

Abstract
 The direct conversion of various amides to isoquinoline and β-carboline derivatives via mild electrophilic amide activation, with trifluoromethanesulfonic anhydride in the presence of 2-chloropyridine, is described. Low temperature amide activation followed by cyclodehydration upon warming provides the desired products with short overall reaction times. The successful use of non-activated and halogenated phenethylene derived amides, N-vinyl amides, and optically active substrates are noteworthy.
The direct conversion of various amides to isoquinoline and β-carboline derivatives via mild electrophilic amide activation, with trifluoromethanesulfonic anhydride in the presence of 2-chloropyridine, is described. Low temperature amide activation followed by cyclodehydration upon warming provides the desired products with short overall reaction times. The successful use of non-activated and halogenated phenethylene derived amides, N-vinyl amides, and optically active substrates are noteworthy.
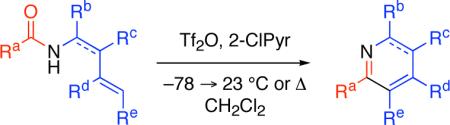
The venerable Bischler-Napieralski reaction offers an important strategy for the synthesis of various azaheterocycles.1,2 Isoquinolines and β-carbolines, including their reduced derivatives, can be found as substructures in many important natural products, pharmaceuticals, and other fine chemicals.3 We have reported the syntheses of pyridine4a and pyrimidine4b derivatives via the intermolecular condensation of readily available N-vinyl and N-aryl amides5 with various nucleophiles. Herein we report mild reaction conditions for the Bischler-Napieralski based synthesis of isoquinoline and β-carboline derivatives from readily available amides.
During our studies concerning the syntheses of pyridines and quinolines via an intermolecular condensation reaction4b we observed a competitive intramolecular cyclization reaction in a single case where a Morgan-Walls6 cyclization pathway was possible. N-Phenethylbenzamide (1, Table 1) was used to further investigate this intramolecular condensation reaction. Consistent with our observations on amide activation for the intermolecular addition of σ- or π-nucleophiles,4 the use of trifluoromethanesulfonic anhydride (Tf2O)7 and 2-chloropyridine8 (2-ClPyr) as the base additive were found to be optimal for a mild cyclodehydration reaction to provide the desired 3,4-dihydroisoquinoline 2 in 95% yield (Table 1, entry 7). The reaction was found to be less sensitive to superstoichiometric quantities of 2-ClPyr as compared to its absence (compare entries 7−9, Table 1), allowing the inclusion of excess base additive for BrØnsted acid sensitive substrates.9 Electrophilic amide activation10 followed by intramolecular π-nucleophilic cyclization and subsequent deprotonation directly provides the desired product 5 (Scheme 1).
Table 1.
Selection of base additive.a
 | |||
|---|---|---|---|
| entry | base additive | equiv | isolated yield (%) |
| 1 | Et3N | 1.2 | 18 |
| 2 | pyridine | 1.2 | 65 |
| 3 | ethyl nicotinate | 1.2 | 51 |
| 4 | 2-bromopyridine | 1.2 | 74 |
| 5 | 2-fluoropyridine | 1.2 | 90 |
| 6 | 3-chloropyridine | 1.2 | 79 |
| 7 | 2-chloropyridine | 1.2 | 95 |
| 8 | 2-chloropyridine | 0 | 43 |
| 9 | 2-chloropyridine | 2.0 | 91 |
Reaction conditions: Tf2O (1.1 equiv), CH2Cl2, 45 °C, 2 h.
Scheme 1.
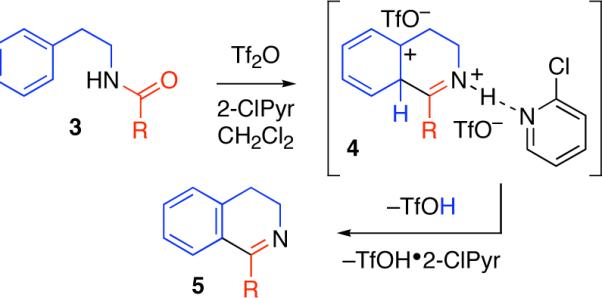
Intramolecular dehydrative cyclization.
