

SUMMARY
The role of bicarbonate (HCO3-) in GABAA receptor-mediated depolarization of human hypothalamic hamartoma (HH) neurons was investigated using cellular electrophysiological and calcium imaging techniques. Activation of GABAA receptors with muscimol (30 μM) provoked neuronal excitation in over 70% of large (18-22 μM) HH neurons in HCO3- buffer. Subsequent perfusion of HCO3--free HEPES buffer produced partial suppression of muscimol-induced excitation. Additionally, 53% of large HH neurons under HCO3--free conditions exhibited reduced intracellular calcium accumulation by muscimol. These results suggest that HCO3- efflux through GABAA receptors on a subpopulation of large HH neurons may contribute to membrane depolarization and subsequent activation of L-type calcium channels.
Keywords: Hypothalamic hamartoma, bicarbonate, GABAA receptor, depolarization, L-type calcium channel
Introduction
GABA (γ-aminobutyric acid) is the major inhibitory neurotransmitter in the mammalian central nervous system. However, in immature neurons, GABA is known to exert an excitatory effect which is mediated by chloride efflux through GABAA receptors. The principal basis of GABA-mediated excitation involves intracellular chloride ([Cl]i) accumulation resulting from the differential expression and activity of the cation chloride co-transporters NKCC1 and KCC2 (Staley, 2006). The potential pro-epileptic activity of GABA has been implicated in both human and experimental animal models of epilepsy (Cohen et al., 2003; Staley, 2006).
Consistent with these observations, activation of GABAA receptors induced neuronal excitation and secondary triggering of L-type voltage-gated calcium channels in most large neurons found in surgically-resected human hypothalamic hamartoma (HH) tissue (Kim et al., 2008). This mechanism may underlie in part the intrinsic epileptogenicity of HH lesions, which have been classically associated with gelastic seizures (Berkovic et al., 1988).
Previous studies have also implicated bicarbonate (HCO3-) in GABAA receptor-mediated depolarization (Staley et al., 1995; Dallwig et al., 1999), Here, we asked whether HCO3- might play a role in GABA-induced excitation in human HH tissue slices using gramicidin-perforated patch recording and calcium imaging techniques.
Methods
HH tissue was obtained from 7 patients (M:F, 3:4; mean age, 7 years 4 months with a median of 6 years, and range, 9 months to 17.1 years; see Table 1) between April and August of 2007. Experimental procedures were modified from Kim et al., (2008). Briefly, tissue specimens were immediately transferred upon surgical resection to ice-cold oxygenated (95% O2/5% CO2) physiological saline (composition in mM: 124 NaCl, 1.3 MgSO4, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2.4 CaCl2, and 10 D-glucose; pH: 7.4). HH brain slices (300 μm) were cut using a vibratome (The Vibratome Company, St. Louis, MO, USA). Each slice was submerged in a recording chamber attached to a Zeiss Axioskop FS2 microscope (Carl Zeiss Microimaging, Inc., Thornwood, NY, USA) and infused with physiological saline (32°C) flowing at a rate of 2-3 ml/min. HH neurons were visualized with differential interference contrast (DIC) optics and infrared illumination. Recording electrodes (resistance, 4-6 MΩ) were backfilled with a solution containing (in mM): 135 KCl, 10 HEPES, 0.5 CaCl2, 2 MgCl2, and 5 EGTA, pH 7.25 (adjusted with KOH) for gramicidin (20 μg/ml) perforated patch recordings. Data were acquired using a Multiclamp 700A amplifier, and were digitized and sampled at 50 μs intervals (Digidata 1322A, pClamp V9.2 software; Axon Instruments, Union City, CA, USA).
Table 1.
