

Abstract
Demyelination is prominent in experimental autoimmune encephalomyelitis (EAE). The receptor p75 and its high affinity ligand proNGF are required for oligodendrocyte death after injury. We hypothesize that bone marrow stromal cells (BMSCs) provide therapeutic benefit in EAE mice by reducing proNGF/p75 expression. PBS or BMSCs (2×10^6) were administered intravenously on the day of EAE onset. Neurological function and demyelination areas were measured. Immunohistochemical staining was used to measure apoptotic oligodendrocytes, expression of proNGF and p75, and the relationship between proNGF and p75 in neural cells. proNGF was used to treat oligodendrocytes in culture with or without BMSCs. EAE mice exhibited neurological function deficit and demyelination, and expression of proNGF and p75 was increased. BMSC treatment improved functional recovery, reduced demyelination area and apoptotic oligodendrocytes, decreased expression of proNGF and p75 compared with PBS treatment. proNGF+ cells colocalized with neural cell markers, while p75 colocalized with an oligodendrocytic marker, and proNGF colocalized with p75. proNGF induced apoptosis of oligodendrocytes in vitro, and p75 antibody blocked this apoptotic activity. BMSCs reduced p75 expression and apoptotic activity in oligodendrocytes with proNGF treatment. BMSC treatment benefits on EAE mice may be fostered by decreasing the cellular expression of proNGF and p75, thereby reducing oligodendrocyte death.
Keywords: proNGF, p75, experimental autoimmune encephalomyelitis, bone marrow stromal cell, oligodendrocyte, apoptosis
Introduction
Oligodendrocytes generate myelin sheaths that enwrap axons. Damage to oligodendrocytes leads to demyelination and neurological functional deficit1, 2. Multiple Sclerosis (MS) is a demyelinating disease of the central nervous system (CNS) with destruction of the myelin sheath as the most prominent pathological hallmark3. Chronic relapsing experimental autoimmune encephalomyelitis (EAE)4 is a CD4+ T cell-mediated autoimmune inflammatory demyelinating disease. EAE can be induced in genetically susceptible SJL/J mice by active immunization with myelin neuroantigens5. Since the disease shares many clinical and histopathologic features with the human MS, EAE has been extensively used as an animal model for MS6.
Apoptosis plays a crucial role in MS/EAE7–12. 30% of MS cases are associated with relatively early loss of oligodendrocytes by apoptosis8. Furthermore, remyelination can occur during the clinical remitting-relapsing course of MS and failure of remyelination in MS could result from the death of oligodendrocyte progenitor cells13–16. Inhibition of apoptosis in oligodendrocytes alleviates the severity of the neurological manifestations17. Recent studies have raised the possibility that apoptotic death of oligodendrocytes may be mediated via intracellular signaling after activation of the p75 receptor, a member of the neurotrophin receptor (NTR) family18, 19.
The p75 receptor is a death-inducing receptor which belongs to the tumor necrosis factor receptor superfamily20, 21, and is induced by various injuries to the nervous system. A high-affinity ligand of the p75 receptor, the precursor form of nerve growth factor (proNGF), facilitates a cell death signaling cascade22, 23. Nerve growth factor (NGF) is normally synthesized as a precursor (proNGF) that can be cleaved to release mature, biologically active ligands22, 24, 25. A biological role for precursor neurotrophins has been proposed22, 26, 27. proNGF can be released as an active form under stimulus conditions22, 28. Binding of proNGF to the p75 receptor, activates caspase pathways and promotes death of oligodendrocytes, neurons and vascular smooth muscle cells that express p7522, 29, 30. Caspase-mediated death of oligodendrocytes contributes to demyelination8. In vitro studies also demonstrate that apoptosis of oligodendrocytes is due in part to an increase in the production of proNGF and its receptor p7529, and disrupted interaction between proNGF and p75 reduces neural cell apoptosis31. To our knowledge, the involvement of proNGF and p75 as a mechanism of oligodendrocyte death has not been demonstrated in a model of EAE.
Bone marrow stromal cells (BMSCs) are a population of multipotential mesenchymal stem and precursor cells32, 33. BMSCs escape immune system surveillance34–36 and can be transplanted as an autograft, allograft and even xenograft37, 38. Functional recovery is evident after BMSC treatment of rodents with EAE39–41. In the present study, we hypothesized that proNGF and p75 are both induced following EAE and are involved in the mechanisms of oligodendrocyte death in EAE mice. We therefore also investigated whether BMSC therapy reduces oligodendrocyte death (demyelination) in the CNS of the EAE mice induced by the proNGF-p75 pathway.
Methods
All experimental procedures have been approved by the Institutional Animal Care and Use Committee of Henry Ford Health System.
Animal Groups and Neurological Functional Measurement
EAE was induced in female SJL/J mice (8–10 week old, Jackson Laboratory, Bar Harbor, ME) by subcutaneous injection with 25ug myelin proteolipid protein (PLP) (p139–151, HSLGKWLGHPDKF, SynPep Corporation, Dublin, CA) dissolved in 50ul complete Freund’s adjuvant (CFA, Difco Laboratories, Livonia, MI). Pertussis toxin (PT, List Biological laboratories, Inc. Campbell, CA) 200ng in 0.2ml phosphate buffered saline (PBS) was injected into the mouse tail vein39, 42 on the day of immunization and 48 hours later. Mice were scored daily for clinical symptoms of EAE, as follows: 0, healthy; 1, loss of tail tone; 2, ataxia and/or paresis of hindlimbs; 3, paralysis of hindlimbs and/or paresis of forelimbs; 4, tetraparalysis; 5, moribund or dead43. Mice were randomly divided into: BMSC treatment group (n=8): mouse BMSCs (2×106 per mouse) were administered intravenously in 0.2ml total fluid volume of PBS on the day of clinical symptom onset (score ≥ 1). PBS treatment group (n=15): PBS (0.2ml) was injected into the tail vein of the EAE mice on the day of clinical symptom onset as EAE controls. With an expectation that there is a higher mortality over time in the PBS group compared with BMSC group, we included more mice in the PBS group. An additional control normal group (n=5) consisted of mice without immunization. We tested the neurological functions of EAE mice treated with BMSCs or PBS daily until 90 days after clinical symptom onset.
Histopathology and Immunohistochemistry
EAE mice treated with PBS or BMSCs were euthanized at 90 days after clinical symptom onset, respectively. Brains (bregma-0.54mm---bregma-1.82mm)44 were immediately removed, preserved in OCT compound, frozen on dry ice and stored at −80°C. They were cut as 10 coronal frozen slides (8μm thick) with 100μm intervals. Brains (bregma +1.54mm---bregma-0.54mm)44 were fixed in 4% of paraformaldehyde and divided into 2 serial blocks which were then embedded in paraffin. Four coronal slides (6μm thick) at intervals of 100um were cut from each block.