A wide range of N-phenethylamide derivatives were found to readily provide the corresponding 3,4-dihydroisoquinoline products (Figure 1, 2, 6−14). Alkoxy and unsubstituted N-phenethylamides provided the desired dihydroisoquinoline products at ambient temperature or with mild heating. The conversion of recalcitrant substrates was found to be optimal via short (5 min) microwave irradiation.11 For example, deactivated halogenated N-phenethylamides did not cyclize at ambient temperature, but provided the desired 3,4-dihydroisoquinolines with microwave irradiation (Figure 1, 9 and 10). The formation of the phenylalaninol–derived dihydroisoquinoline 14 was noted to occur with no loss in optical activity.12 Significantly, sensitive N-vinyl amides13 were used as substrates in this chemistry to directly provide isoquinoline derivatives (Figure 1, 15−18). While (E)-N-styrylcyclohexanecarboxamide did not provide isoquinoline 15, (Z)-N-styrylcyclohexanecarboxamide was converted to the desired isoquinoline 15 in moderate yield (Figure 1). It should be noted that this substrate was sensitive (vide infra) to decomposition/polymerization following electrophilic amide activation. Tri- and tetrasubstituted enamides proved to be excellent substrates for this chemistry and efficiently gave the corresponding azaheterocycles (Figure 1, 16−18). o-Arylaniline derived amides afforded the desired fused tricyclic azaheterocycles, reminiscent of Morgan-Walls cyclization products, in good yield (Figure 1, 19−21). Additionally, the use of tryptamine derived substrates, optimally N-alkyl derivatives, gave the corresponding 3,4-dihydro-β-carbolines (Figure 1, 22−28).
Figure 1.

Synthesis of isoquinoline and β-carboline derivatives.a
Highly deactivated substrates such as N-(4-nitrophenethyl)cyclohexanecarboxamide or N-(4-(trifluoromethyl)phenethyl)benzamide did not provide the corresponding dihydroisoquinolines.14 This is likely due to a more rapid rate of elimination/decomposition upon activation as compared to the desired cyclodehydration reaction. Tryptamine derived amides bearing a sulfonyl group on the indolyl nitrogen were not substrates for this chemistry and unsubstituted indole derivatives led to rapid indolyl nitrogen N-sulfonylation of the starting material under the reaction conditions. It should be noted that in some cases minor side products resulting from oxidation (vide infra) of 3,4-dihydro-β-carboline were observed.15 Additionally, using the phenylalanine derivative 29 as a substrate under the standard reaction conditions competitively gave the oxazole 30 in 84% yield (Equation 1).16,17
 |
(1) |
When 2-vinyl-aniline derived amide 31 was exposed to the standard cyclodehydration reaction conditions described above, a highly efficient condensation reaction ensued to afford 2-phenylquinoline (32, Equation 2) in 99% isolated yield.
 |
(2) |
The direct comparison of the herein described condensation reaction with related protocols further highlights the advantages offered by this chemistry (Table 2).2 The synthesis of 3,4-dihydroisoquinoline 2, isoquinoline 15, and phenanthridine 20 is illustrative. Synthesis of 3,4-dihydroisoquinoline 2 was found to be most efficient using the conditions described here as compared to other reported condensation reaction conditions (Table 2). Sensitive substrates, such as the acid sensitive (Z)-N-styrylcyclohexanecarboxamide, were found to be incompatible with the broadly used conditions involving phosphorus oxychloride (POCl3) in conjunction with heating.2a Similarly, the use of reaction conditions employing oxalyl chloride and iron trichloride did not provide the desired phenanthridine 20 from the corresponding urea substrate.2d
Table 2.
Direct comparison of condensation reaction conditions.
| reaction conditions |
||||
|---|---|---|---|---|
| product | Tf2O (1.2 equiv) 2-ClPyr (This work)a | POCl3 (3.0 equiv) (Ref2a)b | Oxalyl Chloride (1.1 equiv) FeCl3 (Ref2d)c | Tf2O (5.0 equiv) DMAP (Ref2c)d |
 |
95% | 23% | 15% | 71% |
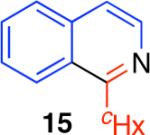 |
63% | 0% | 9% | 42% |
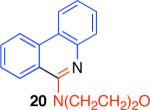 |
86% | 10% | 0% | 63% |
See Figure 1 for reaction conditions.
POCl3 (3.0 equiv), xylenes, 150 °C, 3 h.
1) Oxalyl chloride (1.1 equiv); FeCl3 (1.2 equiv), CH2Cl2, 23 °C, 12 h. 2) MeOH–H2SO4 (19:1), 65 °C, 1 h.
Tf2O (5.0 equiv), DMAP (3.0 equiv), CH2Cl2 23 °C, 16 h.
