

Abstract
Respiratory motoneuron response to hypoxia is reflex in nature and carotid body sensory receptor constitutes the afferent limb of this reflex. Recent studies showed that repetitive exposures to hypoxia evokes long term facilitation of sensory nerve discharge (sLTF) of the carotid body in rodents exposed to chronic intermittent hypoxia (CIH). Although studies with anti-oxidants suggested the involvement of reactive oxygen species (ROS)-mediated signaling in eliciting sLTF, the source of and the mechanisms associated with ROS generation have not yet been investigated. We tested the hypothesis that ROS generated by NADPH oxidase (NOX) mediate CIH-evoked sLTF. Experiments were performed on ex vivo carotid bodies from rats and mice exposed either to 10 d of CIH or normoxia. Acute repetitive hypoxia evoked a ∼12-fold increase in NOX activity in CIH but not in control carotid bodies, and this effect was associated with upregulation of NOX2 mRNA and protein, which was primarily localized to glomus cells of the carotid body. sLTF was prevented by NOX inhibitors and was absent in mice deficient in NOX2. NOX activation by CIH required 5-HT release and activation of 5-HT2 receptors coupled to PKC signaling. Studies with ROS scavengers revealed that H2O2 generated from O2·− contributes to sLTF. Priming with H2O2 elicited sLTF of carotid bodies from normoxic control rats and mice, similar to that seen in CIH-treated animals. These observations reveal a novel role for NOX-induced ROS signaling in mediating sensory plasticity of the carotid body.
Introduction
Repetitive exposures to hypoxia result in long-lasting activation of respiratory motor output known as long-term facilitation (LTF) of breathing (for review, see Powell et al., 1998). LTF is regarded as a form of plasticity of respiratory motoneuron activity that is uniquely expressed in response to repetitive but not by continuous hypoxia (Baker et al., 2001). Respiratory motoneuron response to hypoxia is reflex in nature and carotid body sensory receptor constitutes the afferent limb of this reflex. Recent studies from our laboratory showed that repetitive hypoxia also induces LTF of the carotid body afferent nerve activity [sensory LTF (sLTF)] (Peng et al., 2003, 2006b). However, unlike LTF of the respiratory motor activity, sLTF of the carotid body requires prior conditioning with chronic intermittent hypoxia (CIH). The CIH-induced sLTF was reversible after reexposure to normoxia and could be prevented by treating CIH rats with anti-oxidants (Peng et al., 2003), indicating the involvement of reactive oxygen species (ROS). However, neither the source nor the mechanism(s) by which ROS are generated in CIH-treated carotid bodies has been examined.
The family of NADPH oxidase (NOX) enzymes constitutes one of the major sources of ROS generation. NOX2 (also called gp91phox) is the most extensively studied member of the NOX family. Recent studies have shown that ROS generated by NOX2 mediate long-term potentiation (LTP) of neuronal activity, hippocampal synaptic plasticity, and memory (Kishida et al. 2006; Kishida and Klann, 2007). Zhan et al. (2005) reported that NOX2 mediates CIH-induced changes in sleep behavior. NOX2 is expressed in glomus cells (the putative hypoxic sensing cells) of the carotid body (Youngson et al. 1997; Dinger et al., 2007) and contributes to the induction of LTP elsewhere in the nervous system (Kishida et al. 2006); these findings prompted us to hypothesize that NOX2 and/or closely related NOX isoform mediate sLTF of the carotid body. Here, we demonstrate that CIH leads to regulated NOX2 activation that requires 5-HT/5-HT2 receptors/protein kinase C (PKC) signaling and the resulting ROS, especially H2O2 is critical for evoking sLTF. These observations unravel a novel role for NOX2 and ROS in inducing sensory plasticity of the carotid body.
Materials and Methods
Experiments were approved by the Institutional Animal Care and Use Committee of the University of Chicago and were performed on adult male rats (Sprague Dawley, 200–300 gm), wild type (WT, C57BL/6), hemizygous gp91phox−/Y (from Jackson Laboratories; weights 20–25 gm), and Pet-1−/− mice (from Dr. Deneris, weights 20–25 gm).
Exposure to chronic intermittent hypoxia.
Unrestrained, freely moving animals housed in feeding cages were exposed to chronic intermittent hypoxia (CIH; for 10 d; 8 h · day−1) as previously described (Peng and Prabhakar, 2004). Briefly, animals were placed in a specialized chamber, which was flushed with alternating cycles of pure nitrogen and compressed air. During hypoxia, inspired O2 levels reached 5% O2 (nadir). The gas flows were regulated by timer-controlled solenoid valves. Ambient O2 and CO2 levels in the chamber were continuously monitored by an O2/CO2 analyzer (Alpha Omega Instrument; Series 9500). Control experiments were performed on animals exposed to alternating cycles of compressed room air instead of hypoxia in the same chamber. Acute experiments were performed on rats or mice anesthetized with intraperitoneal injections of urethane (1.2 g · kg −1, Sigma) ∼16 h after terminating CIH.
Measurements of carotid body sensory activity.
Sensory activity from carotid bodies ex vivo was recorded as described previously (Peng and Prabhakar 2004, Peng et al. 2006b). Briefly, carotid bodies along with the sinus nerves were harvested from anesthetized animals. After cleaning the connective tissue, the carotid body along with the sinus nerve was placed in a recording chamber (volume 250 μl) and superfused at a rate of 2 ml · min−1 with warm physiological saline (36°C) containing the following (mm): NaCl (125), KCl (5), CaCl2 (1.8), MgSO4 (2), NaH2PO4 (1.2), NaHCO3 (25), d-Glucose (10), Sucrose (5), bubbled with 95% O2/5% CO2, pH 7.4. To facilitate recording of clearly identifiable action potentials, the sinus nerve was treated with 0.1% collagenase for 5 min. Action potentials (2–4 active units) were recorded from one of the nerve bundles with a suction electrode, amplified (AC-preamplifier, Grass Instrument, P511K; bandwidth of 100–3000 Hz), displayed on an oscilloscope (Tektronix 5B12N), and stored in a computer via an A/D translation board (PowerLab/8P, AD Instruments). “Single” units were selected based on the height and duration of the individual action potentials using a spike discrimination program (Spike Histogram Program, Power Lab, AD Instruments). In each carotid body, at least 2 chemoreceptor units were analyzed.