Double staining for Luxol fast blue and Bielshowsky was used to demonstrate myelin and axons, respectively39. Briefly, for Bielshowsky staining, paraffin slides were placed in 20% silver nitrate in the dark. Ammonium hydroxide was added to stained slides until the tissue turned brown with a gold background, and the slides were then treated with sodium thiosulfate. Slides were stained in Luxol fast blue solution, washed in 95% alcohol, and then placed in lithium carbonate. After staining, myelin and axons appear turquoise and black, respectively.
Immunohistochemical staining was performed on the frozen slides. The following primary antibodies were used to detect the expression of proNGF (rabbit polyclonal antibody, AB9040, 1:400, Chemicon) and p75 (rabbit polyclonal antibody, AB1554, 1:800, Chemicon, CA). ABC kits (PK-6100, Vector, CA) were employed, and nuclei were counterstained with hematoxylin. Apoptosis Detection Kit (S7110, Chemicon) was used to detect apoptotic cells. In addition, the following antibodies were used to measure the cell sources of proNGF and p75: NeuN (1:500, Chemicon, MAB377) for neurons; glial fibrillary acidic protein (GFAP, 1:10000, DAKO, Z0334) for astrocytes; anti-CNPase (1:100, Chemicon, MAB326R) for oligodendrocytes; OX42 (1:300, Accurate Chemical, MCA275R) for microglia cells. Double immunofluorescence labeling was performed to identify the apoptotic oligodendrocytes, and the relationship of proNGF with neural cells and p75, respectively, and p75 with oligodendrocytes. The nuclei were stained with DAPI. Negative control slides for each animal received identical preparations for immunostaining, except that primary antibodies were omitted.
Oligodendrocyte and BMSC cultures
To clarify the relationship between p75 and proNGF expression and apoptosis of oligodendrocytes, we employed an immature oligodendrocyte cell line (N20.1, generously provided by Dr. Anthony Campagnoni, University of California at Los Angeles). N20.1 cells were obtained from mouse primary cultures of oligodendrocytes conditionally immortalized by transformation with a temperature-sensitive large T-antigen45. N20.1 cells grow in Dulbecco's modified Eagle's medium (DMEM)/F12 with 10% fetal bovine serum (FBS) and G418 (100 μg/ml) at 34°C (permissive temperature), and they differentiate into mature oligodendrocytes in DMEM/F12/1%FBS and G418 at 39°C (nonpermissive temperature)46. Therefore, in the present experiments, the N20.1 cell lines were placed in DMEM/F12 high glucose (Invitrogen), with 3.6 g/L Dextrose anhydrous, 3.38 g/L HEPES, 2.16 g/L sodium bicarbonate, 90 mg/L Gentamicin, 1%FBS and 100 μg/ml G418 at 39°C (nonpermissive temperature) for 7 days.
BMSCs were produced from batches of eleven C57/Bl6 mice under sterile conditions (Cognate BioServices, Inc)47. Briefly, hind legs were removed and the marrow was flushed from the cut bones into sterile Alpha Modified Eagles Medium supplemented with 10% fetal bovine serum (pre-screened for mouse BMSC culture) and penicillin/streptomycin. The cells were washed, plated and cultured at 37ºC in a humidified atmosphere containing 5% CO2. After 2 days, the media containing nonadherent cells was aspirated. Cultures were fed every 3–4 days until adherent stromal cells become confluent (approximately 14 days). These cells, designated passage 0 (P0) were expanded and passaged to P3 at which point they were harvested and cryopreserved.
Analysis of Oligodendrocyte Apoptosis
N20.1 cell cultures were divided into 4 experimental groups: 1) normal medium; 2). recombinant human proNGF treatment, 50 and 100ng/ml (Cell Sciences, Inc.) for 6h or 16h; 3). proNGF treatment with BMSC coculture (N20.1 cells:BMSCs=2:1); An insert (0.4μm, BD Biosciences) was used to contain BMSCs. N20.1 cells were plated on the base of the culture wells, and the upper transwell compartments were seeded with BMSCs; 4). proNGF treatment with blocking p75 antibody (1:1000, Chemicon, CA). After 6h treatment, a cell apoptosis assay was performed with TUNEL method (Chemicon, ApopTag® Fluorescein In Situ Apoptosis Detection Kit), according to the manufacture’s instructions. In contrast to normal cells, the nuclei of apoptotic cells have highly condensed chromatin. This can take the form of crescents around the periphery of the nucleus, or the entire nucleus can appear to be one or a group of spherical beads. These morphological changes in the nuclei of apoptotic cells are visualized by fluorescence microscopy. The apoptotic cells were calculated in 10 random fields in each well with 3 wells per group.
Western Blot Analysis
After 16h of proNGF treatment, N20.1 cell cultures were rinsed with PBS, and proteins were extracted in 200μl RIPA lysis buffer. Equal amounts of protein, as determined using the BCA (bicinchoninic acid) protocol (Pierce, Rockford, IL), were loaded on 12% Bis-Tris Gels (Invitrogen, Carlsbad, CA) after being denatured. The proteins were then transferred to Invitrogen PVDF membranes (Invitrogen, Carlsbad, CA). The membranes were blocked for 1h with 5% BSA in TBS-T (10mM Tris–HCl, pH 7.6 and 150mM NaCl, 0.1% Tween-20). Afterward, the membranes were incubated with primary antibodies against p75 (AB1554, Chemicon) or caspase 3 (Cell Signaling, 9661, 1:1000) in 3% BSA at 4 °C overnight, respectively. The membranes were washed with TBS-T and incubated for 1h at room temperature with horseradish peroxidase (HRP)-conjugated secondary antibodies (Bio-Rad Laboratories, Hercules, CA). Following washing, the immunoblots were detected using a SuperSignal West Pico Chemiluminescent Substrate kit (Pierce, Rockford, IL). The experiment was repeated in triplicate. β-actin was used as the internal control. The densities of bands were analyzed by the Gelpro 4.5 program.
