

Abstract
Background
AMP-dependent protein kinase (AMPK) and peroxisome proliferator-activated receptor (PPAR) α facilitate fatty acid oxidation. We have shown that treatment of hepatoma cells with ethanol or feeding ethanol-containing diets to mice inhibited both PPARα and AMPK activity. Importantly, WY-14,643 reversed the development of fatty liver in alcohol-fed mice. Whether WY-14,643, a PPARα agonist, has any effects on AMPK is not known. The aim of this study was to investigate the effect of WY-14,643 on AMPK activity.
Methods
The effect of WY-14,643 on AMPK phosphorylation and activity were examined in rat hepatoma cells (H4IIEC3). The effect of WY-14,643 on upstream kinases of AMPK, PKC-ζ/LKB1, intracellular AMP:ATP ratio, oxidative stress, and AMPK gene expression were studied.
Results
Treatment of the H4IIEC3 cells with WY-14,643 for 24 h led to 60% increase in the phosphorylation of AMPK. The effect of WY-14,643 on AMPK phosphorylation is PKC-ζ/LKB1 independent. WY-14,643 did not alter the levels of intracellular AMP:ATP ratio and it did not increase the levels of reactive oxygen species at 24-h of treatment. WY-14,643-induced AMPK α subunit expression by 2- to 2.5-fold, but there was no change in AMPKα subunit protein at 24 h. The effect of WY-14,643 on AMPK phosphorylation did not altered by the presence of an NADPH oxidase inhibitor.
Conclusions
WY-14,643 induced AMPKα subunit phosphorylation and the activity of the enzyme. This was associated with induction of AMPKα1 and α2 mRNA, but the mechanism for this activation is uncertain.
Keywords: WY-14,643; PPARα agonist; AMPKα
Introduction
AMP-dependent protein kinase (AMPK)1 is a key enzyme in the regulation of energy metabolism and the response to cellular stress. Increases in intracellular AMP activate AMPK via several allosteric mechanisms [1]. The predominant effect is activation of LKB1 that activates AMPK by phosphorylating threonine 172 in the α subunit [1].
AMPK can also be activated by several AMP-independent pathways. Firstly, AMPK can be activated by Ca2+/calmodulin-dependent protein kinase kinase (CaMKK) [2]. In some cell types, over-expression of CaMKKα increased AMPK activity, while pharmacological inhibition of CaMKKα nearly completely abolished AMPK activation [6]. Secondly, a member of the mitogen-activated protein kinase kinase kinase (MAPKKK) family, transforming growth factor-activated kinase (TAK1) can also phosphorylate AMPK [3]. More recently, the ataxia telangiectasia mutated kinase (ATM kinase) was shown to phosphorylate AMPK in response to DNA damage [4].
Other physiological or pathological conditions that activate AMPK include oxidative stress [5]. Peroxynitrite caused phosphorylation of PKC-ζ in bovine aortic endothelial cells, with no effect on cellular AMP levels [6]. Choi et al. reported that H2O2 activated AMPK in NIH-3T3 cells by way of both increased AMP and a tyrosine kinase-dependent pathway [5]. These studies suggest that ROS might activate AMPK in some circumstances by activating other kinase cascades resulting in AMPK phosphorylation.
The activity of AMPK helps determine the fate of fatty acids. It reduces the activity of acetyl-CoA carboxylase (ACC). The net effect of AMPK activation is a reduction in the steady-state level of malonyl-CoA, which in turn increases the access of fatty acyl-CoA esters to the mitochondrial matrix space by removing the inhibition of carnitine palmitoyltransferase (CPT)-I [1]. Another factor regulating fatty acid metabolism is peroxisome proliferator-activated receptor alpha (PPARα). This nuclear factor is a major transcriptional regulator of fatty acid oxidation. Of note, PPARα actions are dependent on interactions with PPARγ coactivator 1α (PGC-1α) which was reported to be activated by phosphorylation by AMPK [7]. Synthetic PPARα agonists, WY-14,643 and clofibrate, have been used extensively to study PPARα-dependent cellular functions, but have been found to have a number of actions that may be independent of this receptor. For instance, in Kupffer cells, WY-14,643 activated NADPH oxidase and increased mRNA for TNFα [8]. WY-14,643 and clofibrate also increased the expression of CPT-1 and CPT-2 in Fao hepatoma cells and rat liver, respectively [9].