Patient Data
| Case | Gender | Age at Surgery | Onset of Seizures | Seizure Types | HH Type | Precocious Puberty | HH volume (cm3) | Surgery Type |
|---|---|---|---|---|---|---|---|---|
| 1 | F | 6Y 2mo | 1mo | Multi (G, CPS) | 4 | Yes | 38.3 | TC |
| 2 | F | 3Y 3mo | 1mo | Multi (G, CPS) | 4 | Yes | 20.3 | TC |
| 3 | F | 5Y 7mo | 4mo | Multi (G, Tonic) | 4 | Yes | 4.8 | TC |
| 4 | F | 6Y | 1mo | Multi (G, CPS) | 2 | No | 4.8 | TC |
| 5 | M | 12Y 9mo | 60mo | Multi (G, CPS) | 3 | No | 3.8 | Endo |
| 6 | M | 9mo | 1mo | Multi (G, IS) | 2 | No | 1.1 | Endo |
| 7 | M | 17Y 1mo | 1mo | Multi (G, GTC) | 2 | No | 0.6 | Endo |
F, female; M, male; Y, year; mo, month; Multi, multiple; G, gelastic seizure; CPS, complex partial seizure; IS, infantile spasms; GTC, generalized tonic-clonic seizures. According to the HH classification system proposed by Delalande et al., (2003), three patients (43%) Type II HH, one (14%) had a Type III HH, and 3 (43%) had a Type IV HH. The mean volume of the HH lesions was 10.5 cm3 (range, 0.6 to 38.3 cm3). Four HH patients underwent resection through a transcallosal (TC) interforniceal approach, and three others by a transventricular endoscopic (Endo) approach. All patients had treatment-resistant epilepsy, and were refractory to at least three anti-epileptic drugs (AEDs; carbamazepine, lamotrigine, levetiracetam, oxcarbazepine, phenobarbital, topiramate, zonisamide). In all cases, diagnosis was confirmed by neuropathological examination. Informed consent was obtained from all patients for the use of surgically-resected tissue for research purposes according to protocols approved by the Institutional Review Board of the Barrow Neurological Institute and St. Joseph’s Hospital and Medical Center, Phoenix, Arizona (U.S.A.).
To deplete intracellular HCO3-, perfusion saline containing 26 mM bicarbonate buffer was replaced by 26 mM HEPES buffer (pH 7.4 adjusted with NaOH) without 95% O2/5% CO2 bubbling (Dallwig et al., 1999). For calcium imaging, HH slices were loaded with the calcium indicator dye fura2-AM (10 μM, molecular probes). After 60 min, each slice was transferred to a recording chamber fixed to an Axioskop FS2 microscope and outfitted with the Zeiss Stallion 2 imaging system (Carl Zeiss Microimaging). Ratiometric excitation was measured using 340 and 380 nm filter sets and controlled with a high-speed filter switching device (Sutter Instruments, Lambda DG-4). Changes in calcium levels before, during and after drug application in HH cells of interest were captured under fluorescence (exposure time, 50-100 ms) every 5-10 s. Changes of fluorescence ratios were measured/analyzed using slide book software (Intelligent Imaging Innovation).
Results
Recently, we reported that the GABAA receptor agonist muscimol (30 μM) induced membrane depolarization in large HH neurons (Kim et al., 2008). To investigate the possibility that HCO3- efflux through GABAA receptor may also contribute to GABA-induced neuronal excitation (Staley et al., 1995; Dallwig et al., 1999), we initially evoked membrane depolarization in large (18-22 μM) HH neurons using HCO3--containing saline (31 out of 43 neurons; Fig 1), and then perfused HH slices with HCO3--free HEPES buffer for 10 min prior to additional muscimol treatment. This protocol (importantly, the absence of 95%O2/5%CO2 bubbling) has previously been shown to render intracellular depletion of HCO3- within 10 min, and hence eliminate the immediate possibility of HCO3- efflux upon GABAA receptor activation, until carbonic anhydrase is able to restore intracellular HCO3- levels (Phillips et al., 1998).
Figure 1.

Effects of HCO3--free HEPES buffer on muscimol-induced HH neuronal excitation. (A) In 4 of 31 large HH neurons, the initial excitatory response to muscimol was fully suppressed by pre-incubation with HEPES buffer, but was not blocked by TTX (8 of 8 cells). Further, the L-type calcium channel blocker nifedipine strongly suppressed muscimol-induced excitation (9 of 10 cells). (B) In 12 out of 31 large HH cells, muscimol-induced neuronal excitation was partially suppressed by a 10 min pre-incubation with HEPES buffer. The effect of HEPES buffer was reversed by subsequent perfusion with bicarbonate-containing buffer. (C) HEPES buffer did not affect muscimol-induced HH neuronal excitation (15 of 31 cells). Two vertical bars in the membrane trace indicate 10 min pre-incubation with HEPES buffer. Horizontal bar in this and following figure indicate the timing of drug infusion.
Under these recording conditions, we observed three distinct effects induced by muscimol. First, in a minority of cells (4 of 31; N = 5 patients; cases 1-5; see Table 1), the initial excitatory response of HH neurons to muscimol was abolished following HEPES buffer application (Fig. 1A). Further, the L-type calcium channel blocker nifedipine (100 μM), but not the sodium channel blocker tetrodotoxin (TTX, 1μM), fully suppressed muscimol-induced membrane depolarization (Fig.1A), consistent with our earlier findings (Kim et al., 2008). Second, in 12 of 31 cells (N = 5 patients), muscimol-evoked neuronal excitation was incompletely but substantially diminished in HCO3−- free HEPES buffer (Fig. 1B). This suppressive effect was reversed by re-exposure to HCO3--containing saline. In contrast, a third group comprising nearly half of the excitable large HH neurons (15 of 31 cells) was unaffected by the switch to HCO3−-free conditions (Fig. 1C). Together, these data indicate that HCO3- efflux through GABAA receptors may contribute to membrane depolarization in the slight majority of large, excitable HH neurons.
