

Abstract
Hsp90 chaperones contain an N-terminal ATP binding site that has been effectively targeted by competitive inhibitors. Despite the myriad of inhibitors, none to date have been designed to bind specifically to just one of the four mammalian hsp90 paralogs, which are cytoplasmic Hsp90α and β, ER GRP94, and mitochondrial Trap-1. Given that each of the hsp90 paralogs is responsible for chaperoning a distinct set of client proteins, specific targeting of one hsp90 paralog may result in higher efficacy and therapeutic control. Specific inhibitors may also help elucidate the biochemical roles of each hsp90 paralog. Here we present side by side comparisons of the structures of yeast Hsp90 and mammalian GRP94, bound to the pan-hsp90 inhibitors Geldanamycin and Radamide. These structures reveal paralog specific differences in the Hsp90 and GRP94 conformations in response to Geldanamycin binding. We also report significant variation in the pose and disparate binding affinities for the Geldanamycin-Radicicol chimera Radamide when bound to the two paralogs, which may be exploited in the design of paralog-specific inhibitors.
Introduction
Cytosolic Hsp90 and endoplasmic reticulum (ER) resident GRP94 are essential molecular chaperones1; 2 that belong to the GHKL protein superfamily, which is characterized by the Bergerat ATP binding fold3; 4. Among the client proteins that require Hsp90 for proper function are cell-cycle proteins such as Cdc2, Cdk4, Cdk6, and Cdk9, signaling kinases such as HRI, Raf-1, mutant p53, Akt, ErbB2, and Her2, and steroid hormone receptors5; 6; 7. The clients of GRP94, which include all Toll-like receptors, certain integrins8, and IgGs9; 10 are cell surface or secreted proteins that are necessary for monitoring the extracellular milieu, mediating cell to cell communication, and effecting an immune response.
Competitive inhibition of the Hsp90 ATP binding site leads to the targeted degradation of client proteins via the ubiquitin proteasome pathway11; 12. Inhibition of Hsp90 results in growth arrest for tumor cells in culture, and tumor growth inhibition or regression in animal models6; 13; 14; 15. While inhibition of Hsp90 leads to degradation of Hsp90 clients, the best understood response to inhibition of GRP94 is the transcriptional up-regulation of ER chaperones16.
The anti-cancer effects of Hsp90 inhibition have driven the development of potent antagonists. Among the first inhibitors to be identified were the natural product ansamycin antibiotics Geldanamycin (Gdm) and Radicicol (Rdc) (Figure 1)17; 18. The scaffolds of these ansamycins and the natural ligand ATP have been successfully exploited for inhibitor design13; 19; 20; 21; 22; 23; 24; 25; 26; 27; 28; 29. Gdm has also been used as a tool to better understand the quaternary structure and regulatory roles of GRP9411; 12.
Figure 1.
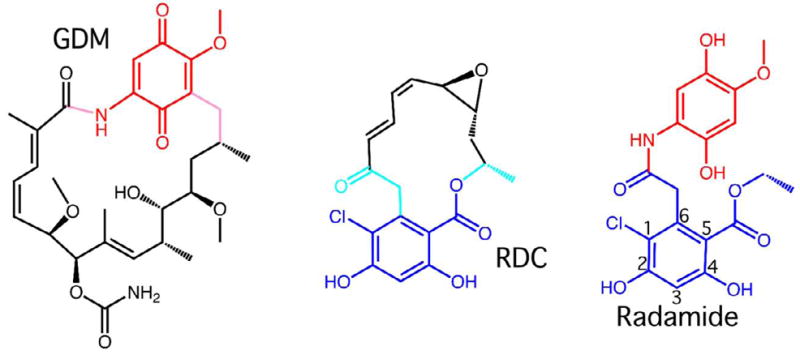
Ansamycin antibiotics Geldanamycin (Gdm) and Radicicol (Rdc), and the chimeric Hsp90 inhibitor Radamide. The quinone in common between Radamide and Gdm is colored in red. The resorcinol ring in common between Radamide and Rdc is colored in dark blue. The positions around the resorcinol ring are numbered.
Despite Gdm’s central place in the arsenal of hsp90 inhibitors, no structural studies have yet been reported for GRP94 in complex with Gdm. To better understand the binding mode of Gdm to GRP94 and to guide the use of Gdm as a probe of GRP94 function, we have determined the structure of the N-terminal regulatory domain of GRP94 bound to Gdm. This structure reveals that GRP94, unlike Hsp90, must undergo a conformational re-arrangement to accommodate Gdm in the ATP binding pocket. Moreover, the limited extent of this rearrangement, compared to that elicited by ATP binding, provides a mechanistic basis for understanding the other known ligand-dependent conformational changes in GRP94.
Finally, to further address differences in how pan-hsp90 inhibitors interact with the GRP94 and Hsp90 ATP binding site we solved the co-crystal structures of the radicicol-geldanamycin chimera Radamide23; 26 (Figure 1), bound both to yeast Hsp82 and GRP94. The Radamide co-crystal structures reveal distinct ligand poses that exploit differences in the ATP binding sites of Hsp90 and GRP94. Direct binding assays with Radamide revealed disparate affinities for the two hsp90 paralogs. Taken together, these results demonstrate that GRP94 and Hsp90, though very similar, interact with a selection of inhibitors in different manners. The observed differences in protein conformation, ligand pose, and binding affinity present a structure-function basis for selective inhibition of these important molecular chaperones.