Induction of sLTF of the carotid body.
The protocols for evoking sLTF of rat carotid body ex vivo were the same as described previously (Peng et al. 2003). Briefly, baseline sensory activity was recorded for 15 min with perfusate (pO2=∼140 mmHg) containing either vehicle or the drugs followed by 10 episodes of repetitive hypoxia (pO2=∼42 mmHg for 30 s), separated by 5 min of normoxia (pO2=∼140 mmHg). Sensory activity was monitored continuously for 60 min after terminating repetitive hypoxia. The protocols evoking sLTF in mice carotid body ex vivo were the same except that 5 episodes of repetitive hypoxia were used.
Measurements of NADPH oxidase activity.
NADPH oxidase activity was determined by monitoring O2·− dependent reduction of ferric cytochrome C at 550 nm as described previously (Mayo and Curnutte, 1990). Briefly, freshly harvested carotid bodies were placed in PBS, 0.9 mm CaCl2, 0.5 mm MgCl2 and 7.5 mm glucose, pH 7.4. The reaction was initiated by addition of cytochrome C (75 μm) to the reaction medium. Increase in the absorbance was monitored at 550 nm for 5 min. The amount of reduced cytochrome C formed during the reaction was determined using the molar extinction coefficient of 21.7μmoles · cm−2. The enzyme activity was expressed as picomoles min−1 per carotid body.
NOX mRNA expression in the carotid body by quantitative real-time PCR.
Real-time RT-PCR was performed using a MiniOpticon system (Bio-Rad Laboratories) with SYBR GreenER two-step qRT-PCR kit (#11764–100, Invitrogen). Briefly, RNA was extracted from rat carotid bodies using TRIZOL and was reverse transcribed using superscript III reverse transcriptase. Primer sequences for real time RT-PCR amplification were as follows: 18 s: [forward (fw)] GTAACCCGTTGAACCCCATT and [reverse (rev)] CCATCCAATCGGTAGTAGCG (Size 151, Gene Bank # X_01117); NOX1: (fw) CACTGTGGCTTTGGTTCTA and (rev) TGAGGACTCCTGCAACTCCT (Size 240, Gene Bank # NM_053683); NOX2: (fw) GTGGAGTGGTGTGAATGC and (rev) TTTGGTGGAGGATGTGATGA (Size 219, Gene Bank # NM_023965); NOX3: (fw) GACCCAACTGGAATGAGGAA and (rev) AATGAACGACCCTAGGATCT (Size 150, Gene Bank # NM_001004216); and NOX4: (fw) CGGGGTGGCTTGTTGAAGTAT and (rev) CGGGGTGGCTTGTTGAAGTAT (Size 205, Gene Bank # NM_053524). Relative mRNA quantification was calculated using the comparative threshold (CT) method using the formula “2−ΔCT” where ΔCT is the difference between the threshold cycle of the given target cDNA between nomoxia and CIH. The CT value was taken as a fractional cycle number at which the emitted fluorescence of the sample passes a fixed threshold above the baseline. Values were compared with an internal standard gene 18S. Purity and specificity of all products were confirmed by omitting the template and by performing a standard melting curve analysis.
Immunohistochemistry.
Anesthetized rats or mice were perfused intracardially with heparinized phosphate buffered saline (PBS; pH 7.4) at a rate of 10 ml min−1 for 10 min followed by buffered Formalde (10% Formalin; Fischer Scientific). Carotid artery bifurcations were removed and placed in 4% paraformaldehyde–PBS for 1 h at 4°C, washed in PBS, and cryoprotected in 30% sucrose–PBS at 4°C for 24 h. Specimens were frozen in Tissue Tek (OCT; VWR Scientific) and serially sectioned at 8 μm. For NOX2 and NOX4 localization, rat carotid body sections were incubated at 37°C for 2 h with either anti-NOX2 antibody (Santa Cruz, # s.c.-5827;1:50) or anti-NOX4 antibody (from Dr. D. Lambeth; 1:500 dilution) followed by FITC and Alexa Fluor 555 conjugated secondary antibodies (Invitrogen). For identification of glomus cells, sections were doubled stained with polyclonal anti-chromogranin-A (CGA) antibody (Abcam, # ab45138; 1:100) or monoclonal anti-tyrosine hydroxylase (TH) antibody (Sigma; 1:4000), established markers of glomus cells. In mice, 5-HT immunolocalization in the carotid body was performed with monoclonal anti-5-HT antibody (Millipore; 1:100). Sections were mounted in DAPI-containing media and visualized using a Nikon fluorescent microscope.
Immunoblot assays.
Immunoblot assays were performed as described previously (Yuan et al. 2008). Briefly, carotid bodies were homogenized in RIPA buffer (1×PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mm sodium orthovanadate, 0.1 mm PMSF, 5 μg ml–1 leupeptin, 5 μg ml–1 aproteinin, and 10 μl · ml−1 protease inhibitor cocktail). Cell lysates were fractionated by 7.5% polyacrylamide-SDS gel electrophoresis and transferred to a polyvinylpyrrolidone difluoride membrane (Immobilon-P, Millipore). The membrane was blocked with Tris-buffered saline (TBS-T) containing 5% nonfat milk at 4°C overnight. Membranes were incubated with anti-gp91phox antibody (Santa Cruz; 1:200) or anti-NOX4 antibody (from Dr. Lambeth; 1;500) or anti-phospho-MARCKS antibody (Protein Tech Group; 1:1000 dilution) or anti-α-tubulin antibody (Sigma; 1:1000) in TBS-T containing 3% nonfat milk. Membranes were treated with donkey anti-goat secondary antibody conjugated with horseradish peroxidase (Santa Cruz; 1:2000) in TBS-T containing 3% nonfat milk. Immune complexes on the membrane were visualized using a chemiluminescence (ECL) detection system (Amersham). The membranes were exposed to Kodak XAR films.