While in all three cases (Table 2) the use of superstoichiometric Tf2O in conjunction with 4-(dimethylamino)pyridine (DMAP) provided the desired product,2c the competing oxidation reaction in more sensitive substrates is a potential complication. For example, using the herein described conditions, electrophilic activation of amide 33 (Equation 3, 97% ee) afforded the desired optically active 3,4-dihydroisoquinoline 34 in 87% yield and 90% ee without undesired oxidation to the corresponding isoquinoline.18 However, electrophilic activation of amide 33 using the reported reaction conditions2c employing excess Tf2O–DMAP gave the desired product 34 in 31% yield, and with only 63% ee, in addition to 26% yield of the corresponding isoquinoline derivative due to oxidation of 34. Additionally, activation of amide 33 via the typical condensation reaction conditions employing POCl3 failed to provide the desired product 34 due to competitive decomposition.
 |
(3) |
As mentioned the 3,4-dihydro-β-carboline condensation products are subject to oxidation with Tf2O, affording the corresponding β-carbolines.15,19 In the case of 3,4-dihydroisoquinoline 34 this was a significant complication when excess Tf2O was used (vide supra). Indeed, exposure of azaheterocycles 2, 6, and 27−28 to Tf2O and 2-ClPyr resulted in the corresponding oxidation products (Figure 2). Electron rich dihydro-β-carbolines are more sensitive to this oxidation reaction as compared to dihydroisoquinolines (Figure 2). For comparison, while oxidation of 3,4-dihydroisoquinoline 2 to isoquinoline 38 required excess reagents and heating to 140 °C, the oxidation of 3,4-dihydro-β-carboline 27 at 23 °C gave β-carboline 36 in 65% yield within 2 h (Figure 2).20
Figure 2.
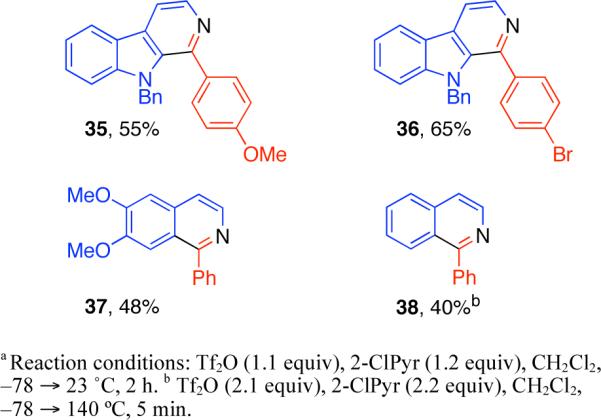
Tf2O–2-ClPyr promoted oxidation of 3,4-dihydro-β-carbolines and 3,4-dihydroisoquinolines.a
The chemistry described herein provides an efficient modified Bischler-Napieralski cyclodehydration reaction to access isoquinolines, β-carbolines, and their 3,4-dihydro derivatives. The successful use of unactivated, halogenated N-phenethylamides, sensitive N-vinyl amides, and optically active substrates is noteworthy. The direct comparison of this chemistry with existing methods as shown in Table 2, and the observations discussed regarding epimerization and oxidation challenges in the context of substrate 33, highlight the advantages offered by this methodology.
Supplementary Material
Acknowledgment
M.M. is a Beckman Young Investigator and an Alfred P. Sloan research fellow. M.D.H. acknowledges an Amgen graduate fellowship. We acknowledge partial financial support by NIH-NIGMS (GM074825), NSF (547905), AstraZeneca, Amgen, Boehringer Ingelheim, GlaxoSmithKline, Lilly, and Merck.
Footnotes
Supporting Information Available Experimental procedures and spectroscopic data for 2, 6−28, 30, 32, and 34−38. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a Bischler A, Napieralski B. Ber. 1893;26:1903. [Google Scholar]; b Whaley WM, Govindachari TR. Org. React. 1951;6:74. [Google Scholar]
- 2.For representative reports, see: Tóth J, Nedves A, Dancsó A, Blaskó G, Töke L, Nyerges M. Synthesis. 2007:1003.Spaggiari A, Davoli P, Blaszczak LC, Prati F. Synlett. 2005:661.Banwell MG, Bissett BD, Busato S, Cowden CJ, Hockless DCR, Holman JW, Read RW, Wu AW. J. Chem. Soc., Chem. Commun. 1995:2551.Larsen RD, Reamer RA, Corley EG, Davis P, Grabowski EJJ, Reider PJ, Shinkai I. J. Org. Chem. 1991;56:6034.Hendrickson JB, Schwartzman SM. Tetrahedron Lett. 1975;16:277.
- 3.For reviews on isoquinolines and their reduced derivatives, see Jones G. In: Comprehensive Heterocyclic Chemistry II. Katritzky AR, Rees CW, Scriven EFV, McKillop A, editors. Vol. 5. Pergamon; Oxford: 1996. p. 167.Bentley KW. Nat. Prod. Rep. 2006;23:444. doi: 10.1039/b509523a.Kartsev VG. Med. Chem. Res. 2004;13:325.Chrzanowska M, Rozwadowska MD. Chem. Rev. 2004;104:3341. doi: 10.1021/cr030692k.Joule JA, Mills K. Heterocyclic Chemistry. 4th ed. Blackwell Science Ltd.; Cambridge MA: 2000. p. 121.Rozwadowska MD. Heterocycles. 1994;39:903. For a review on β-carbolines and their reduced derivatives, see Love BE. Org. Prep. Proced. Int. 1996;28:3.
