

Abstract
Purpose
We have shown recently that glycogen synthase kinase-3 (GSK-3) β regulates nuclear factor-κB (NF-κB) – mediated pancreatic cancer cell survival and proliferation in vitro. Our objective was to determine the localization of GSK-3β in pancreatic cancer cells and assess the antitumor effect of GSK-3 inhibition in vivo to improve our understanding of the mechanism by which GSK-3β affects NF-κB activity in pancreatic cancer.
Experimental Design
Immunohistochemistry and cytosolic/nuclear fractionation were done to determine the localization of GSK-3β in human pancreatic tumors. We studied the effect of GSK-3 inhibition on tumor growth, cancer cell proliferation, and survival in established CAPAN2 tumor xenografts using a tumor regrowth delay assay, Western blotting, bromodeoxyuridine incorporation, and terminal deoxynucleotidyl transferase – mediated dUTP nick end labeling.
Results
We found nuclear accumulation of GSK-3β in pancreatic cancer cell lines and in 62 of 122 (51%) human pancreatic adenocarcinomas. GSK-3β nuclear accumulation is significantly correlated with human pancreatic cancer dedifferentiation. We have found that active GSK-3β can accumulate in the nucleus of pancreatic cancer cells and that inhibition of GSK-3 kinase activity represses its nuclear accumulation via proteasomal degradation within the nucleus. Lastly, we have found that inhibition of GSK-3 arrests pancreatic tumor growth in vivo and decreases NF-κB-mediated pancreatic cancer cell survival and proliferation in established tumor xenografts.
Conclusions
Our results show the antitumor effect of GSK-3 inhibition in vivo, identify GSK-3β nuclear accumulation as a hallmark of poorly differentiated pancreatic adenocarcinoma, and provide new insight into the mechanism by which GSK-3β regulates NF-κB activity in pancreatic cancer.
Despite tremendous scientific efforts, conventional treatment approaches have had little effect on the course of pancreatic cancer. There are numerous factors that contribute to progression of this disease, including constitutively active nuclear factor-κB (NF-κB; ref. 1). Activation of NF-κB in human cancer has been shown to positively influence cancer cell survival, proliferation, invasion, metastasis, and chemoresistance (2, 3). Thus, the identification of the altered molecular pathways regulating NF-κB activity is a major focus of cancer researchers, as these studies will provide valuable knowledge and identify novel targets to antagonize NF-κB activation in human cancer.
The cytoplasmic serine/threonine protein kinase glycogen synthase kinase-3 (GSK-3) was first described as a component of the metabolic pathway for glycogen synthase regulation (4). There are two homologous mammalian isoforms encoded by different genes, GSK-3α and GSK-3β (5). Surprisingly, similar to the disruption of the NF-κB p65 or IκB kinase β genes, ablation of the murine GSK-3β gene resulted in embryonic lethality due to hepatocyte apoptosis and massive liver degeneration (6–8). These findings suggest a role for GSK-3β (but not GSK-3α) in the mechanism of NF-κB activation and suggest that GSK-3β may be a potential therapeutic target in human cancer. Using GSK-3β-deficient mouse embryonic fibroblasts, it was shown that the early steps leading to NF-κB activation following TNF-α treatment (degradation of IκBα and translocation of NF-κB to the nucleus) were unaffected by the loss of GSK-3β, indicating that NF-κB is regulated by GSK-3β at the level of the transcriptional complex (8). Consistent with this idea, we have shown recently that GSK-3β participates in NF-κB-mediated pancreatic cancer cell survival and proliferation in vitro by regulating NF-κB activity at a point downstream of the activation of the IκB kinase complex (9). Taken together, these data rule out an effect of GSK-3β on the cascade of proteins that culminates in phosphorylation of IκBα and its degradation and suggest that GSK-3β may regulate the nuclear activity of NF-κB p65/p50. However, whether GSK-3β can be accumulated in the nuclei of cancer cells where it can contribute to NF-κB transcriptional activity is not known.
The localization of GSK-3β in human cancer cells and the mechanism by which GSK-3β affects NF-κB activity has not yet been determined. Here, we find that GSK-3β is overexpressed in human pancreatic tumors and accumulates in the nuclei of pancreatic cancer cell lines and most poorly differentiated pancreatic adenocarcinomas. Additionally, we show that nuclear accumulation of GSK-3β is dependent on its kinase activity and pharmacologic inhibition of GSK-3 leads to a loss of GSK-3β from the nucleus of pancreatic cancer cells. Furthermore, for the first time, we show that inhibition of GSK-3 affects NF-κB-mediated survival and proliferation of cancer cells in established tumor xenografts and suppresses pancreatic tumor growth in vivo.
Materials and Methods
Reagents, plasmids, and cells
The GSK-3 inhibitor AR-A014418 (10) was obtained from Calbiochem (La Jolla, CA). All other chemicals were obtained from Sigma (St. Louis, MO). GSK-3β expression vectors were a gift from Dr. Jim Woodgett (Ontario Cancer Institute, University Health Network, Toronto, Ontario, Canada). All cell lines were obtained from the American Type Culture Collection (Manassas, VA).