Real-time PCR (RT-PCR) Analysis
After 6h of coculture with proNGF-treated N20.1 cells, the growth factor mRNA expression in BMSCs was measured using SYBR Green RT-PCR method39, 48. Total RNA was isolated from BMSCs using the RNeasy Micro Kit (Qiagene Inc). 1μg of RNA from each sample RNA was used to produce cDNA, following the standard protocol supplied with the SuperScript III RTase (Invitrogen). Quantitative RT-PCR was performed on an ABI 7000 PCR instrument (Applied Biosystems, Foster City, CA). Specificity of the produced amplification product was confirmed by examination of dissociation reaction plots. A distinct single peak indicated that a single DNA sequence will be amplified during PCR. PCR products were run on 2% agarose gels to confirm the correct molecular sizes. Each sample was tested in triplicate using quantitative RT-PCR. Data analysis was used the 2−ΔDelta;CT method49. The following primers for real-time PCR were designed as follows: glyceraldehyde-3-phosphate dehyrogenase (GAPDH, FWD AGAACATCATCCCTGCATCC, REV CACATTGGGGGTAGGAACAC). NGF (FWD CAAGGACGCAGCTTTCTATACTG, REV CTTCAGGGACAGAGTCTCCTTCT), glial cell derived neurotrophic factor (GDNF, FWD GATATTGCAGCGGTTCCTGT, REV AACATGCCTGGCCTACTTTG), brain-derived neurotrophic factor (BDNF, FWD TACTTCGGTTGCATGAAG GCG, REV GTCAGACCTCTCGAACCTGCC).
Quantification and Statistical analysis
Neurological functional tests, histopathological and immunohistochemical results were evaluated by an examiner blinded to the treatment status of each animal. The neurological assessment with a score 1 to 5 (the most severe neurological deficit) was performed before the treatment, and daily up to 90 days. Mice which died earlier than 90 days received a score of 5 daily up to 90 days. Normality of the functional score was evaluated, and data were not normal. Therefore, we used Generalized Estimating Equations (GEE)50 on the ranked data, given that GEE has fewer restrictions on the data distribution. Repeated measure analysis of variance (ANCOVA) including the independent factor of the treatment and dependent factor of the time was employed. The analysis began testing for the treatment by time interaction, followed by testing the main effect of treatment or time, if no interaction was observed at the 0.05 level. If an interaction or main effect of treatment was detected at p<0.05, pair-wise comparisons between the PBS and the BMSC group were made at each day. The mean and standard error of daily functional score were also calculated. In addition, given the characteristic of the EAE model with relatively long relapsing-remitting periods, we also calculated the cumulative neurological deficits up to 30, 60 and 90 days. The nonparametric Smirnov test51 was used to test the percent of mass/distribution (the cumulated neurological deficits) in controls which was not covered in the BMSC treated group, indicating the continuity of the neurological improvements52, 53.
Demyelination area in the white matter of the corpus callosum and striatum in the EAE brain was assessed. The demyelination area was counted on an average of 8 brain slides (bregma +1.54mm---bregma-0.54mm) per mouse (at 40× magnification). Data were obtained by using a 3-CCD color video camera (Sony DXC-970 MD) and interfaced with the Micro Computer Imaging Device (MCID) analysis system (Imaging Research Inc. St. Catharines, Ontario, Canada). The demyelination area is presented as a proportional area. Immunoreactive cell counting in the white matter was performed in coronal sections. The numbers of apoptotic oligodendrocytes (AP+-CNPase+ cells), proNGF+ and p75+ cells were counted on an average of 10 slides (bregma-0.54mm---bregma-1.82mm) per mouse (at 40× magnification). Apoptotic oligodendrocyte data are presented as a percentage, by dividing the AP+-CNPase+ cell number by the total number of CNPase+ cells in the scanned area. The density of proNGF+ and p75+ cells was calculated by dividing the number of positive cells by the scanned area, presented as numbers per mm2.
In order to compare the effects of PBS and BMSC on outcome of proNGF, p75, caspase 3 and demyelination, analysis of variance (ANOVA) was used. If the overall treatment effect was significant at p<0.05, all possible pair-wise comparisons were made. Data are presented as mean ± SE.
Results
1. BMSCs Improved Neurological Functional Recovery
A total of 23 EAE mice were employed in the study; 2/15 EAE mice died in the PBS treatment group and none in the BMSC treatment group. Results from the GEE model showed a significant treatment by day interaction (p<0.001) indicating that BMSC effect depended on time of the follow-up. Pair-wise comparisons showed a significant BMSC effects on day 30 or day 90 (p<0.05), but not on day 60. Significant BMSC effects were retained within the last week prior to sacrifice (Figure 1). These data are consistent with our previous findings39, 40.
Figure 1.

The neurological response of EAE mice treated with BMSCs or PBS. Results show that the average clinical scores in the BMSC treatment group were significantly or marginally decreased over a total duration of 49 days out of the total 90 day disease course.
The analysis of the cumulative neurological deficits (the continuity of the neurological improvements) indicated a relative 66% improvement on cumulative neurological deficits in the BMSC treated group up to 30 days, compared to the PBS treated group (p=0.014 based on nonparametric Smirnov test), and 59% and 50% improvements up to 60 and 90 days with p=0.036 and 0.08, respectively.
2. BMSCs Reduced Demyelination Area and Apoptotic Oligodendrocytes
In EAE mice treated with PBS, demyelination areas were evident in the white matter of the brain accompanied by axonal damage as indicated by Luxol fast blue and Bielshowsky staining 90 days after disease onset (Figure 2B). The percent area of demyelination was significantly reduced in the BMSC treatment group compared with that in the PBS treatment group (p<0.01) (Figure 2C~D). Double fluorescence staining revealed the apoptotic oligodendrocytes in the EAE brain after BMSC treatment were significantly decreased compared with apoptotic oligodendrocytes after PBS treatment (11.4±4.0 vs 26.2±7.0, p<0.01) (Figure 2E~M).
Figure 2.

The staining by Luxol fast blue and Bielshowsky (LFB+B) show the myelin (yellow arrowheads) and axons (red arrows) in the striatum of normal mice (A), EAE mice treated with PBS (B) and BMSCs (C). Some demyelination areas companied with axonal loss. Quantitative data show that demyelination areas (D) and apoptotic oligodendrocytes (M) were significantly decreased on 90 days after EAE onset in the BMSC treatment group compared to that in the PBS group. Double immunofluorescence staining indicated that oligodendrocytes (CY3, red) were colocalized with apoptotic cells (FITC, green) in striatum (ST, E~H) and corpus callosum (CC, I~L). Scale bars in A–C, E~L =50μm.
