

Abstract
Juvenile myelomonocytic leukemia (JMML) is an aggressive childhood myeloproliferative disorder characterized by the overproduction of myelomonocytic cells. JMML incidence approaches 1.2/million persons in the United States (Cancer Incidence and Survival Among Children and Adolescents: United States SEER Program 1975–1995). Although rare, JMML is innately informative as the molecular genetics of this disease implicates hyperactive Ras as an essential initiating event. Given that Ras is one of the most frequently mutated oncogenes in human cancer, findings from this disease are applicable to more genetically diverse and complex adult leukemias. The JMML Foundation (www.jmmlfoundation.org) was founded by parent advocates dedicated to finding a cure for this disease. They work to bring investigators together in a collaborative manner. This article summarizes key presentations from the 2nd International JMML Symposium, on December 7–8, 2007 in Atlanta, GA. A list of all participants is in Supplementary Table.
Keywords: Juvenile myelomonocytic leukemia (JMML), RAS, PTPN11, NF1, Therapy, Diagnosis
Introduction
Juvenile myelomonocytic leukemia (JMML) is a lethal myeloproliferative disease (MPD) of young childhood characterized clinically by overproduction of myelomonocytic cells and by the in vitro phenotype of hematopoietic progenitor hypersensitivity to granulocyte-macrophage colony-stimulating factor (GM-CSF) [1, 2]. In contrast to normal subjects, the morphological composition of progenitor colonies from JMML patients is predominantly macrophages and monocytes [3, 4]. It is notable, however, that progenitor colonies from JMML patients contain monocytic cells along the full spectrum of differentiation, including blast forms, promonocytes, monocytes, and macrophages. This distinct characteristic indicates that JMML is not a disease induced by a complete block in differentiation as observed in acute leukemias, but instead results from shunting of hematopoietic differentiation toward the monocytic pathway, similar to increased granulocytic differentiation in the chronic phase of chronic myeloid leukemia (Figure 1). In addition to monocytic cell overproduction, patients often present with anemia, thrombocytopenia, and 50% of patients also present with elevated fetal hemoglobin, hemoglobin F (Hgb F) [5]. JMML patients can progress to blast crisis, usually with French-American-British (FAB) M4 or M5 morphology, but more frequently succumb to disease due to tissue infiltration of myeloid cells. Standard cytotoxic chemotherapy in JMML is ineffective, producing few durable remissions [6]. Even following the rigorous therapy of allogeneic hematopoietic stem cell transplantation (HSCT), the probability of event-free survival at five years is only 50% [7] and the main cause of treatment failure continues to be leukemia relapse.
Figure 1.

Schematic diagram of A) normal hematopoietic differentiation; B) accumulation of undifferentiated myeloblasts representing acute myeloid leukemia; and C) increased production of monocytic cells along the full spectrum of differentiation, including blast forms, promonocytes, monocytes, and macrophages, as observed in juvenile myelomonocytic leukemia. HSC = hematopoietic stem cell; CMP = common myeloid progenitor; GMP = granulocyte monocyte progenitor.
Activating mutations of the NRAS and KRAS genes and disruption of the tumor suppressor gene NF1 have long been recognized as pathogenic in this disease [8–12]. More recently, somatic mutations in PTPN11, which encodes the protein tyrosine phosphatase, Shp2, have been found in 35% of JMML cases [13–15]. Intriguingly, it is now evident that de novo JMML, as well as several of the neuro-cardio-facio-cutaneous congenital disorders in which a JMML-like MPD has been associated [16–19], are caused by gene mutations (somatic in the former and germline in the latter) contributing to hyperactivation of the Ras – mitogen activated protein kinase pathway (MAPK, Figure 2) [20–31].
Figure 2.
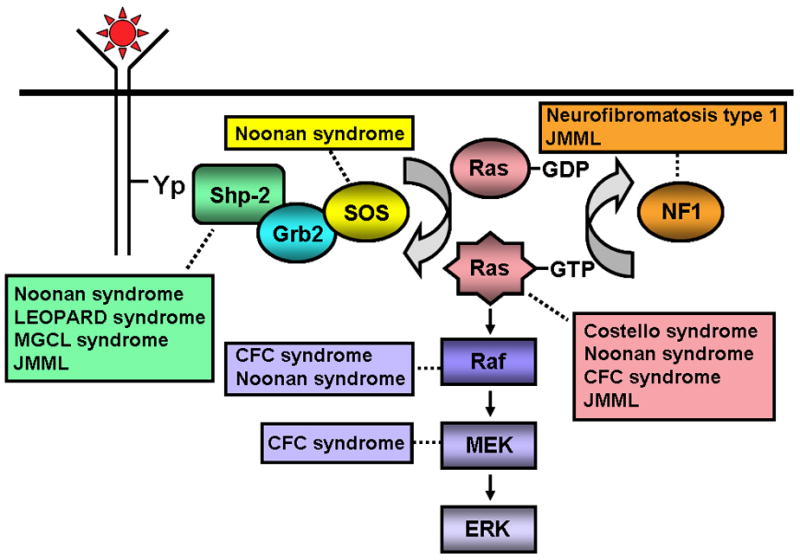
Schematic diagram showing ligand-stimulated Ras activation, the Ras-Erk pathway, and the gene mutations found to date contributing to the neuro-cardio-facio-cutaneous congenital disorders and JMML. NL/MGCL – Noonan-like/multiple giant cell lesion; CFC – Cardia-facio-cutaneous; JMML – juvenile myelomonocytic leukemia.