Immunoblot analysis and antibodies
For immunoblots, cells were lysed as described previously (9). Nuclear/cytosolic fractionation was done by Dignam method (11). Nuclear cell lysate was treated with DNase I (1 unit/μL) to obtain total histone H3 (nuclear marker). Whole-protein extract from tumor tissue was prepared as described (12). Protein sample concentration was quantified and equal amount (50 μg whole, nuclear, or cytosolic protein extract) of protein was loaded in each well of SDS-polyacrylamide gel. Cell or tissue extracts were separated by 10% SDS-PAGE, transferred to polyvinylidene difluoride membrane, and probed as indicated. The following antibodies were used for immunoblot analysis: GSK-3β, β-catenin, cyclin D1, Bcl-2, X-linked inhibitor of apoptosis protein (XIAP), and ORC2 (BD PharMingen, San Diego, CA); NF-κB p65 (Santa Cruz Biotechnology, Santa Cruz, CA); histone H3 (Abcam, Cambridge, MA); and Cu/Zn superoxide dismutase (Stressgen, Victoria, British Columbia, Canada). Bound antibodies were detected as described previously (9).
Analysis of apoptosis and bromodeoxyuridine staining
Carcinoma cells undergoing apoptosis were detected in xenograft tumor tissue sections by the terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) method (13). The apoptotic index of each tumor was calculated as described previously (12). Bromodeoxyuridine incorporation assay was carried out as described (9, 14).
Immunohistochemistry
All studies carried out on human specimens were approved by the Institutional Review Board at the Mayo Clinic (Rochester, MN). The following antibodies were used for immunostaining: GSK-3β and β-catenin; cyclin D1 (Biocare Medical, Concord, CA); Bcl-2 (DakoCytomation, Glostrup, Denmark); XIAP (R&D Systems, Minneapolis, MN); phosphorylated Ser10 histone H3 (Upstate, Lake Placid, NY); and NF-κB p65. GSK-3β immunostaining was done on 122 resected primary pancreatic adenocarcinoma specimens (54 females and 68 males, mean age, 62.7 ± 1). Two pathologists (A.V.O. and T.C.S.) independently reviewed all cases and classified the tumors as well differentiated (n = 22), moderately differentiated (n = 59), or poorly differentiated (n = 41). For each case, the most representative section reflecting the major features of the primary pancreatic tumor (i.e., histologic type) was selected for immunohistochemical examination to determine the expression of GSK-3β. Pancreatic intraepithelial neoplasia (PanIN) lesion specimens were obtained from 47 patients and stained to detect GSK-3β expression. Metastatic lymph node specimens were obtained from 10 pancreatic cancer patients and analyzed by immunohistochemistry for GSK-3β, cyclin D1, and β-catenin expression and NF-κB activation, represented by its nuclear accumulation. Immunohistochemical staining was done as described (12). GSK-3β expression in tumor cells was classified into three patterns as follows: (a) weak cytoplasmic expression similar to normal pancreatic acinar and ductal cells, (b) strong cytoplasmic expression, signified by significant increase of staining intensity in cytoplasm, and (c) nuclear accumulation, defined as staining of >10% of tumor cell nuclei regardless of intensity of cytoplasmic expression. Cyclin D1 and NF-κB nuclear accumulation was defined as positive staining of >10% of cancer cell nuclei throughout the tumor. β-Catenin expression was defined as any of three patterns: (a) membranous expression, similar to that in normal pancreatic ductal cells, (b) loss of membranous staining, signified by a loss of membranous expression found in >50% of cancer cells, and (c) nuclear accumulation, defined as staining of >10% of cancer cell nuclei throughout the tumor.
Statistical analysis
Data were analyzed using the Prism software package (GraphPad, Inc., San Diego, CA). Associations between GSK-3β staining pattern and degree of tumor differentiation were analyzed using Fisher’s exact test for 2 × 2 contingency tables or χ2 test for larger tables. Two-sided tests were used where applicable. χ2 critical value for 0.05 probability level is 3.841, all χ2 values exceeding 3.841 and Ps < 0.05 were considered to indicate statistical significance.
Xenograft tumor model
Female athymic nude mice (8–10 weeks old) were inoculated s.c. with 3 × 106 CAPAN2 pancreatic cancer cells mixed with Matrigel (BD Biosciences). Two weeks after injection, tumors were size matched and mice were randomized into two treatment groups: (a) control DMSO and (b) AR-A014418. Diluent (50 μL DMSO) or AR-A014418 (dissolved in 50 μL DMSO) was injected i.p. in a total volume of 500 μL PBS. Tumors were measured with calipers in three dimensions two times weekly. Tumor volume was calculated using the formula for volume of an ellipsoid: 4/3π × (L / 2) × (W / 2) × (H / 2), where L is the length, W is the width, and H is the height.
Results
GSK-3β is accumulated in the nucleus of pancreatic cancer cells
Recently, we have shown that GSK-3β positively regulates NF-κB activation at a point downstream of the activation of the IκB kinase complex (9), suggesting a role for GSK-3β in the regulation of NF-κB transcriptional activity in pancreatic cancer cells. In view of these results, we sought to determine whether GSK-3β accumulates in nuclei of human pancreatic cancer cells where it might contribute to NF-κB transcriptional activity.