Next, we explored whether the suppressive effects following the switch to HCO3--free perfusate might affect secondary activation of L-type voltage-gated calcium channels. Consistent with our earlier findings (Kim et al., 2008), nifedipine (100 μM) fully abolished the increase in intracellular calcium [Ca 2+]i induced by muscimol (10 of 11 cells; N = 5 patients, cases 3 - 7; Fig. 2A). In contrast, muscimol-evoked elevations in [Ca2+]i were only partially suppressed when slices were subsequently exposed to HCO3--free HEPES buffer (6 of 13 cells; N = 5 patients). Only in 1 of 13 cells did we observe a complete blockade of muscimol-induced rise in [Ca 2+]i. This suppressive effect was easily reversed by subsequent application of HCO3--containing saline (Fig. 2B). As demonstrated in Fig. 2B, HEPES alone did not appear to significantly influence [Ca 2+]i.
Figure 2.
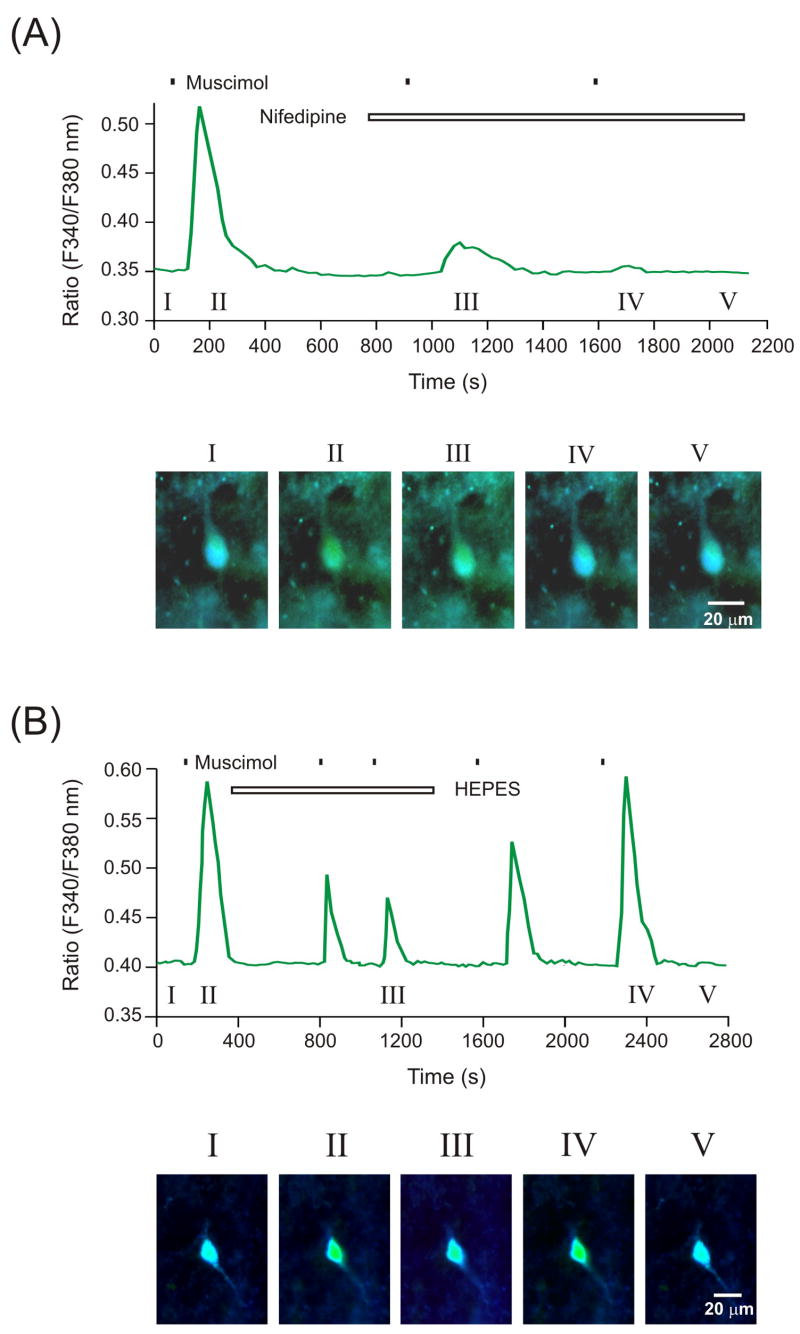
Changes in intracellular calcium levels in large HH neurons following exposure to muscimol, co-applied with either HCO3--free HEPES buffer or nifedipine. (A) Nifedipine prevents the increase in calcium level induced by muscimol. Representative images (panel I to V) showing the change of intracellular calcium fluorescence induced by muscimol with or without nifedipine. (B) The increase in intracellular calcium levels induced by muscimol was partially diminished by application of HEPES buffer (green trace). This effect was completely reversed by re-perfusion of HCO3--containing buffer.