Molecular Genetics
Learning from patients
The clinical presentation of JMML has provided clinicians and researchers with crucial clues to the molecular aberrancies underlying this rare disease. First, young children with the congenital disorder, Neurofibromatosis type 1 (NF1), have an increased incidence of malignant myeloid disorders, including JMML [16, 17]. The characterization of neurofibromin (encoded by NF1) as a Ras GTPase activating protein (GAP) [32, 33] that normally restricts Ras activation suggested that dysregulation of Ras activation contributes to the pathological process of JMML. Furthermore, the finding that loss-of-function NF1 mutations are mostly non-overlapping with gain-of-function KRAS and NRAS mutations in JMML patients strongly implicates Ras hyperactivation in JMML [12]. Similarly, the predisposition of some children with Noonan syndrome (NS), 50% of whom bear germline PTPN11 mutations [20, 34], to develop a JMML-like MPD [18, 19] prompted investigators to test children with non-syndromic, de novo JMML for somatic PTPN11 mutations [13–15]. Taken together, 35% of patients have gain-of-function mutations in PTPN11, 35% gain-of-function mutations in NRAS or KRAS, and another 15% have clinical Neurofibromatosis type 1. Rarely, patients have been described to harbor dual mutations in these molecules and there is active investigation to determine if these mutations exist in separate hematopoietic progenitor cells.
Correlating genotype with phenotype
For potential improved prognostication of JMML, investigators are performing studies to examine if NF1, RAS, and PTPN11 mutational status yields predictive clinical course information for patients. Increased age (> 4 years), increased fetal hemoglobin (HgF, > 15%), and reduced platelets (< 33,000/μL) consistently correlate with a poor outcome (reduced event free survival and overall survival) of JMML patients following allogeneic HSCT; however, the potential correlation of gene mutational status with clinical outcome is unknown. Matsuda and colleagues made the observation that 3 children with JMML bearing mutations of NRAS or KRAS (2 with NRASG12S and 1 with KRASG12S) demonstrated a mild clinical course with spontaneous remission without HSCT [35]. These findings suggested that certain RAS alleles may correlate with favorable clinical outcomes. However, in a follow-up study, Flotho and associates were unable to confirm that certain RAS alleles are associated with longterm survival in the absence of HSCT [36]. In another study directed at examining the hypothesis that mutational status may correlate with JMML clinical features and prognosis, Kojima and colleagues evaluated the clinical course and laboratory findings of 49 JMML patients, 32 of whom bore mutations in NF1, KRAS, NRAS, or PTPN11. In univariate analyses, PTPN11 mutations were associated with older age at diagnosis (> 24 months), increased HgF (> 10%), reduced overall survival, and, importantly, appeared to be an unfavorable prognostic factor predicting relapse following transplantation [37].
Designing accurate murine models
Definition of key mutated genes in JMML has permitted basic researchers to develop in vivo models of JMML. Hematopoietic progenitors from Nf1−/− and KrasG12D mice demonstrate elevated Ras-GTP, hypersensitivity to GM-CSF, and produce MPD in vivo [38–40]. Conditional inactivation of Nf1 in hematopoietic cells induces MPD with 100% penetrance, hypersensitivity to GM-CSF, and resistance to apoptosis [41]. Likewise, wild type mice reconstituted with fetal liver progenitors from Nf1−/− animals develop MPD similar to JMML which is attenuated in mice lacking GM-CSF [42]. Currently, Braun, Shannon, and others are utilizing KrasG12D mice and mice bearing a conditional deletion allele of Ptpn11 [43] to examine the question of whether Shp2 functions upstream, downstream, or in parallel to Ras in GM-CSF-stimulated signaling, in normal hematopoiesis, and in the pathogenesis of JMML. Their studies indicate that Shp2 functions at least partly downstream or parallel to Ras in hematopoietic stem cells, similar to that observed in Drosophila [44], although more recent results indicate that Ras is downstream of Shp2 in myeloid progenitors (B. Braun, personal communication). Furthermore, while Shp2 phosphatase activity is needed for leukemogenesis, it is dispensible for normal hematopoiesis [45–47]. An additional in vivo model of JMML was developed by Neel and colleagues using retroviral transduction of murine hematopoietic stem cells with gain-of-function Ptpn11 mutants (Shp2D61Y and Shp2E76K) followed by transplantation into lethally irradiated recipients [46]. Similar to mice bearing loss-of-function Nf1 or gain-of-function Kras mutations, mice expressing gain-of-function Shp2 also developed MPD in vivo [46]. To remove some of the uncertainty of variable disease onset following adoptive transfer of retrovirally-transduced cells, however, Chan and Neel have developed a conditional knock-in allele of Shp2D61Y (LSL-Shp2D61Y) [48]. Consistent with results obtained from mice transplanted with mutant Shp2-expressing cells, the LSL-Shp2D61Y mice develop MPD by 5 – 7 months following induced expression of Shp2D61Y. However, unlike the transplanted mice, the LSL-Shp2D61Y mice also develop anemia, thus recapitulating more closely the phenotype commonly observed in JMML patients [48]. Using these sophisticated genetic in vivo models, researchers continue to seek unique and novel molecules that can be rationally targeted for improved therapeutic tactics in JMML.
Advances in JMML Diagnosis and Surveillance
Redefining the criteria for diagnosis
In addition to the basic science studies that are being expanded, improved diagnostic, prognostic, and relapse detection techniques are currently under development. The current diagnostic criteria for JMML include absolute monocyte count > 1000/μL, <20% blasts in the bone marrow, and absence of the BCR-ABL fusion gene plus two of the four following criteria: 1) circulating myeloid precursors; 2) WBC > 10,000/μL; 3) increased fetal hemoglobin (HbF) for age; or 4) hematopoietic progenitor hypersensitivity to GM-CSF (Table 1). However, based on the identification of multiple gene mutations in JMML, alternative diagnostic criteria were devised at the JMML Working Group Meeting in Geneva, Switzerland in September, 2006. These proposed criteria have incorporated NF1, RAS, and PTPN11 mutational status or the identification of monosomy 7 into the diagnostic assessment (Table 2).