- 4.a Movassaghi M, Hill MD. J. Am. Chem. Soc. 2006;128:14254. doi: 10.1021/ja066405m. [DOI] [PubMed] [Google Scholar]; b Movassaghi M, Hill MD, Ahmad OK. J. Am. Chem. Soc. 2007;129:10096. doi: 10.1021/ja073912a. [DOI] [PubMed] [Google Scholar]
- 5.For recent advances in synthesis of N-vinyl and N-aryl amides, see: Muci AR, Buchwald SL. Top. Curr. Chem. 2002;219:131.Hartwig JF. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, editor. Wiley-Interscience; New York: 2002. p. 1051.Beletskaya IP, Cheprakov AV. Coordin. Chem. Rev. 2004;248:2337.Dehli JR, Legros J, Bolm C. Chem. Commun. 2005:973. doi: 10.1039/b415954c.
- 6.a Morgan GT, Walls LP. J. Chem. Soc. 1931:2447. [Google Scholar]; b Shabashov D, Daugulis O. J. Org. Chem. 2007;72:7720. doi: 10.1021/jo701387m. [DOI] [PubMed] [Google Scholar]; c Hutchinson I, Stevens MFG. Org. Biomol. Chem. 2007;5:114. doi: 10.1039/b613580n. [DOI] [PubMed] [Google Scholar]
- 7.Charette AB, Grenon M. Can. J. Chem. 2001;79:1694.Baraznenok IL, Nenajdenko VG, Balenkova ES. Tetrahedron. 2000;56:3077. Review.
- 8.a Myers AG, Tom NJ, Fraley ME, Cohen SB, Madar DJ. J. Am. Chem. Soc. 1997;119:6072. [Google Scholar]; b Garcia BA, Gin DY. J. Am. Chem. Soc. 2000;122:4269. [Google Scholar]
- 9.The inhibitory effect of excess 2-ClPyr is more pronounced when using weak σ-nucleophiles (i.e., nitriles, see ref 4a) as compared to stronger nucleophiles (i.e., ynamines, see ref 4b).
- 10.Electrophilic activation of N-alkyl amides may lead to a transient highly electrophilic nitrilium ion (or a pyridinium adduct) that is trapped by the arene ring.
- 11.Amide activation at ambient temperature under standard conditions generally led to the desired product; however, reaction times were often significantly shortened and isolated yields often increased upon heating.
- 12.See Supporting Information for details.
- 13.For representative preparation of enamides, see Jiang L, Job GE, Klapars A, Buchwald SL. Org. Lett. 2003;5:3667. doi: 10.1021/ol035355c. and ref. 5.
- 14.The use of N-(4-nitrophenethyl)cyclohexanecarboxamide as substrate provided 1-nitro-4-vinylbenzene as the major product.
- 15.For example, 27 and 28 were isolated along with 36 (12%, Figure 2) and 35 (6%, Figure 2), respectively. Additionally, minor N-triflouromethanesulfonylated spirocyclic byproducts were detected.
- 16.For competitive oxazole formation under the Bischler-Napieralski reaction conditions, see Liu ZZ, Tang YF, Chen SZ. Chin. Chem. Lett. 2001;12:947.
- 17.While the desired 3,4-dihydroisoquinoline 14 (Figure 1) was prepared from the corresponding O-triisopropylsilyl phenylalaninol derived amide, the use of the non-silylated substrate (S)-N-(1-hydroxy-3-phenylpropan-2-yl)cyclohexanecarboxamide led to competitive oxazoline formation. For a related report, see Whelligan DK, Bolm C. J. Org. Chem. 2006;71:4609. doi: 10.1021/jo060668h.
- 18.Epimerization of 3,4-dihydroisoquinoline 34 can occur within 1 h at room temperature in CH2Cl2 [0.3M] or when stored neat, highlighting the sensitivity of the product.
- 19.For related oxidation reactions, see: Spath E, Lederer E. Chem. Ber. 1930;63B:120.Hufford CD, Sharma AS, Oguntimein BO. J. Pharm. Sci. 1980;69:1180. doi: 10.1002/jps.2600691016.McMahon RM, Thornber CW, Ruchirawat S. J. Chem. Soc., Perkin Trans. 1. 1982:2163.Hilger CS, Fugmann B, Steglich W. Tetrahedron Lett. 1985;26:5975.Andreu I, Cabedo N, Atassi G, Pierre A, Caignard DH, Renard P, Cortesa D, Bermejoa A. Tetrahedron Lett. 2002;43:757. and references therein.
- 20.Using the conditions described in Figure 2, 3,4-dihydro-β-carboline 28 was completely converted to product 35, whereas oxidation of azaheterocycles 2, 6 and 27, gave the corresponding products (Figure 2) along with recovered starting material (8%, 30%, 10%, respectively).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.