Results
Binding of Geldanamycin requires rotation of Gly198 in GRP94
When Gdm is modeled into the ATP binding pocket of the unliganded GRP94 N-terminal domain, a steric clash is observed between the macrocylic amide and the main chain carbonyl oxygen of Gly196 (Figure 2A). This clash is similar to one predicted for the binding of ATP to apo GRP9430. To understand how Gdm is accommodated into GRP94, we solved the co-crystal structure of the GRP94NΔ41-Gdm complex. We used the N-terminal domains for these studies due to the higher resolution afforded by N-terminal domain complexes compared to their full length counterparts 31 and because no structures of intact inhibitor-bound hsp90 chaperones have yet been reported. In addition, no differences have been observed between the dissociation constants for ligands binding to the N-terminal domain of GRP94 compared to the full length counterpart (R.M.I. & D.T.G., unpublished observations), which suggests that their mode of interaction is similar. Finally, the structure of intact GRP94 bound to AMPPNP is consistent with the conformational rearrangements seen in the structure of the isolated N-terminal domain bound to the same ligand 31. As seen in Figure 2, bound Gdm in the complex adopts the compact C-shaped cis conformation previously seen in the complexes with yeast Hsp82 and human Hsp90α 32; 33. As in the Hsp90 complexes, Gdm makes direct and water mediated hydrogen bonds from its carbamate group to the carboxylate of Asp149, direct hydrogen bonds to Asp110, Lys114, Gly196, Gly198, and Phe199, and water mediated contacts to Leu104, Asn107, and Thr254 (Figure 2). The remaining interactions are hydrophobic contacts between Gdm and residues Met154, Leu163, Val197, and Phe199.
Figure 2.

(A) Modeling of Gdm into the nucleotide binding pocket of the un-liganded structure of GRP94NΔ41(slate) based on the position of Gdm in the GRP94NΔ41-Gdm complex results in a steric clash with Gly196. (B) Authentic structure of the GRP94NΔ41-Gdm complex (lime). Van der Waals spheres (blue dots) for the nitrogen of the macrocyclic amide no longer interpenetrate the carbonyl of Gly196 (red and lime dots). Binding of Gdm is accompanied by a rearrangement of the Gly hinge and a small displacement of the preceding Helix5. (C) Stereodiagram of the binding pocket interactions for Gdm in the GRP94NΔ41-Gdm complex. Water molecules are shown as red spheres and hydrogen bonds as dashed lines.
Earlier co-crystal structures of GRP94 in complex with ATP, ADP, and AMP revealed a significant rearrangement in GRP94 involving a large displacement of the Helix 1,4,5 mobile subdomain, compared to the apo enzyme, upon ligand binding34. Surprisingly, despite the fact that similar regions must move to accomodate ATP or Gdm binding to GRP94, Gdm elicits only a small rearrangement of the mobile subdomain (Figure 2B). In particular, the structure of the Gdm-GRP94 complex reveals a ~9 degree rotation at Gly198 in the “Gly hinge” of motif III of the Bergerat fold 4, compared to the ~95 degree rotation at the same position seen in the structure of the GRP94NΔ41-ATP complex34. This rotation at Gly198 in the Gdm-GRP94 complex expands the ligand binding pocket by moving Gly196 ~1Å away from the incoming Gdm, thus avoiding the modeled clash (Figure 2B). As a result of the rotation at Gly198, Helix 5 is slightly displaced and Helix 4 is disordered. This ligand dependent rotation about Gly198 and the displacement of Gly196 are unique to GRP94. These structural rearrangements are also distinct from the minor structural “tweaks” seen in many Hsp90-ligand complexes because of their magnitude and because they are driven by a clear necessity to accommodate incoming Gdm. Adenosine nucleotides and Gdm can be accommodated in the ligand binding pocket of cytoplasmic Hsp90s without structural re-arrangement, and, unsurprisingly, structural distortions have not been observed as a result of these or any ligands binding to cytoplasmic Hsp90s.
Radamide is accommodated differently in GRP94 and yHsp82
To further our understanding of the paralog-specific differences in inhibitor binding we determined the structures of yeast Hsp82N and GRP94NΔ41 in complex with the radicicol-geldanamycin chimera Radamide. Radamide contains a resorcinol ring derived from Radicicol that is linked, via an amide, to the quinone derived from Geldanamycin (Figure 1). Molecules of this design may be new scaffolds for the development of improved hsp90 targeted therapeutics.
In both the Hsp90- and GRP94-Radamide complexes, the Radicicol-derived resorcinol ring makes a direct hydrogen-bonding interaction with the carboxylate of the universally conserved aspartic acid residue required for ATP binding (Asp79 in yHsp82 and Asp149 in GRP94), thereby occupying the ligand binding pocket and blocking ATP binding. This orientation is consistent with the structures of Hsp90 and GRP94 in complex with all of the diaryl resorcinylic inhibitors 20; 35; 36; 37; 38; 39; 40; 41; 42; 43; 44.
The quinone moiety of Radamide sits at the outside of the ligand binding pocket and is well ordered in the yHsp82 and GRP94 complexes. As seen in Figure 3A, the quinone of Radamide contacts yHsp82 through direct hydrogen bonds to Lys44 and Lys98, and additional water mediated contacts to Lys44, Glu88, Asn91, Asn92, and Lys98. In addition there are also water-mediated contacts from the amide linkage to Lys98, Gly121, and Phe124.
Figure 3.

Stereodiagram of the interactions between Radamide and (A) yHsp82 or (B) GRP94. Red spheres represent water molecules, and dashed yellow lines represent hydrogen bonds. In panel (B) the “vertical” orientation of the quinone is solid, and the “horizontal” orientation is semi-transparent. In both (A) and (B) the model density (2Fo-Fc model map contoured at 1.0σ carved at 1.8Å around selected atoms, blue) is displayed for the bound Radamide. The surface representations of Radamide in the ATP binding site of (C) yHsp82 and (D) GRP94 are shown to compare the orientation and trajectory of bound Radamide in the co-crystal complexes. In panel (C) Radamide makes an oblique angle with the back of the ATP binding site and extends slightly into solvent. Radamide bound to GRP94 (D) utilizes the small hydrophobic 5′-extension pocket. The two alternate conformations of Radamide (D) are colored with white carbons for the horizontal orientation of the quinone and grey carbons for the vertical orientation of the quinone.