We have shown that treatment of hepatoma cells with ethanol or feeding ethanol-containing diets to mice inhibited both PPARα [10] and AMPK activity [11]. Furthermore, the amount of AMPKα subunit was decreased approximately 40% in the livers of ethanol-fed mice [11]. WY-14,643 reversed the development of fatty liver in alcohol-fed mice [10]. Since PPARα and AMPK seem to coordinate the regulation of lipid metabolism in the liver, the aim of this study was to investigate the effect of WY-14,643 on AMPK activity.
Materials and methods
Materials
Most chemicals unless otherwise specified were purchased from Sigma Aldrich Chemical Company (St. Louis, MO) or Calbiochem-Novabiochem Corporation (San Diego, CA). Trypsin-EDTA, fetal bovine serum (FBS) and modified Eagle's medium (MEM) were purchased from GIBCO Invitrogen Corporation, Inc (Carlsbad, CA). FBS charcoal-stripped of lipids was purchased from Hyclone Laboratories (Logan, UT). Radioisotopes were purchased from PerkinElmer Life and Analytical Sciences (Wellesley, MA). H4IIEC3 hepatoma cells were from the American Type Culture Collection (Manassas, VA). SAMS peptide (full sequence HMRSAMSGLHLVKRR) a synthetic peptide substrate for AMPK was from Upstate Biotechnology (Charlottesville, VA). PPRE3-tk-luciferase (containing three copies of PPRE from the acyl-CoA oxidase gene ligated to a herpes simplex thymidine kinase promoter upstream of the luciferase gene [6]) and the expression plasmids for murine PPARα were the kind gift of Dr. Ronald Evans (Salk Institute for Biological Studies, San Diego, CA). Antibodies for Western blotting were purchased from Cell Signaling Technology (Danvers, MA).
Measurement of AMPK activity
The AMPK activity assay was measured in cell extracts prepared for immunoblot assays, according to methods described previously [11]. The AMPK activity was expressed as a percentage of control [11].
Real time reverse transcription-PCR
Effects of WY-14,643 on gene expression of AMPKα1 and AMPKα2 subunit genes were measured by real time RT-PCR. Total RNA was isolated from H4IIEC3 cells treated with WY-14,643 for 24 h using RNeasy mini kit from Qiagen (Valencia, CA) following manufacturer's instructions. Initial concentration of RNA was measured by ribogreen RNA quantitation reagent (Molecular Probes, Carlsbad, CA). Real time RT-PCR was performed using Taqman gene expression assays for AMPKα1 and AMPKα2 (pre-designed, pre-optimized probe and primer sets: Applied Biosystems, Foster City, CA) while normalizing with Taqman GAPDH probes (n = 3). For each 25 μl reactions, 2.5 μl of 10× core RT buffer and 50 mM MgCl2, 2 μl of GAUC (5 mM dUTP), 1.25 μl of either AMPKα1 or AMPKα2 gene expression assays, 0.25 μl of Strata ST and Taq DNA polymerase, 0.375 μl of reference dye, and RNase-free water was required to make the volume to 23 μl. Then 2 μl of 100 ng/μl of total RNA from each samples were added to make 25 μl of reaction mixture. Separate reactions were set up for GAPDH quantitation. For each 25 μl reactions, 2.5 μl of 10× core RT buffer and 50 mM MgCl2, 2 μl of GAUC, 0.25 μl of forward and reverse GAPDH primers, GAPDH Taqman probe, Taq DNA polymerase and Strata ST, 0.375 μl of reference dye, and RNase-free water was used to make the volume to 23 μl. Then 2 μl of 50 ng/μl of total RNA from each samples were added to make 25 μl of reaction mixture. In all real time RT-PCR procedures, we made master mixes to reduce errors due to pipetting. Samples were run in triplicate and the results were expressed as a change in fold.