Using immunohistochemical staining for GSK-3β, we found weak cytoplasmic GSK-3β expression in normal human pancreatic ductal and acinar cells (Fig. 1A). Weak cytoplasmic GSK-3β staining of normal pancreatic ductal or acinar cells adjacent to tumor cells was used as an internal staining control. Similar to normal pancreatic ductal cells, weak cytoplasmic expression of GSK-3β was observed in 16 of 18 and 4 of 12 cases of PanIN-1 and PanIN-2 lesions, respectively. GSK-3β weak cytoplasmic staining was significantly related to PanIN-1 and PanIN-2 lesions [relative risk, 13.36; 95% confidence interval (95% CI), 7.240–24.67; odds ratio, 137.0; 95% CI, 27.95–671.4; P < 0.0001]. On the other hand, PanIN-3 lesions, well-differentiated adenocarcinomas (Fig. 1B), and moderately differentiated adenocarcinomas showed strong cytoplasmic expression of GSK-3β in 16 of 17 (94%), 19 of 22 (86%), and 36 of 59 (61%) cases, respectively (Fig. 2A). Significant association was observed between increased malignant phenotype of tumors from PanIN-1 to well-differentiated/moderately differentiated adenocarcinoma and shift from a weak to strong GSK-3β cytoplasmic staining (χ2, 65.28; P < 0.0001).
Fig. 1.
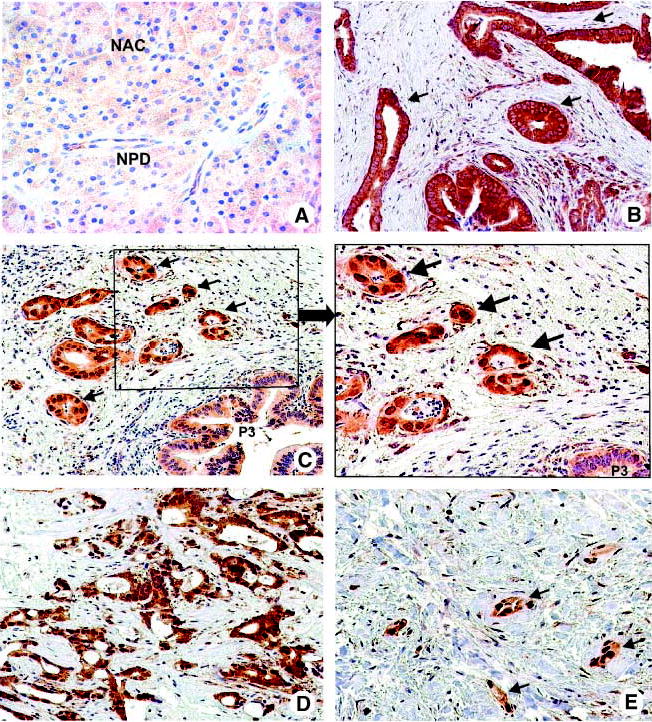
GSK-3β is overexpressed and accumulated in the nucleus of pancreatic cancer cells. A to E, immunohistochemical analysis of GSK-3β expression and localization in normal human pancreas (A) and pancreatic adenocarcinoma (B–E) specimens. A, normal acinar cells (NAC); normal pancreatic duct (NPD). B, arrows, well-differentiated pancreatic adenocarcinoma shows strong cytoplasmic expression of GSK-3β. C, arrows, nuclear accumulation of GSK-3β found in cancer cells of moderately differentiated adenocarcinoma but not in adjacent PanIN-3 (P3) lesion. Right, higher magnification of the delineated inset (left). D, GSK-3β nuclear accumulation in a moderately differentiated pancreatic adenocarcinoma. E, arrows, nuclear accumulation of GSK-3β was found in cancer cells of poorly differentiated pancreatic adenocarcinoma.
Fig. 2.
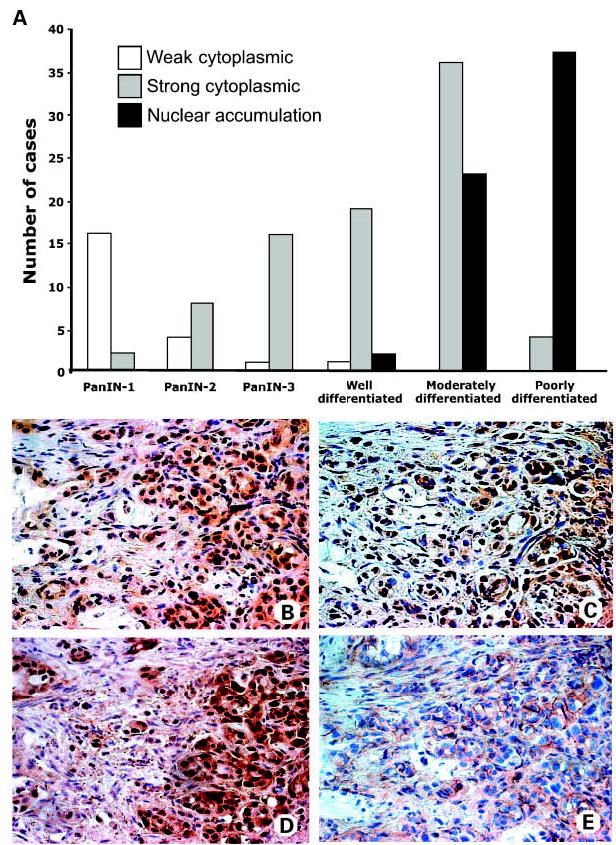
Nuclear accumulation of GSK-3β is associated with the loss of pancreatic cancer differentiation. A, distribution of GSK-3β staining patterns in PanIN lesions and pancreatic carcinomas. B to E, immunohistochemical staining of serial sections from the same metastatic lymph node shows accumulation of GSK-3β (B), NF-κB p65 (C), and cyclin D1 (D) in nuclei of cancer cells, whereas β-catenin (E) shows membranous staining.