Measurement of 5-hydroxytryptamine release.
5-Hydroxytryptamine (5-HT) release from carotid bodies was analyzed by high performance liquid chromatography coupled to an electrochemical detection system (HPLC-ECD) as described previously (Jacono et al., 2005). Briefly, freshly isolated carotid bodies were incubated with 200 μl of Ca2+/Mg2+ free Krebs–Ringer bicarbonate (KRB) medium preequilibrated with 95% O2 + 5% CO2 for 90 min at 4°C. Subsequently, incubation medium was switched to 50 μl of KRB with Ca2+/Mg2+ equilibrated with appropriate gas mixtures resulting in medium PO2s of either 146 ± 7 mmHg (normoxia) or 43 ± 8 mmHg (hypoxia). Each gas challenge was given for 5 min. 5-HT in the medium was analyzed by HPLC-ECD and expressed as femtomoles of 5-HT · min−1 · carotid body−1.
Experimental protocols.
Series 1: In these experiments, the effects of CIH on NOX enzyme activity, mRNAs encoding various NOX isoforms and NOX protein expressions were analyzed in the rat carotid bodies. Control experiments were performed on rats reared under normoxia (n = 6 each; 6 carotid bodies/experiment). For measurements of NOX enzyme activity, carotid bodies were challenged with 10 episodes of repetitive hypoxia (pO2=∼42 mmHg for 30 s. followed by 5 min. of normoxia pO2=∼140 mmHg). Ten minutes after terminating the 10th episode of hypoxia, the carotid bodies were collected and the NOX activity was determined as described above. Series 2: The effects of NOX inhibitors, apocynin, diphenyl iodinium (DPI), 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBFS) on sLTF of the rat carotid body were examined (vehicle control; apocynin; DPI; AEBSF n = 6 each). Series 3: sLTF was examined in wild type (WT) and gp91phox−/Y mice exposed either to normoxia or CIH (WT and gp91phox−/Y, n = 7 each). Series 4: Carotid bodies were harvested from CIH- and normoxia-exposed rats and the effects of acute hypoxia on 5-HT release were examined (control and CIH; n = 3 each and 6 carotid bodies per experiment). Series 5: sLTF of carotid bodies was examined in WT and Pet-1−/− mice exposed either to normoxia or CIH (n = 7 each for WT and Pet-1−/− mice). Series 6: The effects of ketanserin, a 5-HT-2 receptor antagonist or bisindolylmaleimide-1 (Bis-1), a PKC inhibitor on repetitive hypoxia-evoked pMARCKS expression (n = 3; six carotid bodies/experiment) and sLTF (n = 6) were analyzed in carotid bodies harvested from normoxia- and CIH-exposed rats. Series 7: The effects of MnTMPyP (n = 6), a membrane permeable scavenger of O2·−, or PEG-catalase (n = 6), a scavenger of H2O2, on sLTF were examined. Series 8: The effects of exogenous administration of H2O2 on sLTF were examined in rats reared under normoxia with (n = 7) and without (n = 6) H2O2. In 5 additional rat carotid bodies, sensory activity was recorded for 90 min while superfusing carotid bodies with 500 nm H2O2. Parallel experiments were performed examining the effects of exogenous administration of H2O2 on carotid body sLTF in WT, gp91phox−/Y and Pet-1−/− mice (n = 6–7 each). Based on preliminary studies 200 nm H2O2 was chosen in the experiments involving mice carotid bodies.
Drugs and antibodies.
The following drugs or chemicals were used: 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBFS), 2-aminoethyl diphenylborinate (2-APB), apocynin (Acetovanillone), cadmium chloride, diphenyl iodinium (DPI), hydrogen peroxide, ketanserin tartrate, PEG-catalase, and phorbol 12-myristate 13-acetate (PMA) from Sigma; bisindolymadeimide I (Bis-1, Calbiochem); manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride (MnTMPyP, Alexis Chemicals). Stock solutions were made fresh and desired concentrations of the drugs were added to the medium in the reservoirs before the experiment.
Data analysis.
Chemoreceptor activity of “single units” (impulses · s−1)was averaged every 15 min under baseline conditions, every 30 s during and every 5 min following acute hypoxia challenge for 60 min. Changes in chemoreceptor activity were expressed as percentage of baseline values. The data are expressed as mean ± SEM. Statistical significance was assessed by one-way ANOVA followed by Tukey's post hoc test. p values <0.05 were considered significant.
Results
Effects of CIH on NOX expression and enzyme activity in the carotid body
Basal NOX activity was comparable between CIH- and normoxia-treated carotid bodies. Acute repetitive hypoxia resulted in ∼12-fold increase in NOX activity in CIH but not in control carotid bodies (p < 0.01) (Fig. 1A), and apocyanin (500 μm), an inhibitor of NOX, prevented this response. Unlike repetitive exposures, a single episode of hypoxia was ineffective in activating NOX (p > 0.05).
Figure 1.

CIH upregulates NOX enzyme activity, mRNAs in rat carotid body. A, Effect of repetitive hypoxia on NOX activity. Carotid bodies (CB) harvested from control and CIH-exposed rats were challenged with 10 episodes of repetitive hypoxia and NOX activity was determined as described in Materials and Methods. Note that acute repetitive hypoxia increases NOX activity in the CIH but not in control carotid body, and 500 μm apocynin (Apo), a NOX inhibitor, prevented this effect. The data presented are mean ± SEM, n = 3 each with normoxia (open columns) and acute repetitive hypoxia (filled columns). B, CIH upregulates NOX1, NOX2, and NOX4 mRNAs in the carotid body. Results are expressed as fold change with respect to normoxic controls. Data presented are mean ± SEM from three individual experiments performed in triplicate. Asterisks represent p < 0.01 compared with normoxia; n.s., nonsignificant. C, Localization of NOX2 and NOX4 in the rat carotid body. NOX2-like immunoreactivity (left, green color) was seen in the cytoplasm of glomus cells as evidenced by colocalization with chromagranin-A (CGA; denoted by arrows; middle). NOX4-like immunoreactivity (red color; right) is localized in the nuclei of carotid body cells. NOX4 expression can be seen in glomus cells (stained with tyrosine hydroxylase; TH; green color; denoted by arrows) as well as other cells (shown with *). Scale bar, 20 μm. CON, Control.
