doi: 10.1016/s0960-0760(97)00188-x. [DOI] [PubMed] [Google Scholar]
- 22.Widlak P, Palyvoda O, Kumala S, Garrard WT. Modeling apoptotic chromatin condensation in normal cell nuclei. Requirement for intranuclear mobility and action involvement. J Biol Chem. 2002;277:21683–21690. doi: 10.1074/jbc.M201027200. [DOI] [PubMed] [Google Scholar]
- 23.Hughes FM, Jr, Cidlowski JA. Utilization of an in vitro assay to evaluate chromatin degradation by candidate apoptotic nucleases. Cell Death Differ. 1997;4:200–2008. doi: 10.1038/sj.cdd.4400221. [DOI] [PubMed] [Google Scholar]
- 24.Ajiro K. Histone H2B phosphorylation in mammalian apoptotic cells. An association with DNA fragmentation. J Biol Chem. 2000;275:439–443. doi: 10.1074/jbc.275.1.439. [DOI] [PubMed] [Google Scholar]
- 25.Cheung WL, Ajiro K, Samejima K, Kloc M, Cheung P, Mizzen CA, Beeser A, Etkin LD, Chernoff J, Earnshaw WC, Allis CD. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell. 2003;113:507–517. doi: 10.1016/s0092-8674(03)00355-6. [DOI] [PubMed] [Google Scholar]
- 26.Mazzanti M. Ion Permeability of the Nuclear Envelope. News Physiol Sci. 1998;13:44–50. doi: 10.1152/physiologyonline.1998.13.1.44. [DOI] [PubMed] [Google Scholar]
- 27.Mathiasen IS, Sergeev IN, Bastholm L, Elling F, Norman AW, Jaattela M. Calcium and calpain as key mediators of apoptosis-like death induced by vitamin D compounds in breast cancer cells. J Biol Chem. 2002;277:30738–30745. doi: 10.1074/jbc.M201558200. [DOI] [PubMed] [Google Scholar]
- 28.Sergeev IN. Calcium as a mediator of 1,25-dihydroxyvitamin D3-induced apoptosis. J Steroid Biochem Mol Biol. 2004;89–90:419–425. doi: 10.1016/j.jsbmb.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 29.McConkey DJ. Calcium-dependent, interleukin 1-converting enzyme inhibitor-insensitive degradation of lamin B1 and DNA fragmentation in isolated thymocyte nuclei. J Biol Chem. 1996;271:22398–22406. doi: 10.1074/jbc.271.37.22398. [DOI] [PubMed] [Google Scholar]
- 30.Nagata S. Apoptotic DNA fragmentation. Exp Cell Res. 2000;256:12–18. doi: 10.1006/excr.2000.4834. [DOI] [PubMed] [Google Scholar]
- 31.Montague JW, Gaido ML, Frye C, Cidlowski JA. A calcium-dependent nuclease from apoptotic rat thymocytes is homologous with cyclophilin. Recombinant cyclophilins A, B, and C have nuclease activity. J Biol Chem. 1994;269:18877–18880. [PubMed] [Google Scholar]
- 32.Parrish JZ, Xue D. Cuts can kill: the roles of apoptotic nucleases in cell death and animal development. Chromosoma. 2006;115:89–97. doi: 10.1007/s00412-005-0038-0. [DOI] [PubMed] [Google Scholar]
- 33.Counis MF, Torriglia A. DNases and apoptosis. Biochem Cell Biol. 2000;78:405–414. [PubMed] [Google Scholar]
- 34.Shiokawa D, Ohyama H, Yamada T, Takahashi K, Tanuma S. Identification of an endonuclease responsible for apoptosis in rat thymocytes. Eur J Biochem. 1994;226:23–30. doi: 10.1111/j.1432-1033.1994.tb20022.x. [DOI] [PubMed] [Google Scholar]
- 35.Breithaupt TB, Vazquez A, Baez I, Eylar EH. The suppression of T cell function and NF(kappa)B expression by serine protease inhibitors is blocked by N-acetylcysteine. Cell Immunol. 1996;173:124–130. doi: 10.1006/cimm.1996.0258. [DOI] [PubMed] [Google Scholar]