We have observed nuclear GSK-3β in a substantial fraction of the pancreatic carcinomas. Nuclear accumulation of GSK-3β was not observed in any of the PanIN lesions but was found in differentiated adenocarcinomas (well, moderately, and poorly) in 2 of 22 (9%), 23 of 59 (39%), and 37 of 41 (90%) cases, respectively (Fig. 1C–E and Fig. 2A). Nuclear accumulation of GSK-3β was significantly correlated to higher-grade pancreatic adenocarcinomas (χ2, 44.13; P < 0.0001), with the highest expression rate in poorly differentiated adenocarcinomas (90%; relative risk, 2.387; 95% CI, 1.759–3.239; odds ratio, 38.11; 95% CI, 12.43–116.9; P < 0.0001). Our results suggest that induction of GSK-3β overexpression occurs in PanIN lesions and increases toward differentiated adenocarcinomas, whereas nuclear accumulation of GSK-3β is associated with the loss of pancreatic cancer differentiation, providing evidence that GSK-3β nuclear accumulation is a late event in pancreatic tumorigenesis.
To investigate GSK-3β overexpression and nuclear accumulation in metastatic pancreatic cancer cells, we analyzed metastatic lymph nodes from 10 pancreatic cancer patients for the expression and localization of GSK-3β. Similar to the primary tissues, we found localization of GSK-3β in the nuclei of pancreatic cancer cells in metastatic lymph nodes in 8 of 10 cases (Fig. 2B). Using immunostaining of serial sections, we observed NF-κB nuclear accumulation in the same 8 specimens (Fig. 2C) and nuclear accumulation of cyclin D1, a NF-κB target gene, was found in 10 of 10 cases (Fig. 2D). Membranous staining of β-catenin (Fig. 2E) was observed in 8 of 10 cases. Taken together, these data suggest that nuclear accumulation of GSK-3β is a common feature to both the primary pancreatic cancer and the metastatic disease.
Kinase activity correlates with GSK-3β nuclear accumulation in pancreatic cancer cells
To determine whether nuclear accumulation of GSK-3β was only a feature of primary pancreatic cancers, we analyzed GSK-3β localization in pancreatic cancer cell lines and normal human pancreas tissue. Using nuclear/ cytosolic fractionation, we found that all of the pancreatic cancer cell lines show cytoplasmic as well as nuclear localization of both GSK-3β and p65 (Fig. 3A). In contrast to these findings, GSK-3β and p65 were not detected in the nuclear extract of normal human pancreatic cells (Fig. 3A). Thus, nuclear accumulation of GSK-3β seems to be a feature of pancreatic cancer cells.
Fig. 3.
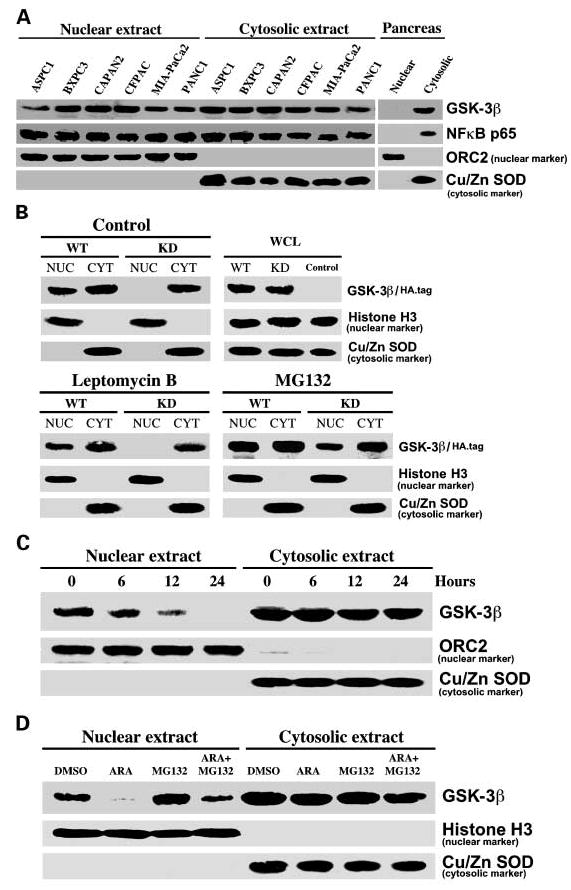
Kinase activity is required for GSK-3β nuclear accumulation in pancreatic cancer cells. A, equivalent amounts (50 μg) of nuclear and cytosolic proteins isolated from the indicated cell lines and normal human pancreas tissue were separated by SDS-PAGE and immunoblotted. B, MIA-PaCa2 cancer cells were transfected with mammalian expression vectors for either HA epitope – tagged (HA.tag) WTor KD GSK-3β. Forty-eight hours after transfection, cells were treated with DMSO (control), leptomycin B (30 ng/mL), or MG132 (10 μmol/L) for 12 hours. Whole-cell lysates (WCL), nuclear (NUC), and cytosolic (CYT) fractions were prepared, separated by SDS-PAGE (50 μg/well), transferred to polyvinylidene difluoride membrane, and probed with the indicated antibodies. Data are representative example of three independent experiments. C, HupT3 pancreatic cancer cells were treated with AR-A014418 (50 μmol/L) for 6, 12, and 24 hours as indicated, nuclear/cytosolic fractions were prepared, and protein expression was analyzed in the two fractions as described in (B). D, CAPAN2 pancreatic cancer cells were treated with AR-A014418 (ARA ; 50 μmol/L), MG132 (10 μmol/L), and combination of these two drugs for 24 hours and nuclear/cytosolic fractions were prepared and analyzed as in (B).