Several NOX isoforms have been identified [see Bedard and Krause (2007) for references]. To determine which of the NOX isoforms are expressed in the rat carotid body, and whether they are affected by CIH, NOX1, 2, 3, and 4 mRNA expressions were examined by quantitative real time PCR in control and CIH-treated carotid bodies. NOX1, 2, 3, and 4 mRNAs could readily be detected in the control carotid body (Fig. 1B). In CIH-treated carotid bodies, NOX 1, 2 and 4 mRNAs were upregulated; whereas NOX3 mRNA expression was unaffected. The magnitude of increase in NOX2 mRNA was more pronounced (∼35-fold) than NOX 1 (∼16-fold) and NOX4 (∼13-fold). Because NOX1 is a homolog of NOX2 (Suh et al. 1999; Bánfi et al. 2000), we chose to examine the localization of NOX2 and NOX4 proteins by immunohistochemistry in control and CIH-treated carotid bodies. Immunoreactive product of NOX2 was localized to the cytoplasm of glomus cells; whereas that of NOX4 was localized to the nucleus of glomus as well as other cells in control and CIH-treated carotid bodies (Fig. 1C,D). Western blot assay revealed a ∼2.0–2.5-fold increase in NOX2 and NOX 4 proteins in CIH compared with control carotid bodies (p < 0.01).
NOX is required for CIH-induced sLTF of the carotid body
The effects of three structurally distinct inhibitors of NOX, i.e., apocynin, DPI and AEBFS on sLTF were examined to assess the potential contribution of NOX activation to sLTF. As shown Figure 2A,B, repetitive hypoxia evoked robust sLTF in CIH-exposed carotid bodies and apocynin prevented this effect. However, hypoxia-evoked sensory excitation (Δimpulses · s−1) was unaffected by apocynin (CIH+vehicle control = +10.8 ± 2.1 vs CIH + apocynin = +8.7 ± 1.7; p > 0.05). Like apocyanin, 3 μm DPI and 500 μm AEBFS also prevented the induction of sLTF (Fig. 2C).
Figure 2.

NOX inhibitors prevent sLTF of the rat carotid body. A, B, Examples of repetitive hypoxia-evoked sLTF of CIH-treated rat carotid bodies in the presence of vehicle (A) or apocynin (B). Arrows in A and B denote application of repetitive hypoxia. Superimposed action potentials of a single fiber from which the data were derived are shown in the insets. imp, Impulses. C, Average data of the effects of apocynin (500 μm), AEBFS (500 μm), and diphenyl iodinium (DPI; 3 μm) on sLTF (sensory activity averaged during 60 min postrepetitive hypoxia period). Data presented are mean ± SEM from six experiments in each group. Asterisk denotes p < 0.05 compared with CIH-exposed carotid bodies treated with vehicle.
Although the above described effects of pharmacological inhibitors suggest a role for NOX in evoking sLTF, they cannot differentiate the contribution of different NOX isoforms. To assess the selective contribution of NOX2, sLTF was monitored in CIH-exposed mice deficient in gp91phox i.e., NOX2 and age matched wild type (WT) controls. As shown in Figure 3, sLTF could be evoked in CIH-treated WT but not in gp91phox−/Y mice. However, hypoxia-evoked sensory excitation could be elicited in both groups of mice (Fig. 3A) (p > 0.05).
Figure 3.

Absence of sLTF of the carotid body in gp91phox−/Y mice exposed to CIH. A, Examples of repetitive hypoxia-evoked sLTF of carotid bodies from CIH-treated WT (top) and gp91phox−/Y (bottom) mice. Arrows denote application of repetitive hypoxia. Superimposed action potentials of a single fiber from which the data were derived are shown in the insets. imp, Impulses. B, Average data showing the magnitude of sLTF of the carotid body from WT and gp91phox−/Y mice exposed to either normoxia (control, open columns) or CIH (filled columns). Data presented are mean ± SEM from WT (control and CIH, n = 7 each) and gp91phox−/Y (control and CIH, n = 7 each) mice. Asterisks denote p < 0.01 compared with control carotid body.
CIH-evoked NOX activation and sLTF require 5-HT and 5-HT2 receptors
The following experiments were performed to address the mechanism(s) underlying NOX activation in CIH-treated carotid bodies. Exogenous applications of 5-HT has been shown to activate NOX in the carotid body via 5-HT2 receptors coupled to protein kinase C (PKC) activation (Peng et al. 2006a). To examine the role of 5-HT in NOX activation, we first determined whether hypoxia releases 5-HT from carotid bodies. As shown in Figure 4, hypoxia (43 ± 8 mmHg) resulted in robust 5-HT release from CIH-treated carotid bodies (basal release during normoxia = 1.6 ± 0.08 vs hypoxia = 8.1 ± 0.1 femtomoles per min per carotid body); whereas it had no effect on 5-HT release from normoxia-treated carotid body (Fig. 4B). The hypoxia-evoked 5-HT release from CIH-treated carotid body was completely prevented by 75 μm 2-APB, a purported ionositol phosphate-3 (IP3) receptor/TRPM2 channel blocker (Pinilla et al. 2005). In contrast, CdCl2 (300 μm), a broad-spectrum voltage-gated Ca2+ channel blocker had no significant effect (Fig. 4B).
Figure 4.