Measurement of reactive oxygen species
H4IIEC3 cells were seeded at 7.3 × 104 cells/well on 24-well plates two days before the experiment and serum starved for 16 h the day before the experiment. The cells were then treated with 100 mM WY-14,643 for 5 min, 3 or 24 h. After the treatment, cells were loaded with 20 μM 2′,7′-dichlorodihydrofluorescein-diacetate (DCFH-DA; Sigma, St. Louis, MO) for 45 min at 37 °C in dark. Subsequently, cells were rinsed with PBS and 500 μl of fresh Earle's salt solution was added to each well. Fluorescence was measured using prewarmed SpectraMax M5 (Molecular Devices, Sunnyvale, CA) spectrofluorometer, with excitation/emission wavelengths of 485/535 nm for 20 min. The slope of the linear part of the graph was used to calculate the rate of increases in fluorescence/min. As a positive control, H2O2 (0.5 mM) was added just before placing the plate into the plate reader. The increase in fluorescence for the first 5 min by ROS production was expressed as % of control.
HPLC analyses of nucleotides
H4IIEC3 cells were split into 10 cm plates. The day before experiment, the cells were serum starved for 16 h in serum-free MEM. On the day of treatment, cells were treated with 100 μM WY-14,643 or 2 mM metformin for 24 h. At the end of treatment, cell culture medium was removed and the cells were rinsed with ice-cold PBS. After the removal of PBS, 300 μl of ice-cold 10% TCA was added directly onto the 10 cm plates. Cells were then scraped with rubber policeman, transferred to Eppendorf tubes and chilled on ice for 10 min. Tubes were vortexed vigorously for 20 s at 4 °C and centrifuged at 14,000 rpm at 4 °C for 2 min. Supernatant was transferred to Eppendorf tubes already containing 0.6 ml of 0.5 M tri-n-octylamine in Freon, vortexed at 4 °C for 20 s, and re-centrifuged at 18,000g at 4 °C for 1 min. The bottom layer was carefully removed, leaving a clean top aqueous layer within the Eppendorf tubes. Tubes were frozen at −80 °C until HPLC analysis. HPLC analysis was performed as previously published [12]. Results were expressed as the AMP/ATP ratio.
Transfection of tissue culture cells
Hepatoma cells were grown to 80−90% confluency in modified Eagle's medium (MEM) supplemented with 10% fetal bovine serum, 100 μg/ml streptomycin, and 63 μg/ml penicillin G. Cells were transfected with 0.5 μg of reporter plasmid (PPRE tk-luc), 0.2 μg of PPARα expression plasmid, 0.2 μg of pRL-TK (Renilla luciferase) as an internal control for transfection efficiency using the Lipofectamine 2000 kit from GIBCO Invitrogen Corporation, Inc (Carlsbad, CA) following the manufacturer's instructions. Luciferase activities were measured using Promega Dual-luciferase reporter assay system (Promega, Madison, WI) following the manufacturer's instructions. The numbers of relative light units was determined with 2-s delay and 10-s reading with the TD 20/20 Luminometer (Turner Designs, Sunnyvale, CA). PPARα expression was expressed as a percentage of control.
Cloning of AMPK alpha promoters
The promoter region between −3000 and −60 of the AMPK alpha2 gene was obtained by polymerase chain reaction amplification using rat hepatocytes genomic DNA as a template with outside primers containing engineered BamH I and Kpn I sites. The primers used were CAGGATCCTTAGCATAAGACCACAAGGTT ACT and CAGGTACCACCGCCTACCCACAGT. The amplified fragment was digested with BamH I and Kpn I. The plasmid 300 AMPKα2 was constructed by ligating digested fragment into the BamH I and Kpn I sites of luciferase expression plasmid, PXP2.
Immunoblot analysis
Immunoblot analyses were performed using 20 μg whole cell extract separated by electrophoresis in a 10% or 6% SDS–polyacrylamide gel and transferred to nitrocellulose filters. Detection of the protein bands was performed using the ECL Plus Kit (Amersham Biosciences, Piscataway, NJ). The protein bands were then quantified on a PhosphoImager and ImageQuant (Amersham Biosciences) software analysis.