Although GSK-3β has no defined nuclear localization signal sequence, GSK-3β is known to shuttle between the cytoplasm and nucleus where it has been proposed to phosphorylate and regulate transcription factors (15, 16). However, whether the kinase activity of GSK-3β contributes to its nuclear accumulation is not known. To determine whether GSK-3β kinase activity contributes to its nuclear accumulation, we transfected MIA-PaCa2 cancer cells with GSK-3β wild-type (WT) or kinase dead (KD; K85M) expression vectors (17). Following nuclear/ cytosolic extraction, we found that, although the WT GSK-3β is readily accumulated in the nucleus of cancer cells, the KD form of GSK-3β was not found in the nucleus but is readily detectable in the cytoplasm of the transfected cells (Fig. 3B). Treatment of cancer cells expressing the KD form of GSK-3β with leptomycin B, a nuclear export inhibitor, did not result in the nuclear accumulation of KD GSK-3β. This data suggest that the loss of the nuclear expression of the catalytic inactive GSK-3β is not a result of active export from the nucleus (Fig. 3B). In contrast, however, we observed nuclear accumulation of KD GSK-3β when cancer cells were treated with the proteasome inhibitor MG132 (Fig. 3B). These data suggest that, although KD GSK-3β can translocate to the cancer cell nucleus from the cytoplasm, the inactive form of GSK-3β is rapidly degraded through a proteosomal pathway in the cancer cell nucleus. Consistent with this notion, we found that pharmacologic inhibition of GSK-3β results in a complete depletion of GSK-3β from the pancreatic cancer cell nucleus by 24 hours of AR-A014418 addition (Fig. 3C). Importantly, the level of GSK-3β protein in the cytoplasm was not significantly changed (Fig. 3C). Similar results were observed in multiple pancreatic cancer cell lines using different GSK-3 inhibitors.6 Moreover, we could rescue GSK-3β nuclear accumulation in AR-A014418-treated CAPAN2 pancreatic cancer cells by addition of MG132 (Fig. 3D). Taken together, these data suggest that active GSK-3β can accumulate in the nuclei of pancreatic cancer cells, whereas kinase inactive GSK-3β is rapidly degraded within the nucleus.
Pharmacologic inhibition of GSK-3 suppresses NF-κB-mediated pancreatic cancer cell proliferation and survival in established tumor xenografts
We have shown previously that pancreatic cancer cell lines are sensitive to the GSK-3 inhibitor AR-A014418 in vitro and that inhibition of GSK-3 leads to a reduction in the expression of several NF-κB target genes, including Bcl-2, cyclin D1, and XIAP (9). To determine whether GSK-3 inhibition also leads to decreased cancer cell survival and proliferation through down-regulation of NF-κB target genes in vivo, we injected either DMSO or 120 mg/kg AR-A014418 i.p. every 12 hours for 2 days into mice with established flank CAPAN2 xenografts (tumor volume, 350-400 mm3). Immunoblot analysis revealed a substantial decrease in polypeptide levels of the NF-κB targets XIAP, Bcl-2, and cyclin D1 in tumor tissues from AR-A014418-treated animals (Fig. 4A). In addition, as measured by TUNEL staining, AR-A014418-treated animals showed a 4-fold increase in the number of apoptotic cells compared with DMSO-treated animals as well as a 2-fold decrease in cancer cell proliferation (Fig. 4B and C). Using immunohistochemical staining, we found a pronounced decrease of histone H3 phosphorylation at Ser10 (a proliferation marker) in tumor xenografts treated with AR-A014418 (Fig. 4D and E). In addition, and consistent with the immunoblot analysis, we found cyclin D1 nuclear expression in 84 ± 19 versus 23 ± 7 cancer cells (mean number of positively stained cells per 100 counted cancer cells) in control and AR-A014418-treated animals, respectively (Fig. 4F and G). Moreover, immunohistochemical staining revealed a significant decrease in XIAP (Fig. 4H and I) and Bcl-2 (Fig. 4J and K) protein expression in tumors from AR-A014418-treated animals. These results suggest that pharmacologic inhibition of GSK-3 leads to inhibition of NF-κB transcriptional activity and decreased pancreatic cancer cell proliferation and survival in established tumor xenografts.
Fig. 4.