Table 1.
Current JMML diagnostic criteria.
| ALL of the following | AT LEAST 2 of the following |
|---|---|
| Absence of the t(9;22) BCR/ABL fusion gene | Circulating myeloid precursors |
| Absolute monocyte count > 1000/μL | White blood count >10,000/μL |
| < 20% blasts in the bone marrow | Increased fetal hemoglobin (HgF) |
| GM-CSF hypersensitivity |
Table 2.
Proposed JMML diagnostic criteria.
| Category 1 | Category 2 | Category 3 |
|---|---|---|
| ALL of the following | At least 1 of the following | At least 2 of the following |
| Splenomegaly | Somatic mutation in RAS or PTPN11 | Circulating myeloid precursors |
| Absolute monocyte count > 1000/μL | Clinical diagnosis of NF1 or NF1 gene mutation | WBC > 10,000/μL |
| Blasts in PB/BM <20% | Monosomy 7 | Increased fetal hemoglobin (HgF) for age |
| Absence of the t(9;22) BCR/ABL fusion gene | Clonal cytogenetic abnormality excluding Monosomy 7 | |
| Age less than 13 years |
While hematopoietic progenitor hypersensitivity to GM-CSF has traditionally been used as a minor diagnostic criteria for JMML (Table 1), it is a time-consuming and cumbersome assay to perform using conventional cell culture methods. To address this problem, Kotecha, Nolan, and Loh have evaluated the correlation between GM-CSF-stimulated activation of STAT5, detected using phosphoflow cytometry for phospho-STAT5, with GM-CSF-stimulated hematopoietic progenitor colony growth in JMML patients and healthy controls [49]. The GM-CSF-stimulated phospho-STAT5 assay yielded results very similar to that of the traditional GM-CSF-stimulated hematopoietic progenitor assay, providing a novel diagnostic test that is 95% specific and 91% sensitive and that reduces the diagnosis time from weeks to days. Additionally, differences in GM-CSF-stimulated phospho-STAT5 levels might be utilized to identify targeted agents with potential efficacy as well as to follow response to treatment, relapse, or transformation to acute myeloid leukemia, which may be particularly relevant in patients receiving experimental or traditional chemotherapeutic agents.
Redefining the criteria for response
In the past, response to chemotherapy for JMML has been defined using a complicated assessment of white blood cell count, platelet count, and organomegaly (Table 3a). However, in 2006, the International JMML Working Group meeting in Geneva discussed this topic. First, there was general agreement on the importance of developing new agents that may be biologically active in the treatment of JMML. The goals of testing these agents would be to determine the biologic or clinical activity as well as to define the toxicity profile. Therefore, the group advocated simplification of the criteria and removal of the marginal response. Also, in order to evaluate new agents in an upfront window it was important for patients to have measurable disease. Therefore, the group agreed that in order to be enrolled on future clinical trials to evaluate new agents, patients would need to exhibit a WBC count > 20,000/μL and splenomegaly greater than 2 cm below the costal margin. Clinical findings from the European Working Group on MDS in Childhood (EWOG-MDS) and the North American JMML Project (NAJP) indicated that spleen size and WBC count were the most significant response parameters to evaluate during pre-transplant chemotherapy. Dr. Cooper reviewed these definitions during the 2007 Symposium, as summarized in Table 3b.
Table 3.
| Table 3a. Current JMML response criteria. | ||
|---|---|---|
| WBC | Organomegaly | |
| Complete Response | Normal | None |
| Partial Response | > 50% reduction | > 50% reduction in size |
| Marginal Response | > 25% ≤ 50% reduction | > 25% ≤ 50% reduction in size |
| > 50% reduction | No change | |
| No change | > 50% reduction in size | |
| Stable Disease | ≤25% reduction | ≤25% reduction in size |
| Progressive Disease | >25% increase | >25% increase in size |
| Table 3b. Proposed JMML response criteria. | ||
|---|---|---|
| Complete clinical response | Partial clinical response | |
| White Blood Count | < 20K | < 50% of initial WBC but total still greater than 20K |
| Splenomegaly | Normalization of spleen size | 25% decrease from initial size |
Following allogeneic stem cell transplantation, response to therapy and detection of minimal residual disease can be assessed by following donor chimerism; however, no good method exists for tracking molecular responses to therapy in children who receive traditional chemotherapy or experimental therapies prior to transplantation. To address these limitations, Archambeault and Loh have developed a novel method of detecting minimal residual disease to follow response to therapy by merging TaqMan chemistry with a mismatched amplification mutation assay (MAMA), referred to as TaqMAMA [50]. This method is based on the preferential amplification of the mutant allele compared to the wild type allele. These assays were developed to follow the most common PTPN11 (c.226G>A, c.214G>A, c.227A>G, and c.1508G>C), KRAS (c.38G>A and c.35G>A), and NRAS (c.38G>A and c.37G>C) mutations found in JMML, were adequately sensitive to detect the mutant alleles, and were very specific for 7 of the 8 mutant alleles examined. Importantly, detection of the mutant allele from peripheral blood cells correlated very well with detection from bone marrow cells, suggesting that tracking disease burden by peripheral blood draws rather than by multiple bone marrow aspirations is feasible using this method. Furthermore, the TaqMAMA method detected disease relapse either earlier or simultaneous to disease relapse detection determined by the conventional autologous cell chimerism method. Collectively, these findings suggest that the TaqMAMA methodology provides a potentially powerful tool for following response to upfront novel therapies and for detecting minimal residual disease following transplantation in children with JMML.