Discussion
The principal finding of this study is that the bicarbonate ion contributes in part to GABAA receptor-mediated neuronal excitation in large (18-22 μM) HH neurons. Further, we found that responses to muscimol application were clearly heterogeneous in the subpopulation of large neurons studied. While the majority of large HH neurons responded to muscimol with membrane depolarization, approximately 30% of tested neurons did not exhibit neuronal excitation upon GABAA receptor activation. At present, we do not have a clear understanding why this heterogeneity exists.
Along similar lines, the percentage of large HH neurons that exhibited either complete blockade or partial suppression of GABA-induced excitation in HEPES buffer did not perfectly match the numbers obtained from cells tested under either perforated patch recording conditions or for calcium imaging. However, approximately 50% of tested neurons (16 of 31 cells) with HEPES buffer showed suppression or block of GABA-induced hyper-excitability. In agreement with this finding, bicarbonate-free HEPES buffer suppressed or blocked GABA-induced calcium influx in approximately the same percentage of cells (7 of 13 cells). While we currently cannot fully reconcile the differences in the data obtained from whole-cell recordings and calcium imaging, one possibility is that incomplete blockade of GABA-induced excitation in HEPES buffer may be influenced by differential distribution of intracellular chloride concentrations among large HH neurons.
It is well established that the key determinant of GABA-mediated neuronal excitation is a reversed chloride electrochemical gradient, which yields a more depolarized membrane reversal potential for muscimol or GABA (Emuscimol/GABA). Importantly, the chloride electrochemical gradient is determined by the relative expression and activity of the cation chloride co-transporters NKCC1 and KCC2 (Palma et al., 2006; Kim et al., 2008). Despite these observations, it is intriguing to note that the NKCC1 blocker bumetanide alone failed to completely block epileptiform activity in the developing brain (Dzhala et al., 2005), suggesting that mechanisms other than chloride flux may be involved.
One alternative factor in the modulation of GABA-mediated excitation is a bicarbonate-driven depolarizing potential. GABAA receptors are known to be permeable to both chloride and HCO3- ions (Bormann et al., 1987), and intracellular accumulation of HCO3- ions from carbonic anhydrase (CA) activity creates an outwardly directed electrochemical gradient for HCO3- (Staley et al., 1995). Moreover, several studies have demonstrated that HCO3--free HEPES buffer is able to suppress GABA-induced membrane depolarization (Staley et al., 1995; Phillips et al., 1998; Dallwig et al., 1999), thus highlighting the relevance of HCO3- to the phenomenon of GABA-mediated neuronal excitation.
It is possible that HCO3- could be affecting neuronal excitability through a mechanism other than L-type calcium channels. A previous study suggested that the intracellular pH (pHi) in excitable cells is higher than that would be expected solely from the normal proton concentration, thus invoking increased intracellular bicarbonate (Thomas et al., 1977). Under such conditions, activation of GABAA receptors would easily lead to bicarbonate efflux through the GABAA receptor ionophore. In support of this, bicarbonate conductance through the GABAA receptor was found to provoke a negative shift of EGABA (Dallwig et al., 1999). Based on these findings, it is reasonable to speculate that changes in pHi under epileptic conditions may in part account for GABA-induced neuronal excitation through bicarbonate efflux. At present, direct evidence for this is lacking.
Clinically, carbonic anhydrase (CA) inhibitors have been successfully used as anticonvulsant agents for over 30 years (mainly in Europe). There are several lines of experimental evidence supporting these clinical observations. First, Staley et al. (1995) observed that acetazolamide decreased GABAA receptor-mediated depolarizing currents induced by muscimol. Second, seizure-like rhythmic activity in hippocampus induced by GABAA receptor-mediated excitation was suppressed by acetazolamide (Fujiwara-Tsukamoto et al., 2003). Finally, indanesulfonamides (which inhibit human CA isoforms) have recently been shown to block maximal electroshock seizures in mice (Thiry et al., 2008). Although these data suggest that CA inhibitors may be helpful in controlling gelastic seizures in HH patients, there are at present no clinical data to support this possibility.