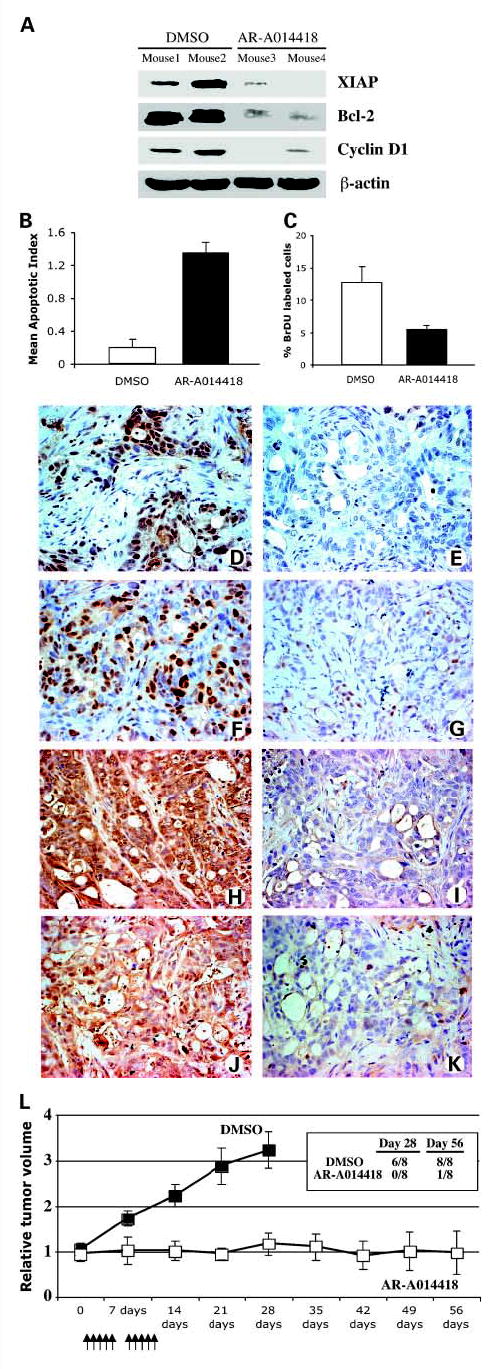
Antitumor effect of GSK-3 inhibition in vivo. Established CAPAN2 xenografts (tumor volume, 350-400 mm3) were treated by i.p. injections with DMSO orAR-A014418 (120 mg/kg; every 12 hours for 2 days) and subsequently injected with bromodeoxyuridine (BrDU; 1mg) 3 hours before sacrifice and tumor harvest. A, tumor proteins were extracted from fresh tumor tissues taken from each mouse as indicated and protein expression analysis was done as indicated in Fig. 3. B, TUNEL staining of tissue sections from DMSO- and AR-A014418-treated animals was done to obtain the apoptotic index. Columns, mean; bars, SE. C, proliferation of cancer cells in DMSO-treated and AR-A014418-treated CAPAN2 xenografts was measured by bromodeoxyuridine labeling. Columns, mean; bars, SE. Sections from DMSO-treated (D, F, H, and J) and AR-A014418-treated (E, G, I, and K) CAPAN2 xenograft tumors were stained for histone H3 phosphorylated Ser10 (D and E), cyclin D1 (F and G), XIAP (H and I), or Bcl-2 (J and K). L, CAPAN2 xenograft tumor regrowth assay. Treatment was initiated on day 0 with i.p. injections of either diluent (DMSO) or 30 mg/kg AR-A014418. Arrows, animals were injected daily, five times weekly for 2 weeks.▪, diluent (DMSO); □, AR-A014418. Points, mean relative tumor volume; bars, SE. Table, the number of animals with tumors grown to more than three times the original starting volume in diluent (DMSO) orAR-A014418-treated mice.
Pharmacologic inhibition of GSK-3 arrests pancreatic tumor growth in vivo
Based on these results, we next evaluated the effect of GSK-3 inhibition on tumor growth using established flank xenografts of CAPAN2 cancer cells. Tumors were size matched and mice were randomized into two treatment groups: (a) control, diluent (DMSO; n = 8 mice) and (b) AR-A014418 (n = 8 mice). Diluent or AR-A014418 (30 mg/kg) was injected i.p. once daily, five times weekly for 2 weeks. We found that tumors grew to more than three times the original starting volume in six of the eight diluent-treated animals by day 14 following the completion of diluent injection (Fig. 4L). In contrast, only one animal in the AR-A014418-treated group realized a three-time tumor growth at 40 days after cessation of therapy, whereas five animals showed regression of the initial tumor and two mice showed minimal tumor growth (Fig. 4L). These results suggest that pharmacologic inhibition of GSK-3 arrests pancreatic tumor growth in vivo.
Discussion
We have shown previously that pharmacologic inhibition of GSK-3 activity or genetic depletion of GSK-3β by RNAi suppresses basal NF-κB transcriptional activity and leads to decreased pancreatic cancer cell proliferation and survival in vitro (9). A recent series of studies have shown that inhibition of GSK-3β also decreases proliferation and survival of colon (18), prostate (19), and hepatocellular cancer cells (20) and acute myeloid leukemia cells (21), thus identifying GSK-3β as a potential therapeutic target in multiple human malignancies. Here, we find that GSK-3β is overexpressed in human pancreatic tumors and accumulates in the nuclei of pancreatic cancer cell lines and most poorly differentiated pancreatic adenocarcinomas. Additionally, we show that GSK-3β kinase activity contributes to its nuclear accumulation in pancreatic cancer cells. Furthermore, for the first time, we show that inhibition of GSK-3 suppresses pancreatic tumor growth in vivo and affects NF-κB-mediated survival and proliferation of cancer cells in established tumor xenografts. Taken together, our results suggest GSK-3β as a potential therapeutic target in the treatment of human pancreatic cancer.