Surprisingly, while the interactions between the resorcinol ring of Radamide and the binding pockets of Hsp82 and GRP94 are largely conserved, the quinone moiety adopts two orthogonal poses when bound to GRP94 that result in novel interactions between the ligand and the protein. The two quinone orientations have been modeled in the crystal structure determination at 50% occupancy. As seen in Figure 3B, in the “vertical” orientation, the quinone makes water-mediated hydrogen bonds to Asn107, Gly196, and Phe199 (Asn37, Gly121, and Phe124 in yHsp82), while in the “horizontal” configuration the quinone makes direct hydrogen bonds to Asn162 and Gly196 (Asn92 and Gly121 in yHsp82). Moreover, not only does the quinone of Radamide make different contacts in the yHsp82 and GRP94 ATP binding sites, it also binds in an orientation that differs between the two paralogs (Figure 3C, D). For yHsp82, the amide linkage of Radamide is bent such that the quinone is pointing out towards solvent. In GRP94, however, the Radamide quinone extends along the back of the ATP binding site and interacts with a small hydrophobic pocket that is unique to GRP9430. This pocket has been shown to accommodate the 5′ extension of the GRP94 specific inhibitor NECA 30, and is blocked in Hsp90 by Lys168 (Figure 3C). Radamide is thus the first ligand other than NECA and its derivatives to utilize the 5′ hydrophobic pocket that is unique to GRP94. Additionally, Radamide is the first example of an inhibitor that has distinct postures in different Hsp90 paralogs.
To test the functional significance of the alternative postures for Radamide in Hsp82 and GRP94, we measured the binding constant by monitoring the change in the intrinsic tryptophan fluorescence of the chaperones as a function of Radamide concentration 45. Non-linear least squares fits to the binding curves yielded Kd values of 0.87 ± 0.14 μM for yHsp82 and 0.52 ± 0.13 μM for GRP94 (Figure 4). It was initially surprising that the binding of Radamide to GRP94 would be tighter given the lack of a single specific binding mode. The option to bind in two poses, however, lowers the entropic penalty for the ligand and may account for the observed tighter binding.
Figure 4.

Tryptophan fluorescence titration for binding of Radamide to full-length yHsp82 and GRP94-NMC. The different poses for Radamide in the yHsp82 and GRP94 binding pockets result in disparate binding affinities. Radamide bound to GRP94 exhibits two alternate conformations and has a Kd of 0.52 ± 13 μM compared to 0.87 ± 14 μM for yHsp82.
Discussion
Like other members of the GHKL superfamily 4, GRP94 contains an ATP binding site in its N-terminal domain that is linked to functionally important conformational rearrangements in the protein 3; 34; 46; 47; 48. In particular, rotation at the Gly hinge in the ATP binding pocket has been observed in each GHKL family member for which structural data exists 3; 34; 46; 47; 49; 50. By competing with ATP for the nucleotide binding pocket, competitive inhibitors such as Gdm, Radicicol, and Radamide have been shown to interrupt the function of Hsp9023; 26; 39; 51 and GRP9416. While the structural details of Gdm binding to Hsp90 have been previously elucidated 32; 33, we have now described the structure of the GRP94-Gdm complex.
Gdm binding requires structural rearrangements in GRP94 but not Hsp90
Binding of Gdm to GRP94 requires a rearrangement of the Gly hinge centered at residue Gly198. The movement of the Gly hinge prevents GRP94 from maintaining the “open” conformation observed in the un-liganded structure 52 or in co-crystal structures of GRP94 with other inhibitors 30. Moreover, the conformational changes instigated by Gdm binding are unique among Hsp90 inhibitors; to date the natural ligand ATP and the adenosine nucleotides are the only other ligands known to produce a rotation at the Gly hinge in any of the GHKL proteins 34.
The movement at the Gly hinge required for Gdm binding to GRP94 has not been observed for Gdm binding to Hsp90. Furthermore, Gdm binding to Hsp90 does not lead to any substantial structural rearrangement 32. This may explain the stronger binding of Gdm 53 and the Gdm derivative 17-AAG 54 to Hsp90 compared to GRP94.
Given the significant differences between the responses of GRP94 and Hsp90 to Gdm binding it seems likely that paralog specific inhibitors based on a Gdm scaffold could be designed. Such inhibitors would be valuable both as tools for probing the biological roles of these chaperones, and possibly for development of novel therapeutics.
Gdm highlights the role of electrostatics in driving ligand-dependent GRP94 conformational changes
The observation that Gdm requires an opening of the Gly hinge in GRP94, but does not result in the “extended open” conformation elicited by ATP, ADP, or AMP binding 34 was initially perplexing. Docking of either Gdm or ATP into the “open” conformation of apo GRP94 results in steric clashes with Gly196 that would require some re-arrangement upon actual ligand binding. Surprisingly, though, with only modest rotation of the phosphate, docking of AMP into the pocket of the GRP94-Gdm complex could be accomplished without van der Waals interpenetration. Since AMP elicits the same “extended open” conformation as ATP, a steric argument alone cannot explain the different conformations observed in the GRP94-Gdm and GRP94-ATP complexes.
An analysis of the electrostatics of the binding pocket provides an explanation for the different magnitude of the protein responses of GRP94 to Gdm and nucleotide binding (Figure 5). The ATP binding pocket in GRP94 is acidic, and a negative surface potential extends across not only the region that interacts with the nucleotide base, but also the region that interacts with the phosphates. Docking of ATP into the binding pocket of apo GRP94 (Figure 5A) would thus not only result in a steric clash as previously described 30; 34, but also an electrostatic repulsion caused by the close interaction of the negatively charged phosphates and the acidic binding pocket. In contrast to ATP, the region of the binding pocket in GRP94 occupied by Gdm consists of the macrocyclic amide and the quinone (Figure 5B). While these groups are polar, they are considerably less so than the formally charged phosphates of ATP. As a result, there would be significantly less electrostatic repulsion upon Gdm binding compared to ATP.
Figure 5.

Modeling of ADP (A), and Gdm (B) into the apo conformations of GRP94NΔ41 and modeling of ADP into the apo conformation of yHsp82N (C). Electrostatic surface potentials were generated using APBS 71, and Gemstone (http://gemstone.mozdev.org). The phosphates of ADP interact with an acidic region of the GRP94NΔ41 ATP binding pocket (A) leading to a modeled electrostatic repulsion. The region of Gdm (B) that interacts with this portion of the GRP94 binding pocket is not charged and therefore no electrostatic repulsion would result from Gdm binding. Yeast Hsp82 unlike GRP94 has a basic patch in its ATP binding pocket (C). This basic region interacts with the negatively charged phosphates, and better compliments ATP than GRP94.