Statistical analysis
All results were expressed as means ± SD. Comparisons between multiple groups were performed with one-way ANOVA, followed by a post hoc LSD test (p ≤ 0.05 was considered significant) using SPSS statistical software program.
Results
Activation of AMPK by WY-14,643 is independent of PKC-ζ/LKB1-phosphorylation
The effects of WY-14,643 on the phosphorylation and the activity of AMPK were examined in H4IIEC3 cells. The level of P-AMPK was normalized to the level of unphosphorylated AMPK to control for possible induction in enzyme mass. WY-14,643 significantly increased the P-AMPK/AMPK ratio, AMPK activity, and phosphorylation of its downstream target protein, ACC by 60%, 100%, and 15%, respectively, after 24 h treatment. Such an effect was not observed with 1−12 h treatment (Fig. 1). Several studies had identified that LKB1 acts as one of the AMPK kinases. Zou et al. showed that PKC-ζ can regulate AMPK activity by increasing the Ser-428 phosphorylation of LKB1, resulting in association of LKB1 with AMPK and consequent AMPK phosphorylation [13]. We therefore asked if WY-14,643 induced AMPK phosphorylation through these two upstream kinases. As previously reported, metformin significantly increased the phosphorylation of PKC-ζ and LKB1 by 29% and 45%, respectively. In contrast, WY-14,643 did not alter the levels of p-PKC-ζ and p-LKB1. Taken together, these data suggested that the activation of AMPK by WY-14,643 is independent of phosphorylation of PKC-ζ and LKB1.
Fig. 1.

Effect of WY-14,643 on AMP phosphorylation and AMP activity in rat hepatoma H4IIEC3 cells. (A and B) Immunoblot analysis. H4IIEC3 cells were treated with WY-14,643 (100 μM) or metformin (2 mM), as indicated. (C) Bar graph demonstrating quantification of the Western blot (B). There were no changes in AMPK phosphorylation with WY-14,643 after 1 to 20-h of treatment. However, at 24 h, WY-14,643 increased the levels of phospho-AMPK and its downstream target protein, p-ACC by 59% and 25%, respectively. There were no effects of WY-14,643 on PKC-ζ and LKB1. (D) AMPK activity measured using a synthetic peptide substrate. The experiments with metformin (2 mM) served as positive controls. The activity of AMPK correlated with the levels of phospho-AMPK from the Western blot analyses. *Significant difference vs control; p < 0.05, by 1-way ANOVA. The results are representative of blots from four separate experiments.
Effects of WY-14,643 on AMPK gene expression
Because WY-14,643 increased the activity of AMPK, we next determined the effect of this compound on the expression of the AMPK α genes. Total RNA was isolated from H4IIEC3 cells after 24 h of treatment with WY-14,643 and one-step real time RT-PCR was performed using AMPKα1 and AMPKα2-specific primers. WY-14,643 increased the abundance of mRNA of AMPKα1 and α2 by 2- to 2.5-fold compared with GAPDH mRNA (Fig. 2). The promoter sequences 3-kilobase upstream from the rat AMPKα1 and α2 genes available in the genomic databases were examined for possible consensus PPARα binding sites; however none were apparent. We cloned 2940 kb of the AMPKα2 promoter into the PXP2 reporter plasmid. When this was transfected into H4IIEC3 cells with the PPARα expression plasmid, the reporter activity was not induced by WY-14,643, while a PPAR response element-containing reporter plasmid PPPRE3-tk-luc serving as a positive was induced (data not shown). We were unable to clone the AMPKα1 promoter sequences despite the use of several sets of PCR primers.
Fig. 2.