Our data suggest that GSK-3β is overexpressed in pancreatic cancer and localizes to the nucleus in the vast majority of moderately and poorly differentiated cancers. Although this study is the first to look at the expression and cellular localization of GSK-3β in human tumor tissues, several reports have used Western blotting to analyze GSK-3β protein levels in other cancers. In fact, using Western blotting for GSK-3β in protein samples from human ovarian cancer specimens, statistical analysis revealed a significant increase of GSK-3β (P < 0.001) expression in the group of ovarian adenocarcinoma compared with the group of normal ovaries and benign adenomas/borderline tumors (22). In a separate study of human colorectal carcinomas, the level of GSK-3β protein expression was found to be significantly higher in the tumor tissue than in their normal counterparts (18 of 20 cases), whereas inactive phosphorylated Ser9 GSK-3β was detected in higher levels in normal tissue than in tumors in 17 of 20 cases (18). Of particular interest, a study using a mouse hepatic carcinogenesis model showed a higher level of GSK-3β expression in mouse liver tumors than in normal liver tissue as shown by Western blotting (23). However, we are unaware of any previous report that assessed the expression and cellular localization of GSK-3β in human tumors using immunohistochemistry.
Although GSK-3β does not contain any identifiable nuclear localization or nuclear export signal sequences, it is known to shuttle from the cytoplasm to the nucleus, where it is thought to participate in the regulation of gene transcription through the phosphorylation of transcription factors (e.g., NFAT and c-Jun; refs. 15, 16). Physiologically, GSK-3β is expressed in the cytoplasm of normal cells, including pancreatic ductal and acinar cells. However, a significant finding of our study is the presence of nuclear accumulation of GSK-3β in pancreatic cancer cell lines and in 62 of 122 (51%) human pancreatic adenocarcinomas. Interestingly, nuclear accumulation of GSK-3β is significantly correlated with pancreatic cancer dedifferentiation. Another finding of our study is that only the active form of GSK-3β is detectable in the nucleus of pancreatic cancer cells. Our results clearly show that down-regulation of inactive GSK-3β in the cancer cell nucleus is independent of its nuclear export. Using the proteasome inhibitor MG132, we could rescue nuclear accumulation of exogenously expressed KD GSK-3β or pharmacologically inhibited endogenous GSK-3β. These data suggest that, although inactive GSK-3β can translocate to the nucleus from the cytoplasm, the inactive form of GSK-3β is rapidly degraded by the proteasome in the nucleus of the cancer cell. Thus, GSK-3β kinase activity is required for its enhanced stabilization within the nucleus. However, the exact mechanism by which GSK-3β becomes overexpressed in human cancers, what regulates its accumulation in the nucleus of pancreatic cancer cells, and what proteasomal pathway is involved in the depletion of inactive GSK-3β in the nucleus is currently not known. The identification of the signals that drive the aberrant nuclear accumulation of active GSK-3β in pancreatic cancer cells is currently under investigation in our laboratory.
The exact mechanism by which GSK-3β affects NF-κB activity is currently unknown. Using GSK-3β-deficient mouse embryonic fibroblasts, it has been shown that the early steps leading to NF-κB activation following TNF-α treatment (degradation of IκBα and translocation of NF-κB to the nucleus) were unaffected by the loss of GSK-3β, indicating that NF-κB is regulated by GSK-3β at the level of the transcriptional complex (8). In addition, we have shown recently that GSK-3β influences NF-κB-mediated gene transcription in pancreatic cancer cells at a point downstream of the activation of the IκB kinase complex (9). Consistent with the putative role of GSK-3β in regulating the nuclear activity of NF-κB, a recent study showed that loss of GSK-3β leads to decreased TNF-α-induced binding of NF-κB to the promoters of a subset of target genes, including antiapoptosis genes (e.g., cIAP2), in GSK-3β-deficient mouse embryonic fibroblasts (24). Of importance, our finding of nuclear accumulation of GSK-3β suggests the possibility that active GSK-3β positively regulates NF-κB transcriptional activity in the nucleus of pancreatic cancer cells. Although, it is possible that GSK-3β controls NF-κB nuclear activity through direct phosphorylation of NF-κB p65, leading to effects on DNA-binding activity or dimerization (25), it is also possible that nuclear GSK-3β may have an effect on chromatin structure, thereby facilitating accessibility of transcription factors, such as NF-κB, at the promoter regions of target genes. Clearly, further studies are required to determine the exact mechanism by which nuclear GSK-3β can regulate NF-κB transcriptional activity.
In summary, our study identifies the overexpression of GSK-3β in pancreatic cancer and its aberrant nuclear accumulation as a hallmark of poorly differentiated pancreatic adenocarcinoma. In addition, we have shown that kinase activity contributes to GSK-3β nuclear accumulation and thereby provides new insight into the mechanism by which GSK-3β regulates constitutive NF-κB activity in pancreatic cancer. Furthermore, we show that inhibition of GSK-3 suppresses pancreatic tumor growth in vivo and affects NF-κB-mediated survival and proliferation of cancer cells in established tumor xenografts. Our work suggests that inhibition of GSK-3 is a promising new approach to pancreatic cancer therapy, holding the potential to arrest tumor growth and induce apoptosis in human pancreatic cancer.
Acknowledgments
We thank Dr. Jann Sarkaria and Brett Carlson for their help with the in vivo studies and Darren Riehle for tissue microarray data acquisition.
Footnotes
A.V. Ougolkov and D.D. Billadeau, unpublished observation.
Grant support: Mayo Foundation, Specialized Program of Research Excellence grant in pancreatic cancer P50 CA102701 (D.D. Billadeau), and Cancer Research Institute Investigator Award (D.D. Billadeau).