The modeled electrostatic repulsion in addition to the steric clash that arises from ATP binding to GRP94 may be sufficient to drive the mobile subdomain past an energetic “tipping-point” that leads to the extended open conformation. Once this required impetus is reached, the extended open conformation can be stabilized by the interactions described previously 34. Since Gdm binding to GRP94 leads only to steric rearrangements but not to electrostatic repulsion, the “tipping-point” is not reached, and GRP94 maintains a conformation close to that of the apo chaperone.
Interestingly, the electrostatics of the yHsp82 binding pocket are different than those of GRP94 and may help to explain the markedly different responses of the two paralogs to nucleotide binding. While the region of the pocket that interacts with the nucleotide base is acidic in both GRP94 and yHsp82, the neighboring phosphate binding regions differ. In yHsp82 this region is basic and better complements the charge of the nucleotide (Figure 5C), while in GRP94 this region is acidic and strongly repels the ligand.
Re-design of the Radamide scaffold may lead to paralog-specific inhibitors
The resorcinol ring has been shown to be a useful scaffold for the design of hsp90 inhibitors. Several diaryl hsp90 inhibitors including the radicicol-geldanamycin chimeras, Radamide and Radester 23; 26, and the diarylpyrazole class of ligands 20; 35; 37; 38; 40; 41, have taken advantage of the resorcinol ring to guide binding. Alignment of several resorcinylic diaryl hsp90 inhibitors shows that the orientation of the second aryl group is highly variable (not shown).
Although NECA interacts preferentially with GRP94, the use of the NECA scaffold for further inhibitor development may be limited by the fact that NECA is also a potent agonist of several cellular Adenosine receptors 55; 56. Previous attempts to design GRP94 specific inhibitors based on a NECA scaffold have failed to produce ligands with higher affinities than the parent molecule(R.M.I. and D.T.G., unpublished), although recently another class of purine-based compounds has shown selective binding to GRP94 (G. Chiosis & D.T.G., in preparation). The variability in the binding orientation of the second aryl group leads to distinct conformations of Radamide when bound to yHsp82 or GRP94. Of particular importance, the conformation of Radamide observed in complex with GRP94 also exploits the GRP94-specific 5′-extension pocket. Radamide may thus serve as a third useful scaffold for the design of high affinity GRP94-specific inhibitors.
Materials and Methods
Expression and Purification
All proteins were overexpressed in E. coli strains BL21(DE3) or BL21 Star (DE3). Canine GRP94(69–337Δ287–327) (GRP94NΔ41), which is 98.5% identical with human GRP94, was over-expressed as a glutathione S-transferase (GST) fusion, purified as described previously 30; 34; 52, and concentrated to 30 mg/mL in preparation for crystallization. The N-terminal domain of yeast Hsp90 residues 1–220 (yHsp82N) was over-expressed as an amino-terminal hexa-Histidine tagged protein and purified by Ni-NTA affinity chromatography followed by dialysis, ion exchange chromatography on Q-sepharose (pH 8.0), and gel filtration chromatography on an S200. The purified protein was concentrated to 33 mg/mL in preparation for crystallization.
Near full-length His-tagged GRP94 28–754(Δ287–327) (GRP94-NMC) was expressed and purified essentially as described 31. Full length yeast His-tagged Hsp82 (residues 1-709) in pDEST17 (Invitrogen) was overexpressed in E. coli BL21 Star (DE3) and purified by Ni-NTA affinity chromatography followed by dialysis, ion exchange chromatography on Q-sepharose (pH 8.0), and gel filtration chromatography on an S200.
Crystallization of Inhibitor bound GRP94NΔ41
Crystals of GRP94NΔ41 in complex with Gdm were grown by hanging-drop vapor diffusion at 18°C. Prior to crystallization, the ligand was added to the concentrated protein in 10mM Tris, pH 7.6, 100mM NaCl, 1mM DTT to a final concentration of 3–10mM. The drops were formed by adding a 1:1 ratio of protein to reservoir (34% w/v PEG400, 60mM MgCl2, 100mM Tris, pH 7.6). Rod shaped crystals with typical dimensions of 400 × 200 × 200 μm appeared after 2–3 days.
Crystals of GRP94Δ41 in complex with Radamide were grown by hanging-drop vapor diffusion at 18°C. The crystallization drops contained 5–10mM ligand (a 5–10 molar excess) and a 1:1 ratio of protein to reservoir (25–30% w/v PEG MME 550, 10mM MgCl2, 100mM Tris, pH 8.0). Diffraction quality crystals appeared after 3–4 days. Typical crystals were rods with edge lengths of 400 × 100× 200 μm.
In each case crystals were harvested with nylon loops and cryo-protected by stepwise capillary transfers of the crystals to solutions that gradually increased the concentration of PEG 400 to 44–46%. If crystals were grown in PEG MME 550 the first capillary transfer was from the mother liquor to a solution that replaced the PEG MME 550 with PEG 400. Following the final equilibration the crystals were flash cooled in liquid nitrogen.
Crystallization of Inhibitor bound yHsp82N
Crystals of yHsp82N in complex with Radamide were grown at 18°C by microbatch under mineral oil. The microbatch drops contained ~10mM Radamide (a 10-fold molar excess) and a 4:1 ratio of protein (33mg/mL Hsp90 in 10mM Tris pH 7.6, 100mM NaCl 1mM DTT) to precipitant (8–9% w/v PEG MME 550, 25% glycerol, 90mM CaCl2, and 30mM Sodium Acetate or Sodium Succinate, pH 5.0). Tetragonal bi-pyramid crystals with a typical edge length of 200 μm appeared after 2–3 days. Crystals were harvested with nylon loops and cryo-protected by equilibration in 50mM Tris pH 7.4, 25% glycerol, 90mM CaCl2, and 13.5% PEG MME 550. Directly following cryo-protection, crystals were flash cooled in liquid nitrogen.