- 36.Grammer TC, Blenis J. The serine protease inhibitors, tosylphenylalanine chloromethyl ketone and tosyllysine chloromethyl ketone, potently inhibit pp70s6k activation. J Biol Chem. 1996;271:23650–23652. doi: 10.1074/jbc.271.39.23650. [DOI] [PubMed] [Google Scholar]
- 37.Jones RA, Johnson VL, Buck NR, Dobrota M, Hinton RH, Chow SC, Kass GE. Fas-mediated apoptosis in mouse hepatocytes involves the processing and activation of caspases. Hepatology. 1998;27:1632–1642. doi: 10.1002/hep.510270624. [DOI] [PubMed] [Google Scholar]
- 38.Dong Z, Saikumar P, Patel Y, Weinberg JM, Venkatachalam MA. Serine protease inhibitors suppress cytochrome c-mediated caspase-9 activation and apoptosis during hypoxia-reoxygenation. Biochem J. 2000;347:669–677. [PMC free article] [PubMed] [Google Scholar]
- 39.Murn J, Urleb U, Mlinaric-Rascan I. Internucleosomal DNA cleavage in apoptotic WEHI 231 cells is mediated by a chymotrypsin-like protease. Genes Cells. 2004;9:1103–1111. doi: 10.1111/j.1365-2443.2004.00794.x. [DOI] [PubMed] [Google Scholar]
- 40.Okada M, Adachi S, Imai T, Watanabe K, Toyokuni SY, Ueno M, Zervos AS, Kroemer G, Nakahata T. A novel mechanism for imatinib mesylate-induced cell death of BCR-ABL-positive human leukemic cells: caspase-independent, necrosis-like programmed cell death mediated by serine protease activity. Blood. 2004;103:2299–2307. doi: 10.1182/blood-2003-05-1605. [DOI] [PubMed] [Google Scholar]
- 41.Mlejnek P, Dolezel P. Apoptosis induced by N6-substituted derivatives of adenosine is related to intracellular accumulation of corresponding mononucleotides in HL-60 cells. Toxicol In Vitro. 2005;19:985–990. doi: 10.1016/j.tiv.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 42.Hughes FM, Jr, Evans-Storms RB, Cidlowski JA. Evidence that non-caspase proteases are required for chromatin degradation during apoptosis. Cell Death Differ. 1998;5:1017–1027. doi: 10.1038/sj.cdd.4400418. [DOI] [PubMed] [Google Scholar]
- 43.Bruin EC, Meersma D, de Wilde J, den Otter I, Schipper EM, Medema JP, Peltenburg LT. A serine protease is involved in the initiation of DNA damage-induced apoptosis. Cell Death Differ. 2003;10:1204–1212. doi: 10.1038/sj.cdd.4401296. [DOI] [PubMed] [Google Scholar]
- 44.Dallaporta B, Hirsch T, Susin SA, Zamzami N, Larochette N, Brenner C, Marzo I, Kroemer G. Potassium leakage during the apoptotic degradation phase. J Immunol. 1998;160:5605–5615. [PubMed] [Google Scholar]
- 45.Hendzel MJ, Nishioka WK, Raymond Y, Allis CD, Bazett-Jones DP, Th’ng JP. Chromatin condensation is not associated with apoptosis. J Biol Chem. 1998;273:24470–24478. doi: 10.1074/jbc.273.38.24470. [DOI] [PubMed] [Google Scholar]
- 46.Wu D, Ingram A, Lahti JH, Mazza B, Grenet J, Kapoor A, Liu L, Kidd VJ, Tang D. Apoptotic release of histones from nucleosomes. J Biol Chem. 2002;277:12001–12008. doi: 10.1074/jbc.M109219200. [DOI] [PubMed] [Google Scholar]
- 47.Gabler C, Blank N, Hieronymus T, Schiller M, Berden JH, Kalden JR, Lorenz HM. Extranuclear detection of histones and nucleosomes in activated human lymphoblasts as an early event in apoptosis. Ann Rheum Dis. 2002;63:1135–1144. doi: 10.1136/ard.2003.011452. [DOI] [PMC free article] [PubMed] [Google Scholar]