Acute hypoxia facilitates 5-HT release from CIH-exposed carotid body. A, Example of 5-HT release from the carotid bodies from CIH exposed rats. Left and right represent 5-HT release during normoxia and acute hypoxia, respectively. Arrows show the elution of 5-HT during normoxia and hypoxia. B, Average data of the basal (open columns) and hypoxia-evoked (filled columns) 5-HT release from carotid bodies derived from rats reared under normoxia or CIH. Data presented are mean ± SEM from three individual experiments performed in triplicate. Asterisks denote p < 0.01 compared with basal release. Note that hypoxia had no effect on 5-HT release from carotid bodies from normoxic rats, whereas it was markedly facilitated in CIH carotid bodies and this effect was prevented by 75 μm 2-APB but not by 300 μm cadmium chloride (Cd2+).
If 5-HT is the upstream signaling molecule for CIH-evoked NOX activation, then the ensuing sLTF should be absent in carotid bodies with impaired 5-HT expression. Mice deficient in the gene encoding the Pet-1 transcription factor exhibit markedly reduced 5-HT expression in the CNS (Hendricks et al. 1999, 2003) and Pet-1−/− mice offer elegant model for exploring the role of 5-HT in CIH-evoked sLTF. We first examined whether 5-HT expression is reduced in the carotid bodies of Pet-1−/− mice. As shown in Figure 5A, many glomus cells expressed 5-HT-like immunoreactivity in WT but not in Pet-1−/− mice. Functional analysis of carotid body activity showed that sLTF was absent in both control WT and Pet-1−/− mice reared under normoxia. However, following CIH, sLTF could readily be elicited only in WT but not in Pet-1−/− mice (Fig. 5B,C).
Figure 5.

Absence of sLTF of the carotid body in Pet-1−/− mice with impaired 5-HT expression in glomus cells. A, 5-HT-like immunoreactivity in glomus cells of the carotid bodies from WT and Pet-1−/− mice. Note the absence of 5-HT-like immunoreactivity in the carotid body from Pet-1−/− mice. B, Representative tracings of the carotid body sensory response to repetitive hypoxia (at arrows) from WT (top two panels) and Pet-1−/− (bottom two panels) mice exposed to either normoxia (control) or CIH. Insets represent superimposed action potentials from a single unit. C, Average data of the magnitude of sLTF (sensory activity averaged during 60 min period of postrepetitive hypoxia) presented as percentage of baseline sensory activity (i.e., before 5 episodes of repetitive hypoxia). Data represent mean ± SEM from WT (Control and CIH, n = 7 each) and Pet-1−/− (Control and CIH, n = 7 each) mice. Asterisks denote p < 0.01 compared with control. Note the absence of sLTF in Pet-1−/− mice treated with CIH.
To assess whether NOX activation and sLTF require activation of 5-HT2 receptors and PKC, carotid bodies harvested from CIH- and normoxia-treated rats were challenged with 10 episodes of repetitive hypoxia and levels of phosphorylated MARCKS (myristoylated alanine-rich C kinase substrate) were monitored as an index of PKC activation (Wen et al. 2006). Repetitive hypoxia increased p-MARCKS levels by ∼3-fold in CIH-treated but not in control carotid bodies (Fig. 6A). Either blockade of 5-HT2 receptors by ketanserin or inhibition of PKC-isoforms with Bis-1 prevented repetitive hypoxia elicited increases in p-MARCKS levels as well as NOX activation in CIH-treated carotid bodies (Fig. 6A,B). More importantly, both ketanserin and Bis-1 also prevented sLTF (Fig. 7). Ketanserin, however, had no effect on dopamine (100 μm) evoked sensory inhibition of the carotid body (data not shown), suggesting that at 1 μm concentration, it selectively affected 5-HT receptors.
Figure 6.

NOX activation by repetitive hypoxia in the CIH-exposed rat carotid bodies requires activation of 5-HT2 receptors and PKC. A, Acute repetitive hypoxia activates PKC via 5-HT2 receptors. The level of phosphorylation of MARCKS (P-MARCKS) was monitored in the rat carotid bodies by Western blot as an index of PKC activation. Top, example of the Western blot. Effect of repetitive hypoxia on P-MARCKS expression in carotid bodies from normoxia-treated control (lane 1), CIH exposed (lane 2), CIH-treated carotid bodies in presence of 1 μm ketanserin (lane 3), or 1 μm Bis-1 (lane 4) or 100 nm PMA (positive control; lane 5). Bottom, Average data of densitometric analysis presented as percentage of control. Note that repetitive hypoxia increased P-MARCKS expression in CIH but not in control carotid bodies, and this effect was prevented by ketanserin as well as Bis-1. B, Effect of repetitive hypoxia on NOX activation in the carotid bodies from normoxia and CIH-treated carotid bodies in the absence and presence of 1 μm ketanserin or 1 μm Bis-1. Note that ketanserin and Bis-1 prevent NOX activation by repetitive hypoxia in CIH-exposed carotid bodies. Data presented in A and B represent mean ± SEM from three individual experiments performed in triplicate. Asterisks represent p < 0.01 compared with controls. Ketan, Ketanserin; CB, carotid bodies.
Figure 7.

5-HT2 receptor antagonist and PKC inhibitor prevent sLTF of the carotid body. A, Examples of the effects of repetitive hypoxia (at arrows) on sensory activity of CIH-treated rat carotid bodies in the presence of 1 μm ketanserin (top) or 1 μm Bis-1 (bottom). Superimposed action potentials of a single fiber from which the data were derived are shown in the insets. Note that ketanserin and Bis-1 prevented repetitive hypoxia-evoked sLTF. imp, Impulses. B, Average data showing the absence of sLTF in presence of ketanserin or Bis-1. Data presented are mean ± SEM from CIH carotid bodies treated with vehicle (n = 7), or in presence of 1 μm ketanserin (n = 6) or 1 μm Bis-1 (n = 6). **p < 0.01 compared with CIH-exposed carotid bodies. Ketan, Ketanserin.