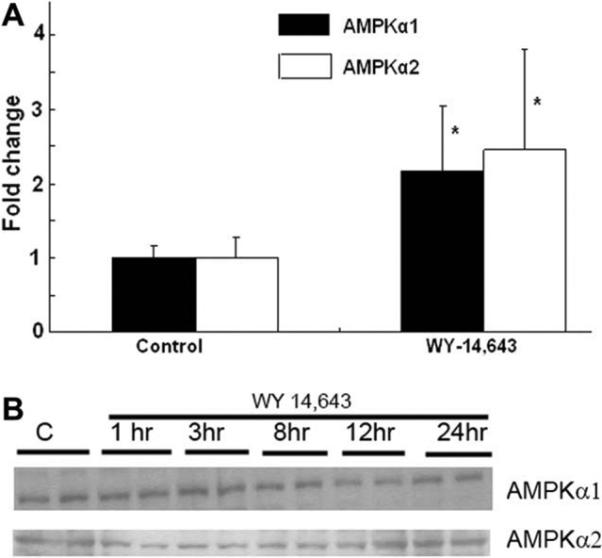
Effects of WY-14,643 on AMPKα1 and AMPKα2 gene expression. WY-14,643 increased the abundance of mRNA of AMPKα1 and α2 by 2- to 2.5-fold. The results were expressed as average fold change ± SD. *p < 0.05 vs control. (B) There were no changes in the levels of AMPKα1 and AMPKα2 subunit proteins after treatment with WY-14,643. The results are representative of blots from three separate experiments.
We next determined whether the changes in the mRNA of AMPKα1 and α2 by WY 14,643 associated with the alteration in protein expressions. Using antibodies selective for AMPKα1 and AMPKα2 isoforms, we did not see an increase in the AMPKα subunit mass for either isoform on Western blots (Fig. 2B).
Effects of WY-14,643 on AMP/ATP and energy charge
The intracellular level of AMP is a sensitive marker of the energy stores of the cell and AMPK cascades have been shown to respond mainly to the intracellular level of AMP or AMP:ATP ratio [1]. We hypothesized that the effect of WY-14,643 on AMPK activity might be mediated through an alteration in the AMP or AMP/ATP ratio. The intracellular levels of nucleotides were measured in the H4IIEC3 cells after 24-h treatment with WY-14,643 (Table 1). The baseline AMP:ATP ratio in H4IIEC3 cells was 0.018 ± 0.002, and they were not significantly different than those in the cells which were treated with WY-14,643 for 24 h (0.020 ± 0.002). Thus, the effects of WY-14,643 on AMPK phosphorylation in this cell line are likely mediated through an AMP-independent pathway.
Table 1.
AMP/ATP ratio after 24-h treatment with WY-14,643 (100 μM) and metformin (2 mM). The results are from several experiments (n = 3) and expressed as average % of control ± SD. *p < 0.05 vs control.
| Treatments | AMP/ATP ratio |
|---|---|
| Control | 0.018 ± 0.002 |
| WY-14,643 | 0.020 ± 0.002 |
| Metformin | 0.024 ± 0.003 |
Effects of WY-14,643 on reactive oxygen species
Previous work suggested that ROS might be involved in the activation of AMPK [5]. The ability of WY-14,643 to stimulate ROS generation was tested using DHCF-acetate. Incubation of H4IIEC3 cells with WY-14,643 for 5 min and 3 h led to 2-fold increase in the formation of ROS. However, no change in ROS production was seen at 24-h treatment (Fig. 3A), This is consistent with the lack of effect of WY-14643 on the phosphorylation of PKC-ζ and LKB1 (discussed below).
Fig. 3.

Effects of WY-14,643 on DHCF oxidation. (A) Cells were treated with WY-14,643 (100 μM) for 5 min, 3 or 24 h. Incubation of H4IIEC3 cells with WY-14,643 for 5 min and 3 h led to 2-fold increase in the formation of ROS. However, no change in ROS production was seen at 24-h treatment. H2O2 (0.5 mM) was used as a positive control. Results are from several experiments (n = 4) and expressed as average % of control ± SD. *p < 0.05 vs control. (B) Pretreatment with apocynin (10 μM) did not affect the baseline p-AMPK and the ability of WY-14,643 to stimulate AMPK phosphorylation after 24-h treatment. H4IIEC3 cells were treated with or without 10 μM apocynin for 2 h and then stimulated with WY-14,643 as indicated. The results are representative of blots from three separate experiments.