Current Therapy
Unfortunately, JMML has proved to be resistant to essentially all chemotherapy regimens examined including single agents as well as combination chemotherapy typically utilized in acute leukemias [6]. The only curative therapy is allogeneic stem cell transplantation which is able to cure approximately 50% of patients; however, the leading cause of death following transplantation continues to be leukemia relapse [7, 51, 52]. Some patients who suffer from relapse are cured from a secondary allogeneic transplant [53]. Two main retrospective studies have been published evaluating the efficacy of allogeneic transplantation in JMML. First, the Japanese Society of Pediatric Hematology (JSPH) published a series of 27 patients diagnosed with JMML based on criteria outlined by the International JMML Working Group. Although the preparative regimens and graft sources varied widely, this retrospective evaluation demonstrated an overall survival of 57.9% and an event free survival of 54.2% at four years after allogeneic transplantation [54]. A second trial with 100 children conducted by the European Working Group on Childhood MDS (EWOG-MDS) and the European Blood and Marrow Transplantation (EMBT) Group used a preparative regimen consisting of busulfan (16 – 20 mg/kg/d for 4 consecutive days), cyclophosphamide (60 mg/kg/d for 2 consecutive days), and melphalan (140 mg/kg, single dose). Graft sources varied from bone marrow, peripheral blood, or cord blood. Similar to the JSPH study, children undergoing allogeneic transplantation achieved an overall survival of 64% and event free survival of 52% [7]. Other analyses from this study revealed no significant difference in event free survival between children receiving grafts from siblings v. unrelated donors or between splenectomized v. non-splenectomized patients at the time of transplant. Event free survival was significantly increased in males and in children < 2 years of age at the time of JMML diagnosis, consistent with the finding of increased survival in children <1 year at the time of diagnosis demonstrated in the JSPH study. However, findings from the EWOG/EBML trial revealed no difference in event free survival between patients with normal v. karyotypic abnormalities while the JSPH trial demonstrated a significant reduction in overall survival in patients with karyotypic abnormalities [7, 54]. Two subsequent studies have been performed by the JPHS. Patients on trial MDS 99 received busulfan (140 mg/m2 × 4 doses), cyclophosphamide (60 mg/kg × 2 doses), and high dose cytarabine (3 gm/m2 × 4 doses). In this study, the investigators observed a 50% disease free survival. Upon subgroup analysis, disease free survival was better in individuals with a normal karyotype as well as patients experiencing either acute or chronic graft v. host disease. In a second trial, a conditioning regimen including busulfan (140 mg/m2 × 4 doses), fludarabine (30 mg/m2 × 4 doses), and LPAM (90 mg/m2 × 2 doses) produced a disease free survival of 75% at six years following transplant. In short, although a rigorous intervention, allogeneic stem cell transplantation provides a real chance of cure for children suffering from JMML. However, scientists and clinicians continue to study JMML aggressively in basic research labs in a concerted effort to define novel therapeutic targets and to develop effective, less toxic, therapeutic interventions.
Experimental Therapies
Although allogeneic transplant does provide a curative modality for JMML, there are significant morbidities associated with this aggressive intervention; thus, improved and less toxic therapies are currently under evaluation. Given the importance of Ras hyperactivation in the pathophysiology of JMML, agents designed to target Ras activity are being evaluated. Ras is first activated at the cell membrane via the addition of a farnesyl group to the newly translated protein. Farnesyl transferase inhibitors (FTIs) prevent Ras translocation to the plasma membrane by inhibiting Ras isoprenylation, thus leading to downregulation of Ras-activated cellular pathways [55]. It was thus hypothesized that inhibiting farnesyl transferase activity may inhibit Ras activation, despite the knowledge that an alternative pathway to Ras activation occurs by geranylgeranylation. In vitro studies demonstrated significant growth inhibition of JMML patient samples by the peptidomimetic FTIs, L-739,749 and L-744,832 [56]. Based on these in vitro findings, Castleberry, Emanuel, and co-workers recently completed a phase II window clinical trial (AML0122) designed to evaluate the dosing and potential effectiveness of R115777, a non-peptidomimetic FTI, in newly diagnosed, previously untreated JMML patients.
Patients received 2 courses of R115777 (200 mg/m2 or 300 mg/m2 by mouth each day for 21 days) followed by two courses of cis-retinoic acid, cytarabine, and fludarabine, splenectomy, and allogeneic transplantation. Following administration of R115777, response to therapy was assessed by white blood cell counts and organomegaly based on current guidelines for JMML response criteria (Table 3a). A total of 11 patients received 200 mg/m2 for two courses. The therapy was well-tolerated and 6 patients achieved a partial response, 1 patient achieved a marginal response, 2 maintained stable disease, and 1 demonstrated progressive disease. A total of 36 patients received 300 mg/m2 for two courses. Among this group, 2 patients achieved a complete remission, 14 achieved a partial response, 7 a marginal response, 2 maintained stable disease, and 3 demonstrated progressive disease. Taken together, 81% of patients demonstrated a clinical response to R115777 (including all with complete, partial, and marginal responses at both doses administered). The main toxicity induced by R115777 was bone marrow suppression. Response or lack of response to the drug was not correlated with patient mutational status (NF1, KRAS, NRAS, PTPN11, or monosomy 7), nor was inhibition of farnesyl transferase activity as measured by the surrogate prenylation of the heat shock molecule, HDJ2 [57]. Unfortunately, preliminary findings presented at the 2nd International JMML Symposium indicated that event-free survival did not appear to be favorably impacted by the use of R115777 compared to the outcomes of other trials; however, this trial is ongoing and the data has not matured. Thus, the event-free survival and overall survival of patients receiving R115777 prior to allogeneic transplantation continues to be evaluated.