ROS generated by NOX mediates IH-induced sLTF of the carotid body
NOX activation leads to generation of ROS, especially O2·−. To determine whether O2·− generated by NOX contributes to sLTF, the effect of MnTMPyP, a membrane permeable scavenger of O2·− was examined. When CIH-treated carotid bodies were continuously challenged with 50 μm MnTMPyP, repetitive hypoxia did not elicit sLTF (CIH+ Vehicle sLTF = +160 ± 11% vs CIH+MnTMPyP=+18 ± 8%; p < 0.01; n = 6 each). However, when applied 15 min after the onset of sLTF, MnTMPyP did not inhibit sLTF (Fig. 8A,C). To test whether H2O2, a dismutated product of O2·− contributes to sLTF, carotid bodies were challenged with 200U/ml PEG-catalase, a membrane permeable scavenger of H2O2 15 min after the onset of sLTF. As shown in Figure 8, B and C, PEG-catalase completely prevented sLTF.
Figure 8.

H2O2 mediates sLTF of the rat carotid bodies. A, B, Examples of CIH-treated rat carotid body sensory response to repetitive hypoxia (at arrows) in the presence of MnTMPyP (50 μm; A) or PEG-catalase (200U/ml; B). Superimposed action potentials of a single fiber from which the data were derived are shown in the insets. imp, Impulses. C, Average data of the magnitude of sLTF during the 15th, 25th, 35th, 45th, and 60th minute of postrepetitive hypoxia period in presence of vehicle (open columns, n = 6), 50 μm MnTMPyP (filled columns, n = 6), or PEG-catalase (200U/ml; gray columns, n = 6). MnTMPyP or PEG catalase was added during postrepetitive hypoxia as indicated by arrow. PEG-catalase but not MnTMPyP prevented sLTF. *p < 0.05 compared with vehicle-treated CIH carotid bodies.
We examined whether priming the carotid body with exogenous H2O2 can mimic the effects of CIH in control normoxia-treated carotid bodies. As shown in Figure 9A, in presence of as little as 500 nm H2O2, repetitive hypoxia evoked robust sLTF in normoxia-treated carotid bodies, similar to that seen with CIH treatment. Control experiments revealed that in the absence of H2O2, repetitive hypoxia was ineffective in evoking sLTF, and superfusion with 500 nm H2O2 alone for 90 min had no effect on the baseline sensory activity (Fig. 9A). Like wise, in presence of 200 nm H2O2, repetitive hypoxia also evoked sLTF in carotid bodies from control WT and gp91phox−/Y as well as Pet-1−/− mice reared under normoxia (Fig. 9B).
Figure 9.

Priming the carotid bodies with H2O2 evokes sLTF. A, Examples of the effects repetitive hypoxia (at arrows) on sensory activity of the carotid bodies from rats reared under normoxia in the presence (top) or in the absence (middle) of 500 nm H2O2. Note sLTF in the presence but not in the absence of H2O2. Continuous exposure with 500 nm H2O2 for 90 min had no effect on baseline sensory activity of the carotid body (bottom). Superimposed action potentials of a single fiber from which the data were derived are shown in the insets. B, Examples of the effects repetitive hypoxia (at arrows) on sensory activity of the carotid bodies from WT (top), gp91phox−/Y (middle), and Pet-1−/− (bottom) mice in the presence of 200 nm H2O2. Note that priming with H2O2 evoked sLTF in all three mice. imp, Impulses.
Discussion
In the present study, we examined the role of NOX in CIH-evoked sensory plasticity of the carotid body manifested as sLTF. The following lines of evidence demonstrate that NOX is essential for evoking sLTF. First, mRNAs encoding NOX1, 2, 3 and 4 isoforms were expressed, and CIH upregulated NOX1, 2 and 4 mRNAs as well as corresponding NOX2 and 4 proteins in rat carotid bodies. Second, NOX activity increased in response to repetitive hypoxia in CIH but not in control carotid bodies. Third, three structurally and functionally distinct inhibitors of NOX (i.e., apocynin, DPI, AEBFS) prevented sLTF.
Although CIH upregulated three NOX isoforms, sLTF was absent in gp91phox knock-out mice, suggesting a major role for NOX2 in mediating sensory plasticity of the carotid body, which was further supported by the observation that it is primarily localized to the cytoplasm of glomus cells, the putative hypoxia sensing cells. In contrast, NOX4 expression was ubiquitous and was confined to the nucleus rather than the cytoplasm. Nuclear localization of NOX4 was also recently reported in cells of pulmonary origin (Pendyala et al. 2009). NOX 4, because of its nuclear localization, might play a role in CIH-evoked transcriptional activation (Yuan et al. 2008), rather than evoking sLTF, a possibility that requires further investigation. Since NOX1 is a homolog of NOX2 (Suh et al. 1999; Bánfi et al. 2000), it might also contribute to sLTF similar to NOX2. It has been proposed that NOX2 functions as an O2 sensor in the carotid body (see Dinger et al., 2007 for references). However, consistent with a previous report (He et al. 2002), we also found that carotid body sensory response to acute hypoxia was preserved in NOX-2 knock-out mice. These findings taken together suggest a novel role for NOX2 in inducing sensory plasticity of the carotid body under the conditions such as CIH, rather than functioning as an O2 sensor.
Despite the upregulation of NOX2 protein by CIH, basal NOX activity was nearly the same as in controls; whereas it could be activated only after acute repetitive hypoxia, suggesting that CIH leads to a regulated rather than constitutive activation of NOX. The following observations demonstrate that 5-HT2 receptors coupled to PKC activation contribute to the regulated NOX activation. First, 5-HT2 receptor antagonist as well as PKC inhibitor prevented NOX activation in CIH-treated carotid bodies. Second, repetitive hypoxia led to PKC activation in CIH but not in control carotid bodies and a 5-HT2 receptor antagonist prevented this effect. Activation of 5-HT2 receptors require release of 5-HT. Indeed, hypoxia evoked robust 5-HT release from CIH but not from control carotid bodies. More importantly, sLTF, which is the consequence of NOX activation by 5-HT2 receptors, was completely absent in Pet-1−/− mice with impaired 5-HT expression in the carotid body. A recent study reported that exogenous 5-HT is a potent inducer of sLTF in control rat carotid bodies, and the effects of 5-HT are mediated via NOX activation by 5-HT2 receptors and PKC (Peng et al. 2006a). These findings taken together with the observations of the present study suggest that 5-HT via 5-HT2 receptor activation mediates regulated NOX2 activation and the ensuing sLTF in CIH-treated carotid bodies.