In order to test whether the increase in the ROS production seen at the earlier time points was required for the activation of AMPK (at 24 h), we performed experiments in the presence or absence of apocynin (10 μM), an inhibitor of NADPH oxidase. The widely used NADPH oxidase inhibitor diphenylene iodonium (DPI) unfortunately is known to activate AMPK [14]. There was no effect of apocynin on baseline AMPK phosphorylation (Fig. 3B). There was also no difference in WY-14,643-induced AMPK phosphorylation at 24-h of treatment with or without pre-treatment with apocynin (Fig. 3B).
Discussion
This study provided evidence that AMPK can be phosphorylated and activated by the PPARα agonist WY-14,643. The mechanism did not involve changes in the phosphorylation of PKC-ζ/LKB1 and it did not depend on the generation of oxidative stress or changes in steady-state intracellular AMP:ATP ratio. We found that WY-14,643 increased expression of AMPK α1 and two subunit mRNAs, but we did not see a corresponding increase in total AMPK subunit (either AMPKα1 or α2) protein levels.
The role of LKB1 in the regulation of AMPK activity has been demonstrated recently. Purified LKB1 is constitutively active, AMP-independent, and can auto-phosphorylate [15]. The exact role of phosphorylation of LKB1 is incompletely understood; however, peroxynitrite-induced oxidative stress activated PKC-ζ and resulted in LBK1 phosphorylation on serine 428 and phosphorylation of AMPK [6]. The phosphorylation of LKB1 results in export of LKB1 from the nucleus, leading to the activation of AMPK. These data support the notion that PKC-ζ acts as an upstream kinase for LKB1 [6]. In our study, WY-14,643 activated AMPK activity without significant changes phosphorylation of PKC-ζ and LKB1 in hepatoma cells (Fig. 1). Traditionally, the activity of AMPK was thought to be particularly sensitive to the level of intracellular AMP. WY-14,643 was shown to inhibit mitochondrial respiration in isolated rat cardiac mitochondria [16] and increase cardiac mitochondrial uncoupling protein (UCP) 2 and UCP3 levels in mice [17] leading to increasing in intracellular AMP. However, we failed to demonstrate a change in the AMP/ATP ratio in the H4IIEC3 cells cultured with WY-14,643 for 24 h. The discrepancy of our results and others suggested that the effect of WY-14,623 on the intracellular level of AMP and ATP is likely a cell-type specific effect. However, we accept that dynamic changes or changes limited to subcellular compartments of the energy charge might not have been detected by our experimental methods. We also acknowledge that it is difficult to be certain what degree of change in this ratio might lead to activation of AMPK, since the system is exquisitely sensitive.
Our group has shown that AMPK can also be activated by the oxidative stress induced by hydrogen peroxide in the H4IIEC3 cell line (exposure to 0.5 mM H2O2 results in about a 50% increased in AMPK activity and phosphorylation, [18]. WY-14,643 had been previously reported to stimulate the production of superoxide by Kupffer cells [19] and also increased the production of ROS in human primary macrophages [20]. It is reported to induce eNOS in some cells, which might increase the production of peroxynitrite. [21]. We next asked whether the effect of WY-14,643 on AMPK phosphorylation in hepatoma cells was related to the generation of oxidative stress. We found that short-term treatment with WY-14,643 (Fig. 3) increased the rate of production of ROS as detected by changes in DHDCF fluorescence. However, the rate of ROS production was the same as that of controls after 24-h treatment, and the magnitude of the acute increase in ROS was far smaller than that following addition of H2O2. This may explain why there was no change in AMPK phosphorylation or activity after 1 h of exposure to WY-14,643 (Fig. 1). It is plausible that the levels of ROS which were generated by WY-14,643 were not high enough to stimulate AMPK phosphorylation acutely, but set in motion cellular responses that resulted in the increase in p-AMPK To test this latter hypothesis, we incubated the cells with WY-14,643 in the presence of apocynin, an NADPH oxidase inhibitor. We did not see any difference in WY-14,643-induced AMPK phosphorylation at 24-h treatment in the presence or absence of apocynin. Taken together, in our experimental model it is unlikely that the increased rate of ROS production led to activation of AMPK.