Data Collection, Structure Determination and Refinement
X-ray diffraction data for the GRP94NΔ41-Gdm co-crystals was collected on an R-AxisIV detector using X-rays from a rotating anode generator. Data for crystals of yHsp82N or GRP94NΔ41 with bound Radamide were collected on a MAR-225 CCD detector at APS beamline 22-BM. Data were reduced and scaled using XDS 57. Initial phases for the co-crystals were obtained by molecular replacement (MR). The search model for GRP94 constructs was the core region (residues 69–166 and 200–337) of apo GRP94NΔ41 (PDBID 1YT1) 52, and the search model for yHsp82 constructs was PDBID 1AH658; 59. The molecular replacement and following intermediate models were manually rebuilt in O or Coot, and refined in CNS, CCP4, or PHENIX 60; 61; 62; 63; 64. Ligand and solvent molecule positions were identified by overlapping peaks of difference density and simulated annealing omit density. Parameter and topology files for Gdm were obtained from HIC-Up, while parameter and topology files for Radamide were generated using the Dundee PRODRG server 65.
In later rounds of refinement, ligand dihedral constraints were relaxed, alternate sidechain conformations were added, and restrained individual B-factors were used. Structure validation was performed using KiNG and MolProbity 66; 67; 68; 69; 70. Data collection and refinement statistics are shown in Table I. Molecular graphics were created using Pymol (DeLano Scientific, San Carlos, CA).
Table I.
Summary of Data Collection and Refinement Statistics
| Diffraction Data | GRP94NΔ41 + GDM | GRP94NΔ41 + Radamide | yHsp82N + Radamide |
|---|---|---|---|
|
Diffraction Data Statistics | |||
| PDB code | 2EXL | 2GFD | 2FXS |
| Source | RaxisII | APS 22-BM | APS 22-BM |
| Space Group | P212121 | P212121 | P4322 |
| a, b, c (Å) | 65.82, 84.36, 95.63 | 65.49, 84.57, 95.58 | 75.20, 75.20, 110.85 |
| Wavelength (Å) | 1.5418 | 0.99997 | 0.99997 |
| Resolution (Å)a | 50–2.30 | 50–2.15 | 50–2.0 |
| (Last Shell) (Å) | 2.4–2.3 | 2.25–2.15 | 2.05–2.00 |
| Unique Reflections | 22646 | 29502 | 21635 |
| Completeness (Last Shell) (%) | 93.2 (46.5) | 99.8 (100) | 99.9 (99.9) |
| Average I/σI (Last Shell) | 12.1 (3.5) | 15.8 (5.6) | 29.8 (5.7) |
| Redundancy | 3.9 | 7.2 | 14.0 |
| Rsymb (Last Shell) (%) | 9.9 (28.8) | 8.4 (38.6) | 6.9 (46.3) |
|
| |||
|
Refinement Statistics | |||
| Refinement Package | CNS 1.1 | CNS 1.1 | CNS 1.1 |
| Resolution range (Å) | 50–2.35 | 50–2.3 | 50–2.0 |
| Reflections | 22516 | 24181 | 21617 |
| Non-solvent atomsc | 3249 | 3352 | 1686 |
| Solvent and hetero- atomsc | 430 | 408 | 321 |
| Molecules in Asymmetric Unit | 2 | 2 | 1 |
| Rms deviation from ideality | |||
| Bond lengths (Å) | .00621 | 0.00596 | .00562 |
| Bond angles (°) | 1.310 | 1.237 | 1.340 |
| Rd value (%) | 22.1 | 21.1 | 19.6 |
| Rd free (%) | 27.6 | 25.4 | 22.6 |
Resolution limit was defined as the highest resolution shell where the average I/σI was >2.
Rmerge = ΣhklΣi|Ii(hkl) - <I(hkl)>|/ΣhklΣII(hkl).
Alternate atoms are counted once.
R = Σ|Fo - Fc|/ΣFo. 10% of reflections were used to calculate Rfree.
Tryptophan Fluorescence Binding Assays
500 μL of GRP94-NMC or full length yHsp82 at a concentration of 0.1 μM in 1× buffer (40 mM Hepes, pH 7.4, 150 mM KCl, 5 mM MgCl2) was titrated with sequential 5 μL additions of Radamide solutions of increasing concentration (0.1 μM, 1 μM, 10 μM, 75 μM, 100 μM, 150 μM, 200 μM, 300 μM, 400 μM, 600 μM, 800 μM, 1200 μM, 1600 μM, 3200 μM). Ligand dilutions were made in 1× buffer from a 3200 μM stock in 50% DMSO such that the carryover DMSO in the titration did not exceed 2.5% (v/v). Fluorescence at 340 nm was monitored in a Jovin-Youbain fluorometer using an excitation wavelength of 275 nm, with 3 nm slits. Correction for the internal absorbance of the Radamide was made to the fluorescence data by applying the negative of the exponential derived from the fit of a similar Radamide titration of a solution of 5 μM L-Trp. Fluorescence data was also corrected for dilution. Corrected binding curves were analyzed using the one-site total binding model in Prism. Errors were estimated from the standard deviation of three replicate titrations.
Electrostatic potential maps for GRP94 and yHsp82
Surface potential maps were generated for the N-terminal domains of both GRP94 and yHsp82. In preparation for charge calculations, the structures were stripped of waters and heterogens and the atoms missing from truncated sidechains were added. The modified structures were then used to calculate the electrostatic potentials using Adaptive Poisson-Boltzman Solver APBS 71, and Gemstone (http://gemstone.mozdev.org).
Coordinates
Structure factors and coordinates have been deposited in the PDB 72. The corresponding PDB codes for these structures are: 2FXS for yHsp82N-Radamide, 2GFD for GRP94NΔ41-Radamide, and 2EXL for GRP94NΔ41-GDM.