3. proNGF is Induced by EAE and Inhibited by BMSC Treatment
Our in vivo data clearly show that BMSCs improve neurological function and reduce oligodendrocyte apoptosis in EAE mice; therefore, we investigated whether BMSCs inhibit an important apoptotic pathway mediated by proNGF and p75. ANOVA showed overall significant group differences. Pair-wise comparison showed that proNGF+ cells significantly increased in the white matter of the brain in the PBS treated EAE mice compared with the normal mice (p<0.01). The expression of proNGF was significantly decreased in the BMSC treatment group compared with that of the PBS group (p<0.01) (Figure 3A~C). Double staining revealed that proNGF positive signals were colocalized with NeuN, GFAP and OX42, implying that proNGF could be released from neurons, astrocytes and microglia cells, respectively (Figure 4A~L).
Figure 3.
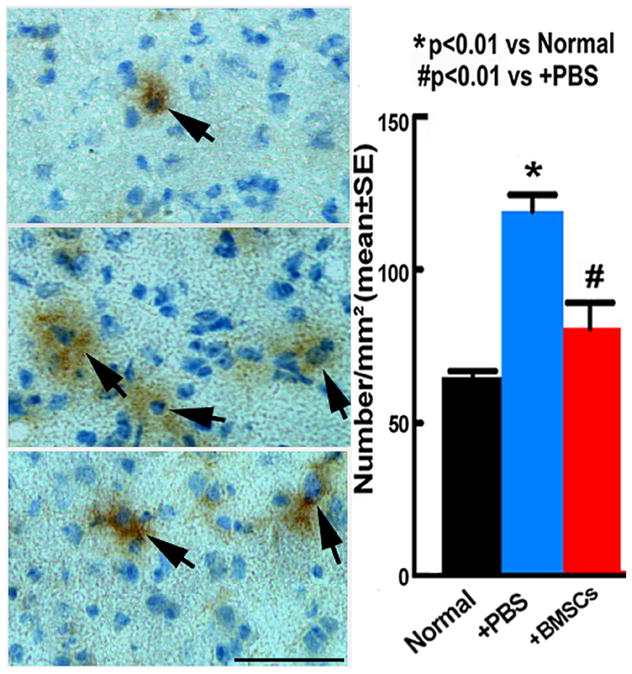
Immunohistochemical staining (DAB, brown; hematoxylin, blue) show proNGF+ cells in the white matter of the EAE brain treated with PBS (A) and BMSCs (B). Quantitative data show proNGF content increased in the EAE brain of PBS treatment compared with the normal brain, the BMSC treatment significantly decreased proNGF level on 90 days after onset compared with the PBS treatment (C). Scale bars in A–B=50μm.
Figure 4.

Double immunofluorescence staining revealed that proNGF+ cells (FITC, green) were reactive for NeuN (CY3, red) (A~D), GFAP (CY3, E~H) and OX42 (CY3, I~L). Scale bars in A~D, I~L=50μm, E~H=100μm.
4. p75 is Induced by EAE and is Inhibited by BMSC Treatment
90 days after EAE onset, pair-wise comparison showed that p75+ cells significantly increased in the white matter of the EAE brain with or without the BMSC treatment compared with the normal mice (p<0.01). p75+ cells significantly decreased in the BMSC treatment group compared with the PBS group (p<0.01) (Figure 5A~C). Two sets of double staining revealed that p75 positive signals were colocalized with oligodendrocytes (Figure 5D~G), and proNGF positive signals were colocalized with p75 and lead to nuclear morphology change (Figure 5H~K), suggesting that proNGF binds with p75 in these cells and induced apoptosis. These data suggest that p75+ cells undergo cell death via binding with proNGF.
Figure 5.

Immunohistochemical staining (DAB, brown; hematoxylin, blue) show p75+ cells in the white matter of the EAE brain treated with PBS (A) and BMSCs (B). Quantitative data show p75 content increased in the EAE brain with the PBS or BMSC treatment compared with the normal brain, the BMSC treatment significantly decreased p75 level on 90 days compared with the PBS treatment (C). Double immunofluorescence staining revealed that some CNPase+ cells (FITC, green) were reactive with p75 (CY3, red) (D~G), p75+ cells (CY3) were reactive for proNGF (FITC) (H~K). DAPI staining shows the blue normal nuclei (F) and apoptotic nuclei which are shrunken and condensed (J). Scale bars in A–B, D–G=50μm, H~K=25μm.
5. BMSC Treatment Reduced Apoptosis of Oligodendrocytes Induced by proNGF and p75
Since in vivo data suggest that up-regulated proNGF and p75 expression in brain after EAE induce oligodendrocyte death in the lesion areas, and BMSC treatment decreases proNGF and p75 expression leading to reduced demyelination, we performed an in vitro study to clarify the relationships among apoptosis of N20.1 cells, proNGF, p75 and BMSC treatment. After proNGF treatment, apoptotic N20.1 cells and the expression of p75 and caspase 3 protein in N20.1 cells were significantly increased compared with the normal N20.1 cells (p<0.01, Figure 6 and 7A~B). N20.1 cells underwent apoptosis with shrunken or condensed nuclei, in the form of crescents around the periphery of the nucleus, or the entire nucleus which appears to be featureless with bright spherical beads. These data imply that proNGF promotes N20.1 cell apoptosis and upregulates expression of p75 and caspase 3 in the N20.1 cells. The p75 blocking antibody disrupts the binding of proNGF with p75, and thereby inhibited the p75/caspase pathway and reduced N20.1 cell apoptosis (p<0.01), even though the p75 expression did not significantly decrease in the N20.1 cells compared with the proNGF treatment group. The apoptotic activity and expression of p75 and caspase 3 after the proNGF treatment was significantly reduced by BMSC treatment (p<0.05, Figure 7A~B). The expression of NGF and GDNF mRNA in BMSCs were significantly increased after coculture of BMSCs with proNGF-treated N20.1 cells compared with that of BMSC monoculture (p<0.01), while the BDNF mRNA expression was not significantly changed (Figure 7C).
Figure 6.

A. TUNEL staining 6h after proNGF treatment. The green fluorescein labels apoptotic nuclei, and the blue fluorescein labels N20.1 cell nuclei. White arrows indicate apoptotic N20.1 cells in the normal group (a), the proNGF treatment group (b), and in the anti-p75 blocking antibody treatment group (c), with BMSCs treatment group (d). B. Quantification of N20.1 cell apoptosis. The apoptotic N20.1 cells were significantly increased after proNGF treatment (50ng/ml or 100ng/ml) compared with the normal cells (p<0.01); and decreased after cocultured with BMSCs or anti-p75 blocking antibody treatment (p<0.01). Scale bars in a–d =50μm.
Figure 7.