It is intriguing that hypoxia released 5-HT from CIH but not from control carotid bodies. Considerable evidence suggests that activation of voltage-gated Ca2+ channels and subsequent elevations in [Ca2+]i in glomus cells are critical for hypoxia-evoked transmitter release from the carotid body [see Prabhakar (2000) for references]. However, CdCl2, a broad spectrum voltage-gated Ca2+ channel blocker had no effect; whereas 2-APB, a purported blocker of IP-3 receptors and/or TRPM2 channels completely prevented hypoxia-evoked 5-HT release from CIH-treated carotid bodies. These findings indicate that CIH recruits additional Ca2+ signaling pathways such as mobilization of intracellular Ca2+ for evoking 5-HT release by hypoxia. The absence of NOX activation and sLTF in carotid bodies reared under normoxia is conceivably be due to the inability of hypoxia to release 5-HT, which is necessary for NOX activation.
The finding that O2·− generated by NOX mediates LTP in the CNS (Kishida and Klann, 2007) prompted us to investigate whether it also contributes to sLTF in the carotid body. The observations that MnTMPyP, a scavenger of O2·− was effective only when given before the onset, whereas PEG-catalase, a scavenger of H2O2 prevented sLTF although given after its onset suggest that H2O2 generated from O2·− contributes to sLTF. Furthermore, priming the carotid bodies from control normoxic rats with nanomolar concentrations of exogenous H2O2 mimicked the effects of CIH by evoking sLTF. Although CIH was ineffective in eliciting sLTF in gp91phox−/Y and Pet-1−/− mice, priming with exogenous H2O2 elicited sLTF by repetitive hypoxia in both groups of mice indicating that H2O2 is downstream to 5-HT dependent NOX2 activation. The signaling cascade leading to CIH-induced NOX activation and the ensuing sLTF identified in this study is summarized in Figure 10.
Figure 10.
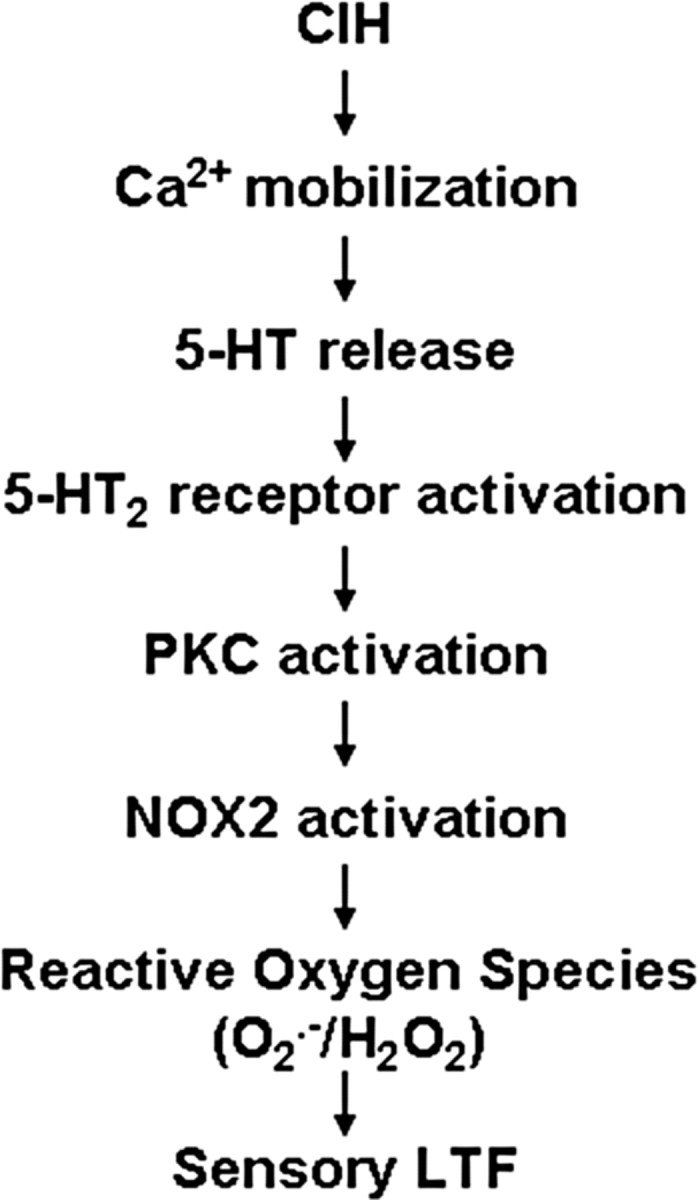
Schematic presentation of 5-HT and NOX signaling cascade mediating sLTF of the CIH-treated carotid bodies.
What might be the functional significance of CIH-induced sLTF of the carotid body? Sleep disordered breathing is characterized by transient repetitive cessations of breathing (apneas) and leads to periodic decreases in arterial pO2 or CIH. Individuals affected by recurrent apneas have increased risk for developing several cardio-respiratory comorbidities including increased sympathetic tone and respiratory abnormalities (Shahar et al. 2001). The augmented carotid body afferent activity triggers reflex activation of sympathetic nervous system and breathing. Therefore, it is conceivable that CIH-evoked sLTF of the carotid body via ROS generated by 5-HT-NOX2 signaling contributes to the autonomic morbidities associated with recurrent apneas.
Footnotes
This work was supported by National Institutes of Health–National, Heart, Lung, and Blood Institute Grants HL-90554, HL-76537, HL-86493 (N.R.P.), and HL-08533 (V.N.). We are grateful to Dr. D. J. Lambeth (Department of Pathology, Emory University School of Medicine, Atlanta, GA) for the generous gift of NOX4 antibody.