Acknowledgments
We thank Dr. John York and Drs. David and Jane Richardson for use of facilities. Geldanamycin was the generous gift of Dr. Christopher Nicchitta. We also thank Dr. Elizabeth Sacho for helpful discussions and critical reading of the manuscript. This work was supported in part by grant CA095130 (to D.T.G.) from the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maloney A, Workman P. HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert Opin Biol Ther. 2002;2:3–24. doi: 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- 2.Pearl LH. Hsp90 and Cdc37 -- a chaperone cancer conspiracy. Curr Opin Genet Dev. 2005;15:55–61. doi: 10.1016/j.gde.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 3.Ban C, Yang W. Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell. 1998;95:541–52. doi: 10.1016/s0092-8674(00)81621-9. [DOI] [PubMed] [Google Scholar]
- 4.Dutta R, Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci. 2000;25:24–8. doi: 10.1016/s0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- 5.Shao J, Grammatikakis N, Scroggins BT, Uma S, Huang W, Chen JJ, Hartson SD, Matts RL. Hsp90 regulates p50(cdc37) function during the biogenesis of the activeconformation of the heme-regulated eIF2 alpha kinase. J Biol Chem. 2001;276:206–14. doi: 10.1074/jbc.M007583200. [DOI] [PubMed] [Google Scholar]
- 6.Chiosis G, Vilenchik M, Kim J, Solit D. Hsp90: the vulnerable chaperone. Drug Discov Today. 2004;9:881–8. doi: 10.1016/S1359-6446(04)03245-3. [DOI] [PubMed] [Google Scholar]
- 7.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 2003;228:111–33. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 8.Randow F, Seed B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat Cell Biol. 2001;3:891–6. doi: 10.1038/ncb1001-891. [DOI] [PubMed] [Google Scholar]
- 9.Melnick J, Aviel S, Argon Y. The endoplasmic reticulum stress protein GRP94, in addition to BiP, associates with unassembled immunoglobulin chains. J Biol Chem. 1992;267:21303–6. [PubMed] [Google Scholar]
- 10.Melnick J, Dul JL, Argon Y. Sequential interaction of the chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature. 1994;370:373–5. doi: 10.1038/370373a0. [DOI] [PubMed] [Google Scholar]
- 11.Chu F, Maynard JC, Chiosis G, Nicchitta CV, Burlingame AL. Identification of novel quaternary domain interactions in the Hsp90 chaperone, GRP94. Protein Sci. 2006;15:1260–9. doi: 10.1110/ps.052065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiosis G, Aguirre J, Nicchitta CV. Synthesis of Hsp90 dimerization modulators. Bioorg Med Chem Lett. 2006;16:3529–32. doi: 10.1016/j.bmcl.2006.03.092. [DOI] [PubMed] [Google Scholar]
- 13.Sausville EA, Tomaszewski JE, Ivy P. Clinical development of 17-allylamino, 17-demethoxygeldanamycin. Curr Cancer Drug Targets. 2003;3:377–83. doi: 10.2174/1568009033481831. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto Y, Machida H, Kubota N. Preferential sensitization of tumor cells to radiation by heat shock protein 90 inhibitor geldanamycin. J Radiat Res (Tokyo) 2005;46:215–21. doi: 10.1269/jrr.46.215. [DOI] [PubMed] [Google Scholar]
- 15.Blagosklonny MV, Fojo T, Bhalla KN, Kim JS, Trepel JB, Figg WD, Rivera Y, Neckers LM. The Hsp90 inhibitor geldanamycin selectively sensitizes Bcr-Abl-expressing leukemia cells to cytotoxic chemotherapy. Leukemia. 2001;15:1537–43. doi: 10.1038/sj.leu.2402257. [DOI] [PubMed] [Google Scholar]
- 16.Lawson B, Brewer JW, Hendershot LM. Geldanamycin, an hsp90/GRP94-binding drug, induces increased transcription of endoplasmic reticulum (ER) chaperones via the ER stress pathway. J Cell Physiol. 1998;174:170–8. doi: 10.1002/(SICI)1097-4652(199802)174:2<170::AID-JCP4>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 17.Neckers L, Schulte TW, Mimnaugh E. Geldanamycin as a potential anti-cancer agent: its molecular target and biochemical activity. Invest New Drugs. 1999;17:361–73. doi: 10.1023/a:1006382320697. [DOI] [PubMed] [Google Scholar]
- 18.Schulte TW, Akinaga S, Murakata T, Agatsuma T, Sugimoto S, Nakano H, Lee YS, Simen BB, Argon Y, Felts S, Toft DO, Neckers LM, Sharma SV. Interaction of radicicol with members of the heat shock protein 90 family of molecular chaperones. Mol Endocrinol. 1999;13:1435–48. doi: 10.1210/mend.13.9.0339. [DOI] [PubMed] [Google Scholar]
- 19.Biamonte MA, Shi J, Hong K, Hurst DC, Zhang L, Fan J, Busch DJ, Karjian PL, Maldonado AA, Sensintaffar JL, Yang YC, Kamal A, Lough RE, Lundgren K, Burrows FJ, Timony GA, Boehm MF, Kasibhatla SR. Orally active purine-based inhibitors of the heat shock protein 90. J Med Chem. 2006;49:817–28. doi: 10.1021/jm0503087. [DOI] [PubMed] [Google Scholar]
- 20.Cheung KM, Matthews TP, James K, Rowlands MG, Boxall KJ, Sharp SY, Maloney A, Roe SM, Prodromou C, Pearl LH, Aherne GW, McDonald E, Workman P. The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3,4-diarylpyrazole class of Hsp90 inhibitors. Bioorg Med Chem Lett. 2005;15:3338–43. doi: 10.1016/j.bmcl.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 21.Chiosis G, Lucas B, Shtil A, Huezo H, Rosen N. Development of a purine-scaffold novel class of Hsp90 binders that inhibit the proliferation of cancer cells and induce the degradation of Her2 tyrosine kinase. Bioorg Med Chem. 2002;10:3555–64. doi: 10.1016/s0968-0896(02)00253-5. [DOI] [PubMed] [Google Scholar]
- 22.Chiosis G, Timaul MN, Lucas B, Munster PN, Zheng FF, Sepp-Lorenzino L, Rosen N. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem Biol. 2001;8:289–299. doi: 10.1016/s1074-5521(01)00015-1. [DOI] [PubMed] [Google Scholar]