In addition to directly targeting Ras, agents that target Ras effectors within the MAPK cascade, such as RAF-1 and MEK, are also being evaluated in myeloid leukemias. Previous in vitro and in vivo studies demonstrated that treatment of JMML cells with a DNA enzyme designed to target RAF1 mRNA, thus reducing RAF1 expression, resulted in reduced hypersensitivity of JMML cells to GM-CSF and reduced mortality of NOD-SCID mice transplanted with JMML cells, respectively [58]. Based on these findings, a phase II window clinical trial is under development to evaluate response rate and acute toxicity of JMML patients to sorafenib (BAY 43-9006), a RAF kinase inhibitor.
Additionally, Lauchle and colleagues have evaluated the effect of CI-1040, a selective MEK inhibitor, on Nf1−/− MPD [41] and Nf1−/− AML (induced by infection of Mx1-Cre, Nf1flox/flox mice [41] with a retroviral insertional mutagen, MOL4070LTR) [59]. While Nf1 mutant MPD cells were not more sensitive to CI-1040 inhibition compared to WT cells, the Nf1 mutant AML cells demonstrated significant sensitivity to CI-1040 inhibition. Similarly, in vivo administration of CI-1040 to Nf1 mutant mice with MPD did not improve survival; however, CI-1040 treatment of mice transplanted with Nf1 mutant AML cells demonstrated significantly increased survival compared to vehicle-treated mice. Interestingly, AML clones that demonstrated resistance to CI-1040 were found to have novel retroviral integration sites, suggesting that cooperating mutations can modulate response to CI-1040 [59].
Conclusions
Tremendous clinical and scientific advances have been made in JMML over the last 20 years; however, regrettably, up to 50% of JMML patients continue to succumb to this lethal malignancy. The goals continue to be improved understanding of the disease at a molecular level in an effort to identify rational therapeutic targets and efficient and successful translation of our mechanistic understanding into superior, less toxic therapeutic modalities for JMML. Furthermore, ideally, JMML will serve as a model disease leading to improved therapies for leukemias and solid tumors, in general, bearing Ras hyperactivation as a central pathogenic mechanism.
Supplementary Material
Acknowledgments
This work was supported by the U.S. National Institutes of Health (5RO1HL082981-03, RJC; 3R13CA132568-02 and 5K22CA113577-03, MLL) and the Leukemia and Lymphoma Society (LLS 2157-08, MLL). MLL is a Clinical Scholar of the Leukemia Lymphoma Society. The authors gratefully acknowledge the administrative assistance of Linda S. Henson.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Emanuel PD, Bates LJ, Castleberry RP, Gualtieri RJ, Zuckerman KS. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood. 1991;77:925–929. [PubMed] [Google Scholar]
- 2.Freedman MH, Cohen A, Grunberger T, Bunin N, Luddy RE, Saunders EF, Shahidi N, Lau A, Estrov Z. Central role of tumour necrosis factor, GM-CSF, and interleukin 1 in the pathogenesis of juvenile chronic myelogenous leukaemia. Br J Haematol. 1992;80:40–48. doi: 10.1111/j.1365-2141.1992.tb06398.x. [DOI] [PubMed] [Google Scholar]
- 3.Estrov Z, Zimmerman B, Grunberger T, Chao J, Teshima IE, Chan HS, Freedman MH. Characterization of malignant peripheral blood cells of juvenile chronic myelogenous leukemia. Cancer Res. 1986;46:6456–6461. [PubMed] [Google Scholar]
- 4.Altman AJ, Palmer CG, Baehner RL. Juvenile “chronic granulocytic” leukemia: a panmyelopathy with prominent monocytic involvement and circulating monocyte colony-forming cells. Blood. 1974;43:341–350. [PubMed] [Google Scholar]
- 5.Emanuel PD. Juvenile myelomonocytic leukemia. Curr Hematol Rep. 2004;3:203–209. [PubMed] [Google Scholar]
- 6.Bergstraesser E, Hasle H, Rogge T, Fischer A, Zimmermann M, Noellke P, Niemeyer CM. Non-hematopoietic stem cell transplantation treatment of juvenile myelomonocytic leukemia: a retrospective analysis and definition of response criteria. Pediatric blood & cancer. 2007;49:629–633. doi: 10.1002/pbc.21038. [DOI] [PubMed] [Google Scholar]
- 7.Locatelli F, Nollke P, Zecca M, Korthof E, Lanino E, Peters C, Pession A, Kabisch H, Uderzo C, Bonfim CS, Bader P, Dilloo D, Stary J, Fischer A, Revesz T, Fuhrer M, Hasle H, Trebo M, van den Heuvel-Eibrink MM, Fenu S, Strahm B, Giorgiani G, Bonora MR, Duffner U, Niemeyer CM. Hematopoietic stem cell transplantation (HSCT) in children with juvenile myelomonocytic leukemia (JMML): results of the EWOG-MDS/EBMT trial. Blood. 2005;105:410–419. doi: 10.1182/blood-2004-05-1944. [DOI] [PubMed] [Google Scholar]
- 8.Brodeur GM. The NF1 gene in myelopoiesis and childhood myelodysplastic syndromes. N Engl J Med. 1994;330:637–639. doi: 10.1056/NEJM199403033300912. [DOI] [PubMed] [Google Scholar]