References
- Baker TL, Fuller DD, Zabka AG, Mitchell GS. Respiratory plasticity: differential actions of continuous and episodic hypoxia and hypercapnia. Respir Physiol. 2001;129:25–35. doi: 10.1016/s0034-5687(01)00280-8. [DOI] [PubMed] [Google Scholar]
- Bánfi B, Maturana A, Jaconi S, Arnaudeau S, Laforge T, Sinha B, Ligeti E, Demaurex N, Krause KH. A mammalian H+ channel generated through alternative splicing of the NADPH oxidase homolog NOH-1. Science. 2000;287:138–142. doi: 10.1126/science.287.5450.138. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Dinger B, He L, Chen J, Liu X, Gonzalez C, Obeso A, Sanders K, Hoidal J, Stensaas L, Fidone S. The role of NADPH oxidase in carotid body arterial chemoreceptors. Respir Physiol Neurobiol. 2007;157:45–54. doi: 10.1016/j.resp.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Chen J, Dinger B, Sanders K, Sundar K, Hoidal J, Fidone S. Characteristics of carotid body chemosensitivity in NADPH oxidase-deficient mice. Am J Physiol Cell Physiol. 2002;282:C27–C33. doi: 10.1152/ajpcell.2002.282.1.C27. [DOI] [PubMed] [Google Scholar]
- Hendricks T, Francis N, Fyodorov D, Deneris ES. The ETS domain factor Pet-1 is an early and precise marker of central serotonin neurons and interacts with a conserved element in serotonergic genes. J Neurosci. 1999;19:10348–10356. doi: 10.1523/JNEUROSCI.19-23-10348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks TJ, Fyodorov DV, Wegman LJ, Lelutiu NB, Pehek EA, Yamamoto B, Silver J, Weeber EJ, Sweatt JD, Deneris ES. Pet-1 ETS gene plays a critical role in 5-HT neuron development and is required for normal anxiety-like and aggressive behavior. Neuron. 2003;37:233–247. doi: 10.1016/s0896-6273(02)01167-4. [DOI] [PubMed] [Google Scholar]
- Jacono FJ, Peng YJ, Kumar GK, Prabhakar NR. Modulation of the hypoxic sensory response of the carotid body by 5-hydroxytryptamine: role of the 5-HT2 receptor. Respir Physiol Neurobiol. 2005;145:135–142. doi: 10.1016/j.resp.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Kishida KT, Klann E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid Redox Signal. 2007;9:233–244. doi: 10.1089/ars.2007.9.ft-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida KT, Hoeffer CA, Hu D, Pao M, Holland SM, Klann E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol Cell Biol. 2006;26:5908–5920. doi: 10.1128/MCB.00269-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo LA, Curnutte JT. Kinetic microplate assay for superoxide production by neutrophils and other phagocytic cells. Methods Enzymol. 1990;186:567–575. doi: 10.1016/0076-6879(90)86151-k. [DOI] [PubMed] [Google Scholar]
- Pendyala S, Gorshkova IA, Usatyuk P, He D, Pennathur A, Lambeth JD, Thannickal VJ, Natarajan V. Role of NOX4 and NOX2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid Redox Signal. 2009;11:1–18. doi: 10.1089/ars.2008.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol. 2004;96:1236–1242. doi: 10.1152/japplphysiol.00820.2003. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci U S A. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Yuan G, Jacono FJ, Kumar GK, Prabhakar NR. 5-HT evokes sensory long-term facilitation of rodent carotid body via activation of NADPH oxidase. J Physiol. 2006a;576:289–295. doi: 10.1113/jphysiol.2006.116020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Yuan G, Ramakrishnan D, Sharma SD, Bosch-Marce M, Kumar GK, Semenza GL, Prabhakar NR. Heterozygous HIF-1alpha deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol. 2006b;577:705–716. doi: 10.1113/jphysiol.2006.114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinilla PJ, Hernández AT, Camello MC, Pozo MJ, Toescu EC, Camello PJ. Non-stimulated Ca2+ leak pathway in cerebellar granule neurones. Biochem Pharmacol. 2005;70:786–793. doi: 10.1016/j.bcp.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Powell FL, Milsom WK, Mitchell GS. Time domains of the hypoxic ventilatory response. Respir Physiol. 1998;112:123–134. doi: 10.1016/s0034-5687(98)00026-7. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. Oxygen sensing by the carotid body chemoreceptors. J Appl Physiol. 2000;88:2287–2295. doi: 10.1152/jappl.2000.88.6.2287. [DOI] [PubMed] [Google Scholar]
- Shahar E, Whitney CW, Redline S, Lee ET, Newman AB, Javier Nieto F, O'Connor GT, Boland LL, Schwartz JE, Samet JM. Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am J Respir Crit Care Med. 2001;163:19–25. doi: 10.1164/ajrccm.163.1.2001008. [DOI] [PubMed] [Google Scholar]
- Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- Wen Y, Gu J, Li SL, Reddy MA, Natarajan R, Nadler JL. Elevated glucose and diabetes promote interleukin-12 cytokine gene expression in mouse macrophages. Endocrinology. 2006;147:2518–2525. doi: 10.1210/en.2005-0519. [DOI] [PubMed] [Google Scholar]
- Youngson C, Nurse C, Yeger H, Curnutte JT, Vollmer C, Wong V, Cutz E. Immunocytochemical localization on O2-sensing protein (NADPH oxidase) in chemoreceptor cells. Microsc Res Tech. 1997;37:101–106. doi: 10.1002/(SICI)1097-0029(19970401)37:1<101::AID-JEMT10>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Yuan G, Nanduri J, Khan S, Semenza GL, Prabhakar NR. Induction of HIF-1alpha expression by intermittent hypoxia: Involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J Cell Physiol. 2008;217:674–685. doi: 10.1002/jcp.21537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan G, Serrano F, Fenik P, Hsu R, Kong L, Pratico D, Klann E, Veasey SC. NADPH oxidase mediates hypersomnolence and brain oxidative injury in a murine model of sleep apnea. Am J Respir Crit Care Med. 2005;172:921–929. doi: 10.1164/rccm.200504-581OC. [DOI] [PMC free article] [PubMed] [Google Scholar]