- 23.Clevenger RC, Blagg BS. Design, synthesis, and evaluation of a radicicol and geldanamycin chimera, radamide. Org Lett. 2004;6:4459–62. doi: 10.1021/ol048266o. [DOI] [PubMed] [Google Scholar]
- 24.He H, Zatorska D, Kim J, Aguirre J, Llauger L, She Y, Wu N, Immormino RM, Gewirth DT, Chiosis G. Identification of potent water soluble purine-scaffold inhibitors of the heat shock protein 90. J Med Chem. 2006;49:381–90. doi: 10.1021/jm0508078. [DOI] [PubMed] [Google Scholar]
- 25.Immormino RM, Kang Y, Chiosis G, Gewirth DT. Structural and quantum chemical studies of 8-aryl-sulfanyl adenine class Hsp90 inhibitors. J Med Chem. 2006;49:4953–60. doi: 10.1021/jm060297x. [DOI] [PubMed] [Google Scholar]
- 26.Shen G, Blagg BS. Radester, a novel inhibitor of the Hsp90 protein folding machinery. Org Lett. 2005;7:2157–60. doi: 10.1021/ol050580a. [DOI] [PubMed] [Google Scholar]
- 27.Vilenchik M, Solit D, Basso A, Huezo H, Lucas B, He H, Rosen N, Spampinato C, Modrich P, Chiosis G. Targeting wide-range oncogenic transformation via PU24FCl, a specific inhibitor of tumor Hsp90. Chem Biol. 2004;11:787–97. doi: 10.1016/j.chembiol.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 28.Wang M, Shen G, Blagg BS. Radanamycin, a macrocyclic chimera of radicicol and geldanamycin. Bioorg Med Chem Lett. 2006;16:2459–62. doi: 10.1016/j.bmcl.2006.01.086. [DOI] [PubMed] [Google Scholar]
- 29.Wright L, Barril X, Dymock B, Sheridan L, Surgenor A, Beswick M, Drysdale M, Collier A, Massey A, Davies N, Fink A, Fromont C, Aherne W, Boxall K, Sharp S, Workman P, Hubbard RE. Structure-activity relationships in purine-based inhibitor binding to HSP90 isoforms. Chem Biol. 2004;11:775–85. doi: 10.1016/j.chembiol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 30.Soldano KL, Jivan A, Nicchitta CV, Gewirth DT. Structure of the N-terminal domain of GRP94. Basis for ligand specificity and regulation. J Biol Chem. 2003;278:48330–8. doi: 10.1074/jbc.M308661200. [DOI] [PubMed] [Google Scholar]
- 31.Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol Cell. 2007;28:41–56. doi: 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem. 1999;42:260–6. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 33.Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell. 1997;89:239–50. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- 34.Immormino RM, Dollins DE, Shaffer PL, Soldano KL, Walker MA, Gewirth DT. Ligand-induced conformational shift in the N-terminal domain of GRP94, an Hsp90 chaperone. J Biol Chem. 2004;279:46162–71. doi: 10.1074/jbc.M405253200. [DOI] [PubMed] [Google Scholar]
- 35.Barril X, Beswick MC, Collier A, Drysdale MJ, Dymock BW, Fink A, Grant K, Howes R, Jordan AM, Massey A, Surgenor A, Wayne J, Workman P, Wright L. 4-Amino derivatives of the Hsp90 inhibitor CCT018159. Bioorg Med Chem Lett. 2006;16:2543–8. doi: 10.1016/j.bmcl.2006.01.099. [DOI] [PubMed] [Google Scholar]
- 36.Bishop SC, Burlison JA, Blagg BS. Hsp90: a novel target for the disruption of multiple signaling cascades. Curr Cancer Drug Targets. 2007;7:369–88. doi: 10.2174/156800907780809778. [DOI] [PubMed] [Google Scholar]
- 37.Brough PA, Aherne W, Barril X, Borgognoni J, Boxall K, Cansfield JE, Cheung KM, Collins I, Davies NG, Drysdale MJ, Dymock B, Eccles SA, Finch H, Fink A, Hayes A, Howes R, Hubbard RE, James K, Jordan AM, Lockie A, Martins V, Massey A, Matthews TP, McDonald E, Northfield CJ, Pearl LH, Prodromou C, Ray S, Raynaud FI, Roughley SD, Sharp SY, Surgenor A, Walmsley DL, Webb P, Wood M, Workman P, Wright L. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: potential therapeutic agents for the treatment of cancer. J Med Chem. 2008;51:196–218. doi: 10.1021/jm701018h. [DOI] [PubMed] [Google Scholar]
- 38.Brough PA, Barril X, Beswick M, Dymock BW, Drysdale MJ, Wright L, Grant K, Massey A, Surgenor A, Workman P. 3-(5-Chloro-2,4-dihydroxyphenyl)-pyrazole-4-carboxamides as inhibitors of the Hsp90 molecular chaperone. Bioorg Med Chem Lett. 2005;15:5197–201. doi: 10.1016/j.bmcl.2005.08.091. [DOI] [PubMed] [Google Scholar]
- 39.Drysdale MJ, Brough PA, Massey A, Jensen MR, Schoepfer J. Targeting Hsp90 for the treatment of cancer. Curr Opin Drug Discov Devel. 2006;9:483–95. [PubMed] [Google Scholar]
- 40.Dymock BW, Barril X, Brough PA, Cansfield JE, Massey A, McDonald E, Hubbard RE, Surgenor A, Roughley SD, Webb P, Workman P, Wright L, Drysdale MJ. Novel, potent small-molecule inhibitors of the molecular chaperone Hsp90 discovered through structure-based design. J Med Chem. 2005;48:4212–5. doi: 10.1021/jm050355z. [DOI] [PubMed] [Google Scholar]
- 41.Kreusch A, Han S, Brinker A, Zhou V, Choi HS, He Y, Lesley SA, Caldwell J, Gu XJ. Crystal structures of human HSP90alpha-complexed with dihydroxyphenylpyrazoles. Bioorg Med Chem Lett. 2005;15:1475–8. doi: 10.1016/j.bmcl.2004.12.087. [DOI] [PubMed] [Google Scholar]
- 42.McDonald E, Jones K, Brough PA, Drysdale MJ, Workman P. Discovery and development of pyrazole-scaffold Hsp90 inhibitors. Curr Top Med Chem. 2006;6:1193–203. doi: 10.2174/156802606777812086. [DOI] [PubMed] [Google Scholar]
- 43.Sharp SY, Boxall K, Rowlands M, Prodromou C, Roe SM, Maloney A, Powers M, Clarke PA, Box G, Sanderson S, Patterson L, Matthews TP, Cheung KM, Ball K, Hayes A, Raynaud F, Marais R, Pearl L, Eccles S, Aherne W, McDonald E, Workman P. In vitro biological characterization of a novel, synthetic diaryl pyrazole resorcinol class of heat shock protein 90 inhibitors. Cancer Res. 2007;67:2206–16. doi: 10.1158/0008-5472.CAN-06-3473. [DOI] [PubMed] [Google Scholar]