Western blot analysis (A, B) shows that expression of p75 and caspase 3 proteins in N20.1 cells after 16h proNGF treatment. After proNGF treatment (group 2) p75 and caspase 3 protein level were significantly increased compared to the normal N20.1 cells (group 1, p<0.01). After combined with anti p75 antibody treatment (group 3), p75 protein level was no significantly changed, however, caspase 3 protein level was significantly decreased compared to the proNGF group (p<0.01). BMSC treatment (group 4) inhibited increased expression level of p75 and caspase 3 in N20.1 cells after proNGF treatment (p<0.05). RT-PCR analysis (C) shows that the mRNA expression of NGF and GDNF in BMSCs were significantly increased after cocultured with proNGF-treated N20.1 cells compared to normal group (p<0.01), while BDNF level was not changed.
Discussion
MS/EAE are associated with complex interactions between inflammatory and local cellular elements, ultimately leading to oligodendrocyte apoptosis and demyelination10. Many experimental treatments are designed to reduce apoptosis of oligodendrocytes and relieve EAE7, 8, 54–56. In the present study, we focus on the potential role of proNGF/p75 in mediating oligodendrocyte apoptosis and the effect of BMSCs on this death process. proNGF binds to its high affinity receptor p75, activates caspases22, 23 and induces apoptosis57, 58. proNGF induces p75-mediated death of oligodendrocytes following spinal cord injury29. Using a demyelination disease model of EAE, we investigated the oligodendrocyte cell death associated with proNGF and p75 in vivo. The massive oligodendrocyte death which leads to destruction of myelin sheath is the most prominent pathological hallmark of MS/EAE. Reduced death of oligodendrocytes should translate into more myelin and greater functional recovery. Our in vivo study demonstrates that treatment of EAE SJL/J mice with 2×106 BMSCs on the day of disease onset significantly improved functional recovery up to 90 days after EAE onset, consistent with previous reports39, 40. Improved functional outcome was associated with reduced demyelination areas and apoptotic oligodendrocytes, and decreased expression of proNGF and p75 in the white matter of the EAE brain. This is the first study which reveals that proNGF and p75 may be involved in the EAE pathological changes, and that BMSC treatment has the capacity to reduce oligodendrocyte death via interfering with the proNGF-p75 pathway. Furthermore, our in vitro data clearly showed that the binding of proNGF with p75 activated caspase 3, induced apoptosis of N20.1 cells, and disruption of this binding protects N20.1 cells from apoptosis. BMSC treatment reduced expression of p75 and caspase 3 and decreased apoptosis of N20.1 cells after proNGF treatment.
How do proNGF+ cells significantly decrease in the EAE brain after BMSC treatment? Double immunofluorescence staining data indicate that proNGF is released from neurons, astrocytes and microglia27. Astrocytes and microglia are activated by inflammatory processes in MS or EAE59, 60, and thereby may express and release proNGF. The increased proNGF released into extracellular space, may also potentiate oligodendrocyte cell death via binding to the p75 receptor expressed on these cells. Our previous study revealed that the BMSC treatment significantly decreased inflammatory infiltrates in EAE brain39. Furthermore, extensive studies revealed that the immunoregulatory properties of MSCs effectively interfere with the pathogenic autoimmune response and induce T-cell unresponsiveness61, 41. These effects may decrease the number of activated astrocytes and microglia, and finally reduce the proNGF expression. Moreover, the ratio of proNGF converted to NGF may be increased in the EAE brain after the BMSC treatment, since we observed more NGF+ cells40 but fewer proNGF+ cells in the BMSCs treated EAE mice than in the PBS treated mice.
NGF exerts an anti-inflammatory effect62 and promotes myelin repair in the EAE animal63. Our previous study found BMSC treatment increased NGF expression and improved neurological functional outcome in the EAE CNS40, and these results are consistent with our present data that proNGF decreased after BMSC treatment. The mechanisms underlying the BMSC benefits on the EAE animal are complicated, involving multi-systemic, cellular and molecular actions. The neuroprotective and neurorestorative roles of BMSCs may come from both sides, i.e. increase of protective factors and a decrease damaging factors, such as increasing NGF level and decreasing proNGF level, respectively. The present study implies that BMSC treatment is a desirable cell therapy for EAE mice and its therapeutic benefit arises from the down-regulation of proNGF and up-regulation of NGF.
The regulatory mechanisms how proNGF is released and converts to NGF remain unclear. It has been proposed that proNGF is synthesized and cleaved intracellularly to release NGF22, 24, 25, 64–66. However, some studies have revealed that proNGF is released upon stimulation, and that the maturation of proNGF largely occurs in the extracellular space with the involvement of a complex protease cascade22, 23, 28, 67. In the present study, we found a cytoplasmic location of proNGF and a cell surface binding with p75, implying that proNGF is present in both the intracellular and extracellular space.
p75 is induced by various injuries to the nervous system29 and induces apoptosis through its death domain, i.e, caspase 3, indicating the activation of a common effector pathway of apoptosis57. Three sets of double immunofluorescence staining show that some oligodendrocytes undergo apoptosis, oligodendrocytes express p75, and proNGF binds to p75 inducing cell apoptosis in the CNS of EAE mice, implying that the binding of proNGF to these p75+-oligodendrocytes mediate oligodendrocyte cell death after EAE. Consistent with this hypothesis are data that oligodendrocytes undergoing apoptosis express p75, and the absence of p75 results in a decreased oligodendrocyte apoptosis and an increased oligodendrocyte survival after spinal cord injury29. Although the mechanisms by which BMSC treatment decrease p75 expression after EAE are unknown, data from our in vitro study suggest that BMSC treatment induces the production of growth factors (i.e. NGF, GDNF, BDNF), which support neural cell survival and inhibit apoptosis68–72, and thereby attenuate oligodendrocyte damage and lead to reduced p75 expression in oligodendrocytes.
In conclusion, BMSC treatment of EAE mice improves functional recovery that may be fostered by decreasing the cellular expression of proNGF and p75, increasing the mature ratio of proNGF, thereby reducing oligodendrocyte death induced by the proNGF-p75 pathway.
Acknowledgments
The authors thank Dr. Mark Katakowski and Qinge Lu for their technical assistance and Deborah Jewell for secretarial support.