- 9.Side L, Taylor B, Cayouette M, Conner E, Thompson P, Luce M, Shannon K. Homozygous inactivation of the NF1 gene in bone marrow cells from children with neurofibromatosis type 1 and malignant myeloid disorders. N Engl J Med. 1997;336:1713–1720. doi: 10.1056/NEJM199706123362404. [DOI] [PubMed] [Google Scholar]
- 10.Shannon KM, O’Connell P, Martin GA, Paderanga D, Olson K, Dinndorf P, McCormick F. Loss of the normal NF1 allele from the bone marrow of children with type 1 neurofibromatosis and malignant myeloid disorders. N Engl J Med. 1994;330:597–601. doi: 10.1056/NEJM199403033300903. [DOI] [PubMed] [Google Scholar]
- 11.Sheng XM, Kawamura M, Ohnishi H, Ida K, Hanada R, Kojima S, Kobayashi M, Bessho F, Yanagisawa M, Hayashi Y. Mutations of the RAS genes in childhood acute myeloid leukemia, myelodysplastic syndrome and juvenile chronic myelocytic leukemia. Leuk Res. 1997;21:697–701. doi: 10.1016/s0145-2126(97)00036-2. [DOI] [PubMed] [Google Scholar]
- 12.Kalra R, Paderanga DC, Olson K, Shannon KM. Genetic analysis is consistent with the hypothesis that NF1 limits myeloid cell growth through p21ras. Blood. 1994;84:3435–3439. [PubMed] [Google Scholar]
- 13.Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hahlen K, Hasle H, Licht JD, Gelb BD. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–150. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 14.Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH, Cheng JW, Lee CM, Stokoe D, Bonifas JM, Curtiss NP, Gotlib J, Meshinchi S, Le Beau MM, Emanuel PD, Shannon KM. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. 2004;103:2325–2331. doi: 10.1182/blood-2003-09-3287. [DOI] [PubMed] [Google Scholar]
- 15.Kratz CP, Niemeyer CM, Castleberry RP, Cetin M, Bergstrasser E, Emanuel PD, Hasle H, Kardos G, Klein C, Kojima S, Stary J, Trebo M, Zecca M, Gelb BD, Tartaglia M, Loh ML. The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood. 2005;106:2183–2185. doi: 10.1182/blood-2005-02-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bader JL, Miller RW. Neurofibromatosis and childhood leukemia. J Pediatr. 1978;92:925–929. doi: 10.1016/s0022-3476(78)80362-x. [DOI] [PubMed] [Google Scholar]
- 17.Shannon KM, Watterson J, Johnson P, O’Connell P, Lange B, Shah N, Steinherz P, Kan YW, Priest JR. Monosomy 7 myeloproliferative disease in children with neurofibromatosis, type 1: epidemiology and molecular analysis. Blood. 1992;79:1311–1318. [PubMed] [Google Scholar]
- 18.Bader-Meunier B, Tchernia G, Mielot F, Fontaine JL, Thomas C, Lyonnet S, Lavergne JM, Dommergues JP. Occurrence of myeloproliferative disorder in patients with Noonan syndrome. J Pediatr. 1997;130:885–889. doi: 10.1016/s0022-3476(97)70273-7. [DOI] [PubMed] [Google Scholar]
- 19.Choong K, Freedman MH, Chitayat D, Kelly EN, Taylor G, Zipursky A. Juvenile myelomonocytic leukemia and Noonan syndrome. J Pediatr Hematol Oncol. 1999;21:523–527. [PubMed] [Google Scholar]
- 20.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 21.Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, Bollag G, van der Burgt I, Musante L, Kalscheuer V, Wehner LE, Nguyen H, West B, Zhang KY, Sistermans E, Rauch A, Niemeyer CM, Shannon K, Kratz CP. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38:331–336. doi: 10.1038/ng1748. [DOI] [PubMed] [Google Scholar]
- 22.Carta C, Pantaleoni F, Bocchinfuso G, Stella L, Vasta I, Sarkozy A, Digilio C, Palleschi A, Pizzuti A, Grammatico P, Zampino G, Dallapiccola B, Gelb BD, Tartaglia M. Germline missense mutations affecting KRAS Isoform B are associated with a severe Noonan syndrome phenotype. Am J Hum Genet. 2006;79:129–135. doi: 10.1086/504394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA, Li L, Yassin Y, Tamburino AM, Neel BG, Kucherlapati RS. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007;39:70–74. doi: 10.1038/ng1926. [DOI] [PubMed] [Google Scholar]
- 24.Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, Pandit B, Oishi K, Martinelli S, Schackwitz W, Ustaszewska A, Martin J, Bristow J, Carta C, Lepri F, Neri C, Vasta I, Gibson K, Curry CJ, Siguero JP, Digilio MC, Zampino G, Dallapiccola B, Bar-Sagi D, Gelb BD. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007;39:75–79. doi: 10.1038/ng1939. [DOI] [PubMed] [Google Scholar]
- 25.Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, Pogna EA, Schackwitz W, Ustaszewska A, Landstrom A, Bos JM, Ommen SR, Esposito G, Lepri F, Faul C, Mundel P, Lopez Siguero JP, Tenconi R, Selicorni A, Rossi C, Mazzanti L, Torrente I, Marino B, Digilio MC, Zampino G, Ackerman MJ, Dallapiccola B, Tartaglia M, Gelb BD. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39:1007–1012. doi: 10.1038/ng2073. [DOI] [PubMed] [Google Scholar]
- 26.Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, Amo R, Kamisago M, Momma K, Katayama H, Nakagawa M, Fujiwara Y, Matsushima M, Mizuno K, Tokuyama M, Hirota H, Muneuchi J, Higashinakagawa T, Matsuoka R. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet. 2007;39:1013–1017. doi: 10.1038/ng2078. [DOI] [PubMed] [Google Scholar]