- 44.Sharp SY, Prodromou C, Boxall K, Powers MV, Holmes JL, Box G, Matthews TP, Cheung KM, Kalusa A, James K, Hayes A, Hardcastle A, Dymock B, Brough PA, Barril X, Cansfield JE, Wright L, Surgenor A, Foloppe N, Hubbard RE, Aherne W, Pearl L, Jones K, McDonald E, Raynaud F, Eccles S, Drysdale M, Workman P. Inhibition of the heat shock protein 90 molecular chaperone in vitro and in vivo by novel, synthetic, potent resorcinylic pyrazole/isoxazole amide analogues. Mol Cancer Ther. 2007;6:1198–211. doi: 10.1158/1535-7163.MCT-07-0149. [DOI] [PubMed] [Google Scholar]
- 45.Epps DE, Raub TJ, Caiolfa V, Chiari A, Zamai M. Determination of the affinity of drugs toward serum albumin by measurement of the quenching of the intrinsic tryptophan fluorescence of the protein. J Pharm Pharmacol. 1999;51:41–8. doi: 10.1211/0022357991772079. [DOI] [PubMed] [Google Scholar]
- 46.Ali MM, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–7. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Corbett KD, Berger JM. Structure of the topoisomerase VI-B subunit: implications for type II topoisomerase mechanism and evolution. Embo J. 2003;22:151–63. doi: 10.1093/emboj/cdg008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sacho EJ, Kadyrov FA, Modrich P, Kunkel TA, Erie DA. Direct visualization of asymmetric adenine nucleotide-induced conformational changes in MutL alpha. Mol Cell. 2008;29:112–21. doi: 10.1016/j.molcel.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shiau AK, Harris SF, Southworth DR, Agard DA. Structural Analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell. 2006;127:329–40. doi: 10.1016/j.cell.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 50.Huai Q, Wang H, Liu Y, Kim HY, Toft D, Ke H. Structures of the N-terminal and middle domains of E. coli Hsp90 and conformation changes upon ADP binding. Structure. 2005;13:579–90. doi: 10.1016/j.str.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 51.Chiosis G, Caldas Lopes E, Solit D. Heat shock protein-90 inhibitors: a chronicle from geldanamycin to today’s agents. Curr Opin Investig Drugs. 2006;7:534–41. [PubMed] [Google Scholar]
- 52.Dollins DE, Immormino RM, Gewirth DT. Structure of unliganded GRP94, the endoplasmic reticulum Hsp90. Basis for nucleotide-induced conformational change. J Biol Chem. 2005;280:30438–47. doi: 10.1074/jbc.M503761200. [DOI] [PubMed] [Google Scholar]
- 53.Xu W, Mimnaugh E, Rosser MF, Nicchitta C, Marcu M, Yarden Y, Neckers L. Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J Biol Chem. 2001;276:3702–8. doi: 10.1074/jbc.M006864200. [DOI] [PubMed] [Google Scholar]
- 54.Ge J, Normant E, Porter JR, Ali JA, Dembski MS, Gao Y, Georges AT, Grenier L, Pak RH, Patterson J, Sydor JR, Tibbitts TT, Tong JK, Adams J, Palombella VJ. Design, synthesis, and biological evaluation of hydroquinone derivatives of 17-amino-17-demethoxygeldanamycin as potent, water-soluble inhibitors of Hsp90. J Med Chem. 2006;49:4606–15. doi: 10.1021/jm0603116. [DOI] [PubMed] [Google Scholar]
- 55.Hutchison KA, Fox IH. Purification and characterization of the adenosine A2-like binding site from human placental membrane. J Biol Chem. 1989;264:19898–903. [PubMed] [Google Scholar]
- 56.Cristalli G, Lambertucci C, Taffi S, Vittori S, Volpini R. Medicinal chemistry of adenosine A2A receptor agonists. Curr Top Med Chem. 2003;3:387–401. doi: 10.2174/1568026033392282. [DOI] [PubMed] [Google Scholar]
- 57.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Cryst. 1993;26:795–800. [Google Scholar]
- 58.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Applied Crystallography. 1997;30:1022–25. [Google Scholar]
- 59.Prodromou C, Roe SM, Piper PW, Pearl LH. A molecular clamp in the crystal structure of the N-terminal domain of the yeast Hsp90 chaperone. Nat Struct Biol. 1997;4:477–82. doi: 10.1038/nsb0697-477. [DOI] [PubMed] [Google Scholar]
- 60.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 61.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 62.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 63.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 64.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 65.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–63. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 66.Richardson JS, Arendall WB, 3rd, Richardson DC. New Tools and Data for Improving Structures, Using All-Atom Contacts. In: Carter CWJ, Sweet RM, editors. Methods in Enzymology. Vol. 412. Academic Press; San Diego: 2003. pp. 374–385. [DOI] [PubMed] [Google Scholar]
- 67.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50:437–50. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 68.Davis IW, Murray LW, Richardson JS, Richardson DC. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–9. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davis IW, Arendall WB, 3rd, Richardson DC, Richardson JS. The backrub motion: how protein backbone shrugs when a sidechain dances. Structure. 2006;14:265–74. doi: 10.1016/j.str.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 70.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–83. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–41. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berman HM, Bhat TN, Bourne PE, Feng Z, Gilliland G, Weissig H, Westbrook J. The Protein Data Bank and the challenge of structural genomics. Nat Struct Biol. 2000;7(Suppl):957–9. doi: 10.1038/80734. [DOI] [PubMed] [Google Scholar]