Notwithstanding the lack of current evidence for using CA inhibitors in HH patients, it is important to note that nifedipine is more effective than either bumetanide or HEPES buffer in blocking muscimol-induced neuronal excitation in large HH neurons. The present study further supports our recently reported findings (Kim et al., 2008) and implicates a mechanistic role for HCO3- in GABAA receptor-mediated neuronal excitation in large HH neurons, one that contributes importantly to subsequent activation of L-type calcium channels, and possibly intrinsic seizure genesis in HH tissue.
Acknowledgments
The authors wish to thank the patients and their families, and our pediatric neurosurgeon, Harold Rekate, for making this work possible. This work was supported by NIH grant NS 057786 and the Barrow Neurological Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berkovic SF, Andermann F, Melanson D, Ethier RE, Feindel W, Gloor P. Hypothalamic hamartomas and ictal laughter: evolution of a characteristic epileptic syndrome and diagnostic value of magnetic resonance imaging. Ann Neurol. 1988;23:429–439. doi: 10.1002/ana.410230502. [DOI] [PubMed] [Google Scholar]
- Bormann J, Hamill OP, Sakmann B. Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J Physiol. 1987;385:243–286. doi: 10.1113/jphysiol.1987.sp016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Le Duigou C, Miles R. Mesial temporal lobe epilepsy: a pathological replay of developmental mechanisms? Biol Cell. 2003;95:329–333. doi: 10.1016/s0248-4900(03)00081-9. [DOI] [PubMed] [Google Scholar]
- Dallwig R, Deitmer JW, Backus KH. On the mechanism of GABA-induced currents in cultured rat cortical neurons. Pflugers Arch. 1999;437:289–297. doi: 10.1007/s004240050782. [DOI] [PubMed] [Google Scholar]
- Delalande O, Fohlen M. Disconnecting surgical treatment of hypothalamic hamartoma in children and adults with refractory epilepsy and proposal of a new classification. Neurol Med Chir (Tokyo) 2003;43:61–68. doi: 10.2176/nmc.43.61. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE, Staley KJ. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- Fujiwara-Tsukamoto Y, Isomura Y, Nambu A, Takada M. Excitatory gaba input directly drives seizure-like rhythmic synchronization in mature hippocampal CA1 pyramidal cells. Neuroscience. 2003;119:265–275. doi: 10.1016/s0306-4522(03)00102-7. [DOI] [PubMed] [Google Scholar]
- Kim DY, Fenoglio KA, Simeone TA, Coons SW, Wu J, Chang Y, Kerrigan JF, Rho JM. GABAA receptor-mediated activation of L-type calcium channels induces neuronal excitation in surgically resected human hypothalamic hamartomas. Epilepsia. 2008;49:861–871. doi: 10.1111/j.1528-1167.2007.01455.x. [DOI] [PubMed] [Google Scholar]
- Palma E, Amici M, Sobrero F, Spinelli G, Di Angelantonio S, Ragozzino D, Mascia A, Scoppetta C, Esposito V, Miledi R, Eusebi F. Anomalous levels of Cl- transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc Natl Acad Sci U S A. 2006;103:8465–8468. doi: 10.1073/pnas.0602979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips I, Martin KF, Thompson KS, Heal DJ. GABA-evoked depolarisations in the rat cortical wedge: involvement of GABAA receptors and HCO3- ions. Brain Res. 1998;798:330–332. doi: 10.1016/s0006-8993(98)00479-x. [DOI] [PubMed] [Google Scholar]
- Staley KJ. Wrong-way chloride transport: is it a treatable cause of some intractable seizures? Epilepsy Curr. 2006;6:124–127. doi: 10.1111/j.1535-7511.2006.00119.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–981. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- Thiry A, Rolin S, Vullo D, Frankart A, Scozzafava A, Dogne JM, Wouters J, Supuran CT, Masereel B. Indanesulfonamides as carbonic anhydrase inhibitors and anticonvulsant agents: Structure-activity relationship and pharmacological evaluation. Eur J Med Chem. 2008 doi: 10.1016/j.ejmech.2008.02.018. In press. [DOI] [PubMed] [Google Scholar]
- Thomas RC. The role of bicarbonate, chloride and sodium ions in the regulation of intracellular pH in snail neurones. J Physiol. 1977;273(1):317–38. doi: 10.1113/jphysiol.1977.sp012096. [DOI] [PMC free article] [PubMed] [Google Scholar]