- 27.Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, Filocamo M, Kato K, Suzuki Y, Kure S, Matsubara Y. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37:1038–1040. doi: 10.1038/ng1641. [DOI] [PubMed] [Google Scholar]
- 28.Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, Verloes A, Okamoto N, Hennekam RC, Gillessen-Kaesbach G, Wieczorek D, Kavamura MI, Kurosawa K, Ohashi H, Wilson L, Heron D, Bonneau D, Corona G, Kaname T, Naritomi K, Baumann C, Matsumoto N, Kato K, Kure S, Matsubara Y. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet. 2006;38:294–296. doi: 10.1038/ng1749. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, McCormick F, Rauen KA. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287–1290. doi: 10.1126/science.1124642. [DOI] [PubMed] [Google Scholar]
- 30.Sarkozy A, Conti E, Seripa D, Digilio MC, Grifone N, Tandoi C, Fazio VM, Di Ciommo V, Marino B, Pizzuti A, Dallapiccola B. Correlation between PTPN11 gene mutations and congenital heart defects in Noonan and LEOPARD syndromes. J Med Genet. 2003;40:704–708. doi: 10.1136/jmg.40.9.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keren B, Hadchouel A, Saba S, Sznajer Y, Bonneau D, Leheup B, Boute O, Gaillard D, Lacombe D, Layet V, Marlin S, Mortier G, Toutain A, Beylot C, Baumann C, Verloes A, Cave H. PTPN11 mutations in patients with LEOPARD syndrome: a French multicentric experience. J Med Genet. 2004;41:e117. doi: 10.1136/jmg.2004.021451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356:713–715. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- 33.DeClue JE, Papageorge AG, Fletcher JA, Diehl SR, Ratner N, Vass WC, Lowy DR. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell. 1992;69:265–273. doi: 10.1016/0092-8674(92)90407-4. [DOI] [PubMed] [Google Scholar]
- 34.Tartaglia M, Kalidas K, Shaw A, Song X, Musat DL, Van Der Burgt I, Brunner HG, Bertola DR, Crosby A, Ion A, Kucherlapati RS, Jeffery S, Patton MA, Gelb BD. PTPN11 Mutations in Noonan Syndrome: Molecular Spectrum, Genotype- Phenotype Correlation, and Phenotypic Heterogeneity. Am J Hum Genet. 2002;70:1555–1563. doi: 10.1086/340847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsuda K, Shimada A, Yoshida N, Ogawa A, Watanabe A, Yajima S, Iizuka S, Koike K, Yanai F, Kawasaki K, Yanagimachi M, Kikuchi A, Ohtsuka Y, Hidaka E, Yamauchi K, Tanaka M, Yanagisawa R, Nakazawa Y, Shiohara M, Manabe A, Kojima S, Koike K. Spontaneous improvement of hematologic abnormalities in patients having juvenile myelomonocytic leukemia with specific RAS mutations. Blood. 2007;109:5477–5480. doi: 10.1182/blood-2006-09-046649. [DOI] [PubMed] [Google Scholar]
- 36.Flotho C, Kratz CP, Bergstrasser E, Hasle H, Stary J, Trebo M, van den Heuvel-Eibrink MM, Wojcik D, Zecca M, Locatelli F, Niemeyer CM. Genotype-phenotype correlation in cases of juvenile myelomonocytic leukemia with clonal RAS mutations. Blood. 2008;111:966–967. doi: 10.1182/blood-2007-09-111831. author reply 967–968. [DOI] [PubMed] [Google Scholar]
- 37.Yoshida N, Yagasaki H, Yoshimi A, Takahashi Y, Xu Y, Tanaka M, Watanabe N, Matsumoto K, Kato K, Ueyama J, Inada H, Goto H, Yabe M, Mimaya J, Kikuchi A, Manabe A, Kojima S. Correlation of Clinical Features with the Mutational Status of GM-CSF Signaling Pathway-Related Genes in Children with Juvenile Myelomonocytic Leukemia. Blood. 2007;110:457a. doi: 10.1203/PDR.0b013e3181961d2a. [DOI] [PubMed] [Google Scholar]
- 38.Braun BS, Tuveson DA, Kong N, Le DT, Kogan SC, Rozmus J, Le Beau MM, Jacks TE, Shannon KM. Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc Natl Acad Sci U S A. 2004;101:597–602. doi: 10.1073/pnas.0307203101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Largaespada DA, Brannan CI, Jenkins NA, Copeland NG. Nf1 deficiency causes Ras-mediated granulocyte/macrophage colony stimulating factor hypersensitivity and chronic myeloid leukaemia. Nat Genet. 1996;12:137–143. doi: 10.1038/ng0296-137. [DOI] [PubMed] [Google Scholar]
- 40.Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P, Lange BJ, Freedman MH, McCormick F, Jacks T, Shannon K. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet. 1996;12:144–148. doi: 10.1038/ng0296-144. [DOI] [PubMed] [Google Scholar]
- 41.Le DT, Kong N, Zhu Y, Lauchle JO, Aiyigari A, Braun BS, Wang E, Kogan SC, Le Beau MM, Parada L, Shannon KM. Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood. 2004;103:4243–4250. doi: 10.1182/blood-2003-08-2650. [DOI] [PubMed] [Google Scholar]
- 42.Birnbaum RA, O’Marcaigh A, Wardak Z, Zhang YY, Dranoff G, Jacks T, Clapp DW, Shannon KM. Nf1 and Gmcsf interact in myeloid leukemogenesis. Molecular cell. 2000;5:189–195. doi: 10.1016/s1097-2765(00)80415-3. [DOI] [PubMed] [Google Scholar]