This work was supported by NIH grants P01 NS42345 and P01 NS23393 and the Benson Ford Foundation and the Wollowick Foundation (CB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bjartmar C, Wujek JR, Trapp BD. Axonal loss in the pathology of ms: Consequences for understanding the progressive phase of the disease. J Neurol Sci. 2003;206:165–171. doi: 10.1016/s0022-510x(02)00069-2. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120 ( Pt 3):393–399. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- 3.Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 4.Trotter JL, Clark HB, Collins KG, Wegeschiede CL, Scarpellini JD. Myelin proteolipid protein induces demyelinating disease in mice. J Neurol Sci. 1987;79:173–188. doi: 10.1016/0022-510x(87)90271-1. [DOI] [PubMed] [Google Scholar]
- 5.Brown AM, McFarlin DE. Relapsing experimental allergic encephalomyelitis in the sjl/j mouse. Lab Invest. 1981;45:278–284. [PubMed] [Google Scholar]
- 6.Ben-Chetrit E, Brocke S. Experimental models of multiple sclerosis. Springer; US: 2005. [Google Scholar]
- 7.Lev N, Barhum Y, Melamed E, Offen D. Bax-ablation attenuates experimental autoimmune encephalomyelitis in mice. Neurosci Lett. 2004;359:139–142. doi: 10.1016/j.neulet.2004.01.076. [DOI] [PubMed] [Google Scholar]
- 8.Cudrici C, Niculescu T, Niculescu F, Shin ML, Rus H. Oligodendrocyte cell death in pathogenesis of multiple sclerosis: Protection of oligodendrocytes from apoptosis by complement. J Rehabil Res Dev. 2006;43:123–132. doi: 10.1682/jrrd.2004.08.0111. [DOI] [PubMed] [Google Scholar]
- 9.Akassoglou K, Bauer J, Kassiotis G, Pasparakis M, Lassmann H, Kollias G, Probert L. Oligodendrocyte apoptosis and primary demyelination induced by local tnf/p55tnf receptor signaling in the central nervous system of transgenic mice: Models for multiple sclerosis with primary oligodendrogliopathy. Am J Pathol. 1998;153:801–813. doi: 10.1016/S0002-9440(10)65622-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ercolini AM, Miller SD. Mechanisms of immunopathology in murine models of central nervous system demyelinating disease. J Immunol. 2006;176:3293–3298. doi: 10.4049/jimmunol.176.6.3293. [DOI] [PubMed] [Google Scholar]
- 11.Okuda Y, Sakoda S. [the role of apoptosis in autoimmune encephalomyelitis] Nippon Rinsho. 2003;61:1323–1328. [PubMed] [Google Scholar]
- 12.Pender MP, Nguyen KB, McCombe PA, Kerr JF. Apoptosis in the nervous system in experimental allergic encephalomyelitis. J Neurol Sci. 1991;104:81–87. doi: 10.1016/0022-510x(91)90219-w. [DOI] [PubMed] [Google Scholar]
- 13.Carroll WM, Jennings AR, Ironside LJ. Identification of the adult resting progenitor cell by autoradiographic tracking of oligodendrocyte precursors in experimental cns demyelination. Brain. 1998;121 ( Pt 2):293–302. doi: 10.1093/brain/121.2.293. [DOI] [PubMed] [Google Scholar]
- 14.Keirstead HS, Blakemore WF. The role of oligodendrocytes and oligodendrocyte progenitors in cns remyelination. Adv Exp Med Biol. 1999;468:183–197. doi: 10.1007/978-1-4615-4685-6_15. [DOI] [PubMed] [Google Scholar]
- 15.Scolding NJ, Franklin RJ. Remyelination in demyelinating disease. Baillieres Clin Neurol. 1997;6:525–548. [PubMed] [Google Scholar]
- 16.Blakemore WF, Keirstead HS. The origin of remyelinating cells in the central nervous system. J Neuroimmunol. 1999;98:69–76. doi: 10.1016/s0165-5728(99)00083-1. [DOI] [PubMed] [Google Scholar]
- 17.Hisahara S, Araki T, Sugiyama F, Yagami K, Suzuki M, Abe K, Yamamura K, Miyazaki J, Momoi T, Saruta T, Bernard CC, Okano H, Miura M. Targeted expression of baculovirus p35 caspase inhibitor in oligodendrocytes protects mice against autoimmune-mediated demyelination. Embo J. 2000;19:341–348. doi: 10.1093/emboj/19.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. 1996;383:716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- 19.Dowling P, Ming X, Raval S, Husar W, Casaccia-Bonnefil P, Chao M, Cook S, Blumberg B. Up-regulated p75ntr neurotrophin receptor on glial cells in ms plaques. Neurology. 1999;53:1676–1682. doi: 10.1212/wnl.53.8.1676. [DOI] [PubMed] [Google Scholar]
- 20.Ladiwala U, Lachance C, Simoneau SJ, Bhakar A, Barker PA, Antel JP. P75 neurotrophin receptor expression on adult human oligodendrocytes: Signaling without cell death in response to ngf. J Neurosci. 1998;18:1297–1304. doi: 10.1523/JNEUROSCI.18-04-01297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hempstead BL, Salzer JL. Neurobiology. A glial spin on neurotrophins. Science. 2002;298:1184–1186. doi: 10.1126/science.1078709. [DOI] [PubMed] [Google Scholar]
- 22.Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- 23.Nykjaer A, Lee R, Teng KK, Jansen P, Madsen P, Nielsen MS, Jacobsen C, Kliemannel M, Schwarz E, Willnow TE, Hempstead BL, Petersen CM. Sortilin is essential for prongf-induced neuronal cell death. Nature. 2004;427:843–848. doi: 10.1038/nature02319. [DOI] [PubMed] [Google Scholar]
- 24.Heymach JV, Jr, Shooter EM. The biosynthesis of neurotrophin heterodimers by transfected mammalian cells. J Biol Chem. 1995;270:12297–12304. doi: 10.1074/jbc.270.20.12297. [DOI] [PubMed] [Google Scholar]
- 25.Chao MV, Bothwell M. Neurotrophins: To cleave or not to cleave. Neuron. 2002;33:9–12. doi: 10.1016/s0896-6273(01)00573-6. [DOI] [PubMed] [Google Scholar]
- 26.Fahnestock M, Yu G, Michalski B, Mathew S, Colquhoun A, Ross GM, Coughlin MD. The nerve growth factor precursor prongf exhibits neurotrophic activity but is less active than mature nerve growth factor. J Neurochem. 2004;89:581–592. doi: 10.1111/j.1471-4159.2004.02360.x. [DOI] [PubMed] [Google Scholar]
- 27.Srinivasan B, Roque CH, Hempstead BL, Al-Ubaidi MR, Roque RS. Microglia-derived pronerve growth factor promotes photoreceptor cell death via p75 neurotrophin receptor. J Biol Chem. 2004;279:41839–41845. doi: 10.1074/jbc.M402872200. [DOI] [PubMed] [Google Scholar]