References
- 1.Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ. The nuclear factor-κBRelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5:119–27. [PubMed] [Google Scholar]
- 2.Aggarwal BB. Nuclear factor-κB: the enemy within. Cancer Cell. 2004;6:203–8. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Wang CY, Cusack JC, Jr, Liu R, Baldwin AS., Jr Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-κB. Nat Med. 1999;5:412–7. doi: 10.1038/7410. [DOI] [PubMed] [Google Scholar]
- 4.Plyte SE, Hughes K, Nikolakaki E, Pulverer BJ, Woodgett JR. Glycogen synthase kinase-3: functions in oncogenesis and development. Biochim Biophys Acta. 1992;1114:147–62. doi: 10.1016/0304-419x(92)90012-n. [DOI] [PubMed] [Google Scholar]
- 5.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–86. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–70. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 7.Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM. Severe liver degeneration in mice lacking the IκB kinase 2 gene. Science. 1999;284:321–5. doi: 10.1126/science.284.5412.321. [DOI] [PubMed] [Google Scholar]
- 8.Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- 9.Ougolkov AV, Fernandez-Zapico ME, Savoy DN, Urrutia RA, Billadeau DD. Glycogen synthase kinase-3β participates in nuclear factor-κB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005;65:2076–81. doi: 10.1158/0008-5472.CAN-04-3642. [DOI] [PubMed] [Google Scholar]
- 10.Bhat R, Xue Y, Berg S, et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem. 2003;278:45937–45. doi: 10.1074/jbc.M306268200. [DOI] [PubMed] [Google Scholar]
- 11.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–89. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ougolkov A, Zhang B, Yamashita K, et al. Associations among β-TrCP, an E3 ubiquitin ligase receptor, β-catenin, and NF-κB in colorectal cancer. J Natl Cancer Inst. 2004;96:1161–70. doi: 10.1093/jnci/djh219. [DOI] [PubMed] [Google Scholar]
- 13.Tornusciolo DR, Schmidt RE, Roth KA. Simultaneous detection of TDT-mediated dUTP-biotin nick end-labeling (TUNEL)-positive cells and multiple immunohistochemical markers in single tissue sections. Biotechniques. 1995;19:800–5. [PubMed] [Google Scholar]
- 14.Fernandez-Zapico ME, Gonzalez-Paz NC, Weiss E, et al. Ectopic expression of Vav1 reveals an unexpected role in pancreatic cancer tumorigenesis. Cancer Cell. 2005;7:39–49. doi: 10.1016/j.ccr.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 15.Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997;275:1930–4. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- 16.Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Liu S, Yu S, Hasegawa Y, et al. Glycogen synthase kinase 3β is a negative regulator of growth factor-induced activation of the c-Jun N-terminal kinase. J Biol Chem. 2004;279:51075–81. doi: 10.1074/jbc.M408607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shakoori A, Ougolkov A, Yu ZW, et al. Deregulated GSK3β activity in colorectal cancer: its association with tumor cell survival and proliferation. Biochem Biophys Res Commun. 2005;334:1365–74. doi: 10.1016/j.bbrc.2005.07.041. [DOI] [PubMed] [Google Scholar]
- 19.Mazor M, Kawano Y, Zhu H, Waxman J, Kypta RM. Inhibition of glycogen synthase kinase-3 represses androgen receptor activity and prostate cancer cell growth. Oncogene. 2004;23:7882–92. doi: 10.1038/sj.onc.1208068. [DOI] [PubMed] [Google Scholar]
- 20.Erdal E, Ozturk N, Cagatay T, Eksioglu-Demiralp E, Ozturk M. Lithium-mediated downregulation of PKB/ Akt and cyclin E with growth inhibition in hepatocellular carcinoma cells. Int J Cancer. 2005;115:903–10. doi: 10.1002/ijc.20972. [DOI] [PubMed] [Google Scholar]
- 21.DeToni F, Racaud-Sultan C, Chicanne G, et al. A cross-talk between the Wnt and the adhesion-dependent signaling pathways governs the chemosensitivity of acutemyeloidleukemia. Oncogene. 2006;25:3113–22. doi: 10.1038/sj.onc.1209346. [DOI] [PubMed] [Google Scholar]
- 22.Rask K, Nilsson A, Brannstrom M, et al. Wnt-signalling pathway in ovarian epithelial tumours: increased expression of β-catenin and GSK3β. Br J Cancer. 2003;89:1298–304. doi: 10.1038/sj.bjc.6601265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gotoh J, Obata M, Yoshie M, Kasai S, Ogawa K. Cyclin D1 overexpression correlates with β-catenin activation, but not with H-ras mutations, and phosphorylation of Akt, GSK3β, and ERK1/2 in mouse hepatic carcinogenesis. Carcinogenesis. 2003;24:435–42. doi: 10.1093/carcin/24.3.435. [DOI] [PubMed] [Google Scholar]
- 24.Steinbrecher KA, Wilson W, III, Cogswell PC, Baldwin AS. Glycogen Synthase Kinase 3{β} Functions To Specify Gene-Specific, NF-{κ}B-Dependent Transcription. Mol Cell Biol. 2005;25:8444–55. doi: 10.1128/MCB.25.19.8444-8455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwabe RF, Brenner DA. Role of glycogen synthase kinase-3 in TNF-α-induced NF-κB activation and apoptosis in hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2002;283:G204–11. doi: 10.1152/ajpgi.00016.2002. [DOI] [PubMed] [Google Scholar]