- 43.Fornaro M, Burch PM, Yang W, Zhang L, Hamilton CE, Kim JH, Neel BG, Bennett AM. SHP-2 activates signaling of the nuclear factor of activated T cells to promote skeletal muscle growth. J Cell Biol. 2006;175:87–97. doi: 10.1083/jcb.200602029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allard JD, Chang HC, Herbst R, McNeill H, Simon MA. The SH2-containing tyrosine phosphatase corkscrew is required during signaling by sevenless, Ras1 and Raf. Development. 1996;122:1137–1146. doi: 10.1242/dev.122.4.1137. [DOI] [PubMed] [Google Scholar]
- 45.Schubbert S, Lieuw K, Rowe SL, Lee CM, Li X, Loh ML, Clapp DW, Shannon KM. Functional analysis of leukemia-associated PTPN11 mutations in primary hematopoietic cells. Blood. 2005;106:311–317. doi: 10.1182/blood-2004-11-4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mohi MG, Williams IR, Dearolf CR, Chan G, Kutok JL, Cohen S, Morgan K, Boulton C, Shigematsu H, Keilhack H, Akashi K, Gilliland DG, Neel BG. Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell. 2005;7:179–191. doi: 10.1016/j.ccr.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 47.Braun BS, Cheung LS, Chan GG, Yang WA, Archard JA, Neel BG, Shannon KM. A Noncatalytic Ras-independent Function of SHP-2 Is Essential in Hematopoietic Progenitors. Blood. 2007;110:34a. [Google Scholar]
- 48.Chan G, Kalaitzidis D, Mohi MG, Yang W, Kutok JL, Neel BG. Inducible expression of leukemia-associated Shp2 (Ptpn11) affects multiple stages of hematopoiesis and causes a fatal myeloproliferative disorder (MPD) in mice. Blood. 2007;110:457a. doi: 10.1182/blood-2008-10-182626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kotecha N, Flores NJ, Irish JM, Simonds E, Sakai DS, Archambeault S, Diaz-Flores E, Coram M, Shannon KM, Nolan GP, Loh ML. Single Cell Profiling Identifies Aberrant STAT5 Activation in Myeloid Malignancies with Specific Clinical and Biologic Correlates. Cancer Cell. 2008 doi: 10.1016/j.ccr.2008.08.014. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Archambeault S, Flores NJ, Yoshimi A, Kratz CP, Reising M, Fischer A, Noellke P, Locatelli F, Sedlacek P, Flotho C, Zecca M, Emanuel PD, Castleberry RP, Niemeyer CM, Bader P, Loh ML. Development of an allele-specific minimal residual disease assay for patients with juvenile myelomonocytic leukemia. Blood. 2008;111:1124–1127. doi: 10.1182/blood-2007-06-093302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koike K, Matsuda K. Recent advances in the pathogenesis and management of juvenile myelomonocytic leukaemia. Br J Haematol. 2008 doi: 10.1111/j.1365-2141.2008.07104.x. [DOI] [PubMed] [Google Scholar]
- 52.Niemeyer CM, Kratz CP. Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia: molecular classification and treatment options. Br J Haematol. 2008;140:610–624. doi: 10.1111/j.1365-2141.2007.06958.x. [DOI] [PubMed] [Google Scholar]
- 53.Yoshimi A, Mohamed M, Bierings M, Urban C, Korthof E, Zecca M, Sykora KW, Duffner U, Trebo M, Matthes-Martin S, Sedlacek P, Klingebiel T, Lang P, Fuhrer M, Claviez A, Wossmann W, Pession A, Arvidson J, O’Marcaigh AS, van den Heuvel-Eibrink MM, Stary J, Hasle H, Nollke P, Locatelli F, Niemeyer CM. Second allogeneic hematopoietic stem cell transplantation (HSCT) results in outcome similar to that of first HSCT for patients with juvenile myelomonocytic leukemia. Leukemia. 2007;21:556–560. doi: 10.1038/sj.leu.2404537. [DOI] [PubMed] [Google Scholar]
- 54.Manabe A, Okamura J, Yumura-Yagi K, Akiyama Y, Sako M, Uchiyama H, Kojima S, Koike K, Saito T, Nakahata T. Allogeneic hematopoietic stem cell transplantation for 27 children with juvenile myelomonocytic leukemia diagnosed based on the criteria of the International JMML Working Group. Leukemia. 2002;16:645–649. doi: 10.1038/sj.leu.2402407. [DOI] [PubMed] [Google Scholar]
- 55.Rowinsky EK, Windle JJ, Von Hoff DD. Ras protein farnesyltransferase: A strategic target for anticancer therapeutic development. J Clin Oncol. 1999;17:3631–3652. doi: 10.1200/JCO.1999.17.11.3631. [DOI] [PubMed] [Google Scholar]
- 56.Emanuel PD, Snyder RC, Wiley T, Gopurala B, Castleberry RP. Inhibition of juvenile myelomonocytic leukemia cell growth in vitro by farnesyltransferase inhibitors. Blood. 2000;95:639–645. [PubMed] [Google Scholar]
- 57.Castleberry RP, Loh ML, Jayaprakash N, Peterson A, Casey V, Chang M, Widemann B, Emanuel PD. Phase II Window Study of the Farnesyltransferase Inhibitor R115777 (Zarnestra®) in Untreated Juvenile Myelomonocytic Leukemia (JMML): A Children’s Oncology Group Study. Blood. 2005;106:727a. [Google Scholar]
- 58.Iversen PO, Emanuel PD, Sioud M. Targeting Raf-1 gene expression by a DNA enzyme inhibits juvenile myelomonocytic leukemia cell growth. Blood. 2002;99:4147–4153. doi: 10.1182/blood.v99.11.4147. [DOI] [PubMed] [Google Scholar]
- 59.Lauchle JO, Le DT, Kim D, Akagi K, Gorman MF, Tran M, Sebolt-Leopold J, Wolff L, Parada L, Jenkins N, Copeland N, Shannon KM. Mutations That Cooperate with Nf1 Inactivation in Leukemogenesis Influence Therapeutic Response to MEK Inhibition. Blood. 2007;110:183a. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.