- 28.Bruno MA, Cuello AC. Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc Natl Acad Sci U S A. 2006;103:6735–6740. doi: 10.1073/pnas.0510645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo FM, Bresnahan JC, Hempstead BL, Yoon SO. Prongf induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron. 2002;36:375–386. doi: 10.1016/s0896-6273(02)01005-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coulson EJ, Reid K, Murray SS, Cheema SS, Bartlett PF. Role of neurotrophin receptor p75ntr in mediating neuronal cell death following injury. Clin Exp Pharmacol Physiol. 2000;27:537–541. doi: 10.1046/j.1440-1681.2000.03295.x. [DOI] [PubMed] [Google Scholar]
- 31.Harrington AW, Leiner B, Blechschmitt C, Arevalo JC, Lee R, Morl K, Meyer M, Hempstead BL, Yoon SO, Giehl KM. Secreted prongf is a pathophysiological death-inducing ligand after adult cns injury. Proc Natl Acad Sci U S A. 2004;101:6226–6230. doi: 10.1073/pnas.0305755101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 33.Kopen GC, Prockop DJ, Phinney DG. Marrow stromal cells migrate throughout forebrain and cerebellum, and they differentiate into astrocytes after injection into neonatal mouse brains. Proc Natl Acad Sci U S A. 1999;96:10711–10716. doi: 10.1073/pnas.96.19.10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deans RJ, Moseley AB. Mesenchymal stem cells: Biology and potential clinical uses. Exp Hematol. 2000;28:875–884. doi: 10.1016/s0301-472x(00)00482-3. [DOI] [PubMed] [Google Scholar]
- 35.Maitra B, Szekely E, Gjini K, Laughlin MJ, Dennis J, Haynesworth SE, Koc ON. Human mesenchymal stem cells support unrelated donor hematopoietic stem cells and suppress t-cell activation. Bone Marrow Transplant. 2004;33:597–604. doi: 10.1038/sj.bmt.1704400. [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Chen J, Chen XG, Wang L, Gautam SC, Xu YX, Katakowski M, Zhang LJ, Lu M, Janakiraman N, Chopp M. Human marrow stromal cell therapy for stroke in rat: Neurotrophins and functional recovery. Neurology. 2002;59:514–523. doi: 10.1212/wnl.59.4.514. [DOI] [PubMed] [Google Scholar]
- 37.Azizi SA, Stokes D, Augelli BJ, DiGirolamo C, Prockop DJ. Engraftment and migration of human bone marrow stromal cells implanted in the brains of albino rats--similarities to astrocyte grafts. Proc Natl Acad Sci U S A. 1998;95:3908–3913. doi: 10.1073/pnas.95.7.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saito T, Kuang JQ, Bittira B, Al-Khaldi A, Chiu RC. Xenotransplant cardiac chimera: Immune tolerance of adult stem cells. Ann Thorac Surg. 2002;74:19–24. doi: 10.1016/s0003-4975(02)03591-9. discussion 24. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J, Li Y, Chen J, Cui Y, Lu M, Elias SB, Mitchell JB, Hammill L, Vanguri P, Chopp M. Human bone marrow stromal cell treatment improves neurological functional recovery in eae mice. Exp Neurol. 2005;195:16–26. doi: 10.1016/j.expneurol.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, Li Y, Lu M, Cui Y, Chen J, Noffsinger L, Elias SB, Chopp M. Bone marrow stromal cells reduce axonal loss in experimental autoimmune encephalomyelitis mice. J Neurosci Res. 2006;84:587–595. doi: 10.1002/jnr.20962. [DOI] [PubMed] [Google Scholar]
- 41.Zappia E, Casazza S, Pedemonte E, Benvenuto F, Bonanni I, Gerdoni E, Giunti D, Ceravolo A, Cazzanti F, Frassoni F, Mancardi G, Uccelli A. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing t-cell anergy. Blood. 2005;106:1755–1761. doi: 10.1182/blood-2005-04-1496. [DOI] [PubMed] [Google Scholar]
- 42.Youssef S, Stuve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM, Bravo M, Mitchell DJ, Sobel RA, Steinman L, Zamvil SS. The hmg-coa reductase inhibitor, atorvastatin, promotes a th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420:78–84. doi: 10.1038/nature01158. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Li Y, Cui Y, Chen J, Lu M, Elias SB, Chopp M. Erythropoietin treatment improves neurological functional recovery in eae mice. Brain Res. 2005;1034:34–39. doi: 10.1016/j.brainres.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 44.Franklin K, Paxino G. The mouse brain in stereotaxic coordinates. Academic Press; San Diego: 1997. [Google Scholar]
- 45.Verity AN, Bredesen D, Vonderscher C, Handley VW, Campagnoni AT. Expression of myelin protein genes and other myelin components in an oligodendrocytic cell line conditionally immortalized with a temperature-sensitive retrovirus. J Neurochem. 1993;60:577–587. doi: 10.1111/j.1471-4159.1993.tb03188.x. [DOI] [PubMed] [Google Scholar]
- 46.Paez PM, Garcia CI, Davio C, Campagnoni AT, Soto EF, Pasquini JM. Apotransferrin promotes the differentiation of two oligodendroglial cell lines. Glia. 2004;46:207–217. doi: 10.1002/glia.20001. [DOI] [PubMed] [Google Scholar]
- 47.Li Y, McIntosh K, Chen J, Zhang C, Gao Q, Borneman J, Raginski K, Mitchell J, Shen L, Zhang J, Lu D, Chopp M. Allogeneic bone marrow stromal cells promote glial-axonal remodeling without immunologic sensitization after stroke in rats. Exp Neurol. 2006;198:313–325. doi: 10.1016/j.expneurol.2005.11.029. [DOI] [PubMed] [Google Scholar]
- 48.Wang L, Gang Zhang Z, Lan Zhang R, Chopp M. Activation of the pi3-k/akt pathway mediates cgmp enhanced-neurogenesis in the adult progenitor cells derived from the subventricular zone. J Cereb Blood Flow Metab. 2005;25:1150–1158. doi: 10.1038/sj.jcbfm.9600112. [DOI] [PubMed] [Google Scholar]
- 49.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 50.Zeger SL, Liang KY. Longitudinal data analysis for discrete and continuous outcomes. Biometrics. 1986;42:121–130. [PubMed] [Google Scholar]
- 51.Conover WJ. Practical nonparametric statistics. New York: John Wiley and Sons; 1980. [Google Scholar]