An interesting finding from our work was that WY-14,643-induction of AMPK activity was associated with increased expression of AMPK mRNA. There are two isoforms of the α subunit of AMPK; AMPKα1 and AMPKα2. When hepatoma cells were treated with WY-14,643 for 24 h, we observed a 2- to 2.5-fold increase in the level of both AMPKα1 and AMPKα2 mRNA. The exact mechanism of this induction is not clearly understood. One possibility is that it might be partly due a direct PPARα interaction with the AMPKα promoter region. However, we were not able to demonstrate PPARα-responsiveness of a cloned segment of the AMPK α2 promoter in transfection experiments. We were unable to clone the AMPKα1 promoter sequences. However, this effect of WY-14,643 was not accompanied by an increase in AMPKα subunit mass of either AMPKα1 or AMPKα2 on Western blots (Fig. 2B), and even if it were, it is not clear that the mere increase in the amount of AMPK protein would result in increased levels of phosphorylation. Murakami et al. reported that fenofibrate, but not bezafibrate or WY-14,643, induced phosphorylation of AMPK in human umbilical vein endothelial cells [22], and suggested that this was independent from its effects on PPARs. It is possible that WY-14,643 stimulated AMPK phosphorylation through pathways that were not examined in this study, such as CaMKK, ATM, or TAK1.
Our study may have clinical implications, especially for the treatment of alcohol-induced hepatic steatosis. Previous work from our laboratory showed that the ability of PPARα to bind to its DNA consensus sequence and the levels of several PPARα-regulated mRNAs were reduced in the livers of ethanol-fed mice [10]. Similarly, AMPK activity and protein levels were decreased in ethanol-fed mice [13]. It is possible that the impaired PPARα activity resulted in decreased AMPK mRNA levels and, over longer time periods than tested in the present experiments, led to a reduction in AMPK protein expression. Thus, the improvement in liver histology that resulted from administration of WY-14,643 [10] might have been the result of the combination of activation of PPARα and induction of enzymes in the pathway of fat oxidation and export, induction of AMPK mRNA and protein, and activation of pathways leading to increased AMPK phosphorylation and activity. The potential for WY-14,643 to activate AMPK needs to be kept in mind when interpreting the effects of this compound.
Acknowledgments
This study is supported by R01 AA15070 and P60 AA07611 from the NIAAA (D.W.C.), Veterans Administration Young Investigator Award/Indiana Institute for Medical Research (S.L.), K08 AA016570 (S.L.) and Veterans Administration Merit Review Award (H.N.J.).
Footnotes
Abbreviations used: AMPK, AMP-dependent protein kinase; CaMKK, Ca2+/calmodulin-dependent protein kinase kinase; MAPKKK, mitogen-activated protein kinase kinase kinase; TAK1, transforming growth factor-activated kinase; ATM kinase, ataxia telangiectasia mutated kinase; ACC, acetyl-CoA carboxylase; CPT, carnitine palmitoyltransferase; PPARα, peroxisome proliferator-activated receptor alpha; DCFH-DA, 2′,7′-dichlorodihydrofluorescein-diacetate; DPI, diphenylene iodonium; UCP, uncoupling protein.