- 52.Bohning D, Hempfling A, Schelp FP, Schlattmann P. The area between curves (abc)--measure in nutritional anthropometry. Stat Med. 1992;11:1289–1304. doi: 10.1002/sim.4780111004. [DOI] [PubMed] [Google Scholar]
- 53.Lu M, Chase G, Li S. Permutation tests and other statistics for ill-behaved data: Experience of the ninds t-pa stroke trial. Communications in Statistics. 2001;30:1481–1496. [Google Scholar]
- 54.Ousman SS, Tomooka BH, van Noort JM, Wawrousek EF, O'Conner K, Hafler DA, Sobel RA, Robinson WH, Steinman L. Protective and therapeutic role for alphab-crystallin in autoimmune demyelination. Nature. 2007;448:474–479. doi: 10.1038/nature05935. [DOI] [PubMed] [Google Scholar]
- 55.Niculescu T, Weerth S, Niculescu F, Cudrici C, Rus V, Raine CS, Shin ML, Rus H. Effects of complement c5 on apoptosis in experimental autoimmune encephalomyelitis. J Immunol. 2004;172:5702–5706. doi: 10.4049/jimmunol.172.9.5702. [DOI] [PubMed] [Google Scholar]
- 56.Rus H, Cudrici C, Niculescu F. C5b-9 complement complex in autoimmune demyelination and multiple sclerosis: Dual role in neuroinflammation and neuroprotection. Ann Med. 2005;37:97–104. doi: 10.1080/07853890510007278. [DOI] [PubMed] [Google Scholar]
- 57.Wang X, Bauer JH, Li Y, Shao Z, Zetoune FS, Cattaneo E, Vincenz C. Characterization of a p75(ntr) apoptotic signaling pathway using a novel cellular model. J Biol Chem. 2001;276:33812–33820. doi: 10.1074/jbc.M010548200. [DOI] [PubMed] [Google Scholar]
- 58.Casha S, Yu WR, Fehlings MG. Oligodendroglial apoptosis occurs along degenerating axons and is associated with fas and p75 expression following spinal cord injury in the rat. Neuroscience. 2001;103:203–218. doi: 10.1016/s0306-4522(00)00538-8. [DOI] [PubMed] [Google Scholar]
- 59.Ambrosini E, Columba-Cabezas S, Serafini B, Muscella A, Aloisi F. Astrocytes are the major intracerebral source of macrophage inflammatory protein-3alpha/ccl20 in relapsing experimental autoimmune encephalomyelitis and in vitro. Glia. 2003;41:290–300. doi: 10.1002/glia.10193. [DOI] [PubMed] [Google Scholar]
- 60.Heese K, Hock C, Otten U. Inflammatory signals induce neurotrophin expression in human microglial cells. J Neurochem. 1998;70:699–707. doi: 10.1046/j.1471-4159.1998.70020699.x. [DOI] [PubMed] [Google Scholar]
- 61.Gerdoni E, Gallo B, Casazza S, Musio S, Bonanni I, Pedemonte E, Mantegazza R, Frassoni F, Mancardi G, Pedotti R, Uccelli A. Mesenchymal stem cells effectively modulate pathogenic immune response in experimental autoimmune encephalomyelitis. Ann Neurol. 2007;61:219–227. doi: 10.1002/ana.21076. [DOI] [PubMed] [Google Scholar]
- 62.Flugel A, Matsumuro K, Neumann H, Klinkert WE, Birnbacher R, Lassmann H, Otten U, Wekerle H. Anti-inflammatory activity of nerve growth factor in experimental autoimmune encephalomyelitis: Inhibition of monocyte transendothelial migration. Eur J Immunol. 2001;31:11–22. doi: 10.1002/1521-4141(200101)31:1<11::AID-IMMU11>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 63.Villoslada P, Hauser SL, Bartke I, Unger J, Heald N, Rosenberg D, Cheung SW, Mobley WC, Fisher S, Genain CP. Human nerve growth factor protects common marmosets against autoimmune encephalomyelitis by switching the balance of t helper cell type 1 and 2 cytokines within the central nervous system. J Exp Med. 2000;191:1799–1806. doi: 10.1084/jem.191.10.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blochl A, Thoenen H. Localization of cellular storage compartments and sites of constitutive and activity-dependent release of nerve growth factor (ngf) in primary cultures of hippocampal neurons. Mol Cell Neurosci. 1996;7:173–190. doi: 10.1006/mcne.1996.0014. [DOI] [PubMed] [Google Scholar]
- 65.Canossa M, Griesbeck O, Berninger B, Campana G, Kolbeck R, Thoenen H. Neurotrophin release by neurotrophins: Implications for activity-dependent neuronal plasticity. Proc Natl Acad Sci U S A. 1997;94:13279–13286. doi: 10.1073/pnas.94.24.13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Edwards RH, Selby MJ, Garcia PD, Rutter WJ. Processing of the native nerve growth factor precursor to form biologically active nerve growth factor. J Biol Chem. 1988;263:6810–6815. [PubMed] [Google Scholar]
- 67.Fahnestock M, Michalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in alzheimer's disease. Mol Cell Neurosci. 2001;18:210–220. doi: 10.1006/mcne.2001.1016. [DOI] [PubMed] [Google Scholar]
- 68.Xie Y, Tisi MA, Yeo TT, Longo FM. Nerve growth factor (ngf) loop 4 dimeric mimetics activate erk and akt and promote ngf-like neurotrophic effects. J Biol Chem. 2000;275:29868–29874. doi: 10.1074/jbc.M005071200. [DOI] [PubMed] [Google Scholar]
- 69.Dolcet X, Egea J, Soler RM, Martin-Zanca D, Comella JX. Activation of phosphatidylinositol 3-kinase, but not extracellular-regulated kinases, is necessary to mediate brain-derived neurotrophic factor-induced motoneuron survival. J Neurochem. 1999;73:521–531. doi: 10.1046/j.1471-4159.1999.0730521.x. [DOI] [PubMed] [Google Scholar]
- 70.Wang HJ, Cao JP, Yu JK, Gao DS. Role of pi3-k/akt pathway and its effect on glial cell line-derived neurotrophic factor in midbrain dopamine cells. Acta Pharmacol Sin. 2007;28:166–172. doi: 10.1111/j.1745-7254.2007.00494.x. [DOI] [PubMed] [Google Scholar]
- 71.Anitha M, Gondha C, Sutliff R, Parsadanian A, Mwangi S, Sitaraman SV, Srinivasan S. Gdnf rescues hyperglycemia-induced diabetic enteric neuropathy through activation of the pi3k/akt pathway. J Clin Invest. 2006;116:344–356. doi: 10.1172/JCI26295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aloe L, Micera A. A role of nerve growth factor in oligodendrocyte growth and differentiation of eae affected rats. Arch Ital Biol. 1998;136:247–256. [PubMed] [Google Scholar]