References
- 1.Carling D, Sanders MJ, Woods A. Int. J. Obes. (Lond.) 2008;32(Suppl 4):S55–S59. doi: 10.1038/ijo.2008.124. [DOI] [PubMed] [Google Scholar]
- 2.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Momcilovic M, Hong SP, Carlson M. J. Biol. Chem. 2006;281:25336–25343. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- 4.Fu X, Wan S, Lyu YL, Liu LF, Qi H. PLoS ONE. 2008;3:e2009. doi: 10.1371/journal.pone.0002009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi SL, Kim SJ, Lee KT, Kim J, Mu J, Birnbaum MJ, Soo KS, Ha J. Biochem. Biophys. Res. Commun. 2001;287:92–97. doi: 10.1006/bbrc.2001.5544. [DOI] [PubMed] [Google Scholar]
- 6.Xie Z, Dong Y, Zhang M, Cui MZ, Cohen RA, Riek U, Neumann D, Schlattner U, Zou MH. J. Biol. Chem. 2006;281:6366–6375. doi: 10.1074/jbc.M511178200. [DOI] [PubMed] [Google Scholar]
- 7.Jager S, Handschin C, St-Pierre J, Spiegelman BM. Proc. Natl. Acad. Sci. USA. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rusyn I, Yamashina S, Segal BH, Schoonhoven R, Holland SM, Cattley RC, Swenberg JA, Thurman RG. Cancer Res. 2000;60:4798–4803. [PubMed] [Google Scholar]
- 9.Luci S, Geissler S, Konig B, Koch A, Stangl GI, Hirche F, Eder K. Biochem. Biophys. Res. Commun. 2006;350:704–708. doi: 10.1016/j.bbrc.2006.09.099. [DOI] [PubMed] [Google Scholar]
- 10.Fischer M, You M, Matsumoto M, Crabb DW. J. Biol. Chem. 2003;278:27997–28004. doi: 10.1074/jbc.M302140200. [DOI] [PubMed] [Google Scholar]
- 11.You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. Gastroenterology. 2004;127:1798–1808. doi: 10.1053/j.gastro.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 12.Paulik E, Jayaram HN, Weber G. Anal. Biochem. 1991;197:143–148. doi: 10.1016/0003-2697(91)90370-9. [DOI] [PubMed] [Google Scholar]
- 13.Zou MH, Hou XY, Shi CM, Kirkpatick S, Liu F, Goldman MH, Cohen RA. J. Biol. Chem. 2003;278:34003–34010. doi: 10.1074/jbc.M300215200. [DOI] [PubMed] [Google Scholar]
- 14.Hutchinson DS, Csikasz RI, Yamamoto DL, Shabalina IG, Wikstrom P, Wilcke M, Bengtsson T. Cell Signal. 2007;19:1610–1620. doi: 10.1016/j.cellsig.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 15.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Biochem. J. 2007;403:139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zungu M, Felix R, Essop MF. Mitochondrion. 2006;6:315–322. doi: 10.1016/j.mito.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Murray AJ, Panagia M, Hauton D, Gibbons GF, Clarke K. Diabetes. 2005;54:3496–3502. doi: 10.2337/diabetes.54.12.3496. [DOI] [PubMed] [Google Scholar]
- 18.Liangpunsakul S, Wou SE, Zeng Y, Ross RA, Jayaram HN, Crabb DW. Am. J. Physiol. Gastrointest. Liver Physiol. 2008;295:G1173–G1181. doi: 10.1152/ajpgi.90349.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rose ML, Rivera CA, Bradford BU, Graves LM, Cattley RC, Schoonhoven R, Swenberg JA, Thurman RG. Carcinogenesis. 1999;20:27–33. doi: 10.1093/carcin/20.1.27. [DOI] [PubMed] [Google Scholar]
- 20.Teissier E, Nohara A, Chinetti G, Paumelle R, Cariou B, Fruchart JC, Brandes RP, Shah A, Staels B. Circ. Res. 2004;95:1174–1182. doi: 10.1161/01.RES.0000150594.95988.45. [DOI] [PubMed] [Google Scholar]
- 21.Okayasu T, Tomizawa A, Suzuki K, Manaka K, Hattori Y. Life Sci. 2008;82:884–891. doi: 10.1016/j.lfs.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 22.Murakami H, Murakami R, Kambe F, Cao X, Takahashi R, Asai T, Hirai T, Numaguchi Y, Okumura K, Seo H, Murohara T. Biochem. Biophys. Res. Commun. 2006;341:973–978. doi: 10.1016/j.bbrc.2006.01.052. [DOI] [PubMed] [Google Scholar]