

Abstract
The environment of the adult CNS prevents axonal regeneration after injury. This inhibition of axonal regeneration can be blocked by elevating cAMP. Previously, we showed that the cAMP pathway can be activated via pre-treatment with neurotrophins and requires activation of several signaling pathways which converge at activation of the transcription factor, CREB. Here, we show that calcium/calmodulin-dependent kinase IV (CaMKIV) is necessary for the neurotrophin-induced phosphorylation of CREB and the block of myelin-mediated inhibition of axonal growth. Pharmacological inhibition of CaMKIV or over-expression of a dominant-negative mutant form of CaMKIV blocks the neurotrophin effect. Interestingly, CaMKIV activation is not necessary if cAMP levels is already elevated. Finally, calcium flux from intracellular stores is necessary for this CaMKIV signaling. These results demonstrate that CaMKIV is another player in the neurotrophin-induced signaling which leads to axonal regeneration and therefore, is a potential target for therapeutic intervention following injury to the adult CNS.
Introduction
Neurons that are injured following damage to the adult mammalian nervous system are unable to regenerate their severed axons. This is due, in part, to the presence of myelin-associated inhibitors of regeneration and formation of a glial scar (Filbin, 2003; Yamashita et al., 2005). To date, three known myelin-associated inhibitors have been identified, which are myelin-associated glycoprotein (MAG) (McKerracher et al., 1994; Mukhopadhyay et al., 1994), Nogo (Chen et al., 2000; GrandPre et al., 2000; Prinjha et al., 2000) and oligodendrocyte-myelin glycoprotein (OMgp) (Kottis et al., 2002; Wang et al., 2002). A number of studies have shown that inhibition by all the myelin-associated inhibitors is blocked by elevating cAMP and as a consequence, regeneration is promoted both in vitro (Cai et al., 1999) and in vivo (Neumann et al., 2002; Qiu et al., 2002). Elevation of cAMP can be accomplished in several ways, including: application of nonhydrolysable analogues such as dibutyryl-cAMP (db-cAMP) (Andersen et al., 2000; Cai et al., 1999; Monsul et al., 2004; Neumann et al., 2002; Qiu et al., 2002), by inhibition of the enzymes that degrade cAMP, phosphodiesterases (Nikulina et al., 2004; Pearse et al., 2004) or by pre-treatment of neurons with neurotrophins (“priming”) (Cai et al., 1999).
We have shown that this cAMP-dependent block of axonal inhibition involves activation of the transcription factor, CREB. Further evidence suggests that when priming neurons with neurotrophins, such as BDNF, the block of MAG-mediated axonal inhibition requires a “threshold” of CREB activation which requires the activity of not only PKA but also PI3K, Erk and CaMK (Gao et al., 2004). Blocking any one of these pathways greatly reduces, but does not eliminate, activation of CREB by BDNF. However, if the signaling of any one of these pathways is disrupted, the improved axonal growth associated with the priming effect is lost (Cai et al., 1999; Gao et al., 2004; Gao et al., 2003). Therefore, the simultaneous activation of each one of these pathways by BDNF is essential to reach the threshold level of CREB activation necessary for overcoming the MAG-mediated block of axonal regeneration. What is not known, however, is if these signaling pathways work in sequence or in parallel to encourage regeneration in an inhibitory environment. Of particular interest is the role of CaMK in this phenomenon because, as the name implies, calcium is required to activate this enzyme and calcium has been implicated both in the repulsive turning of growth cones in response to MAG as well as in the signaling paradigms which can change axon repulsion to attraction.
The CaMKs have been shown to mediate many signaling events in the CNS including activation of certain adenylyl cyclases (Shaywitz and Greenberg, 1999; Wong et al., 1999) and activation of CREB (Bito et al., 1996; Enslen et al., 1994; Finkbeiner et al., 1997; Matthews et al., 1994; Shaywitz and Greenberg, 1999; Sun et al., 1994). These mechanisms are believed to mediate a variety of neuronal/brain functions such as longterm potentiation (Bach et al., 1995; Giese et al., 1998; Mayford et al., 1996; Silva et al., 1992a; Silva et al., 1992b), synaptic plasticity and learning and memory (Shaywitz and Greenberg, 1999; Wei et al., 2002). The CaMKs consist of a family of at least seven members, of which CaMKII and CaMKIV have been shown to play roles in neuronal signaling. Although we have already shown that a CaMK is involved in the ability of BDNF to overcome inhibition by MAG, we do not know if CaMKII or CaMKIV is responsible for this effect as the pharmacological inhibitor we used blocks both. In addition, we do not know the source of the calcium released in response to BDNF to activate CaMK and if BDNF activates CaMK, PKA and ERK in parallel or sequentially to overcome inhibition by MAG.
Here, we show that it is activation of CaMKIV which is necessary for the neurotrophin-induced CREB activation and subsequent block of MAG-mediated inhibition of neurite outgrowth. We also show that this CaMKIV-mediated effect is dependent on calcium flux from intracellular stores and that BDNF activates CaMKIV via a parallel signaling pathway, separate from that of PKA and ERK. This is the first time that calcium has been implicated in the signaling which leads to overcoming MAG-mediated inhibition of axonal regeneration.
Results and Discussion
CaMKIV is necessary for the BDNF-induced priming effect but not the db-cAMP block of axonal inhibition
In order to address the role of CaMK (and indirectly, the role of calcium) in overcoming inhibition by MAG, we first confirmed that the CaMK inhibitor, KN62, was able to attenuate the phosphorylation of CREB by BDNF and block completely its ability to overcome inhibition (Fig. 1A and B). A significant increase in CREB phosphorylation was observed in cerebellar (Fig. 1A) and dorsal root ganglion (data not shown) neurons following exposure to BDNF. In addition, our findings show that while CaMK inhibition alone has no effect on basal levels of CREB phosphorylation, KN-62 is indeed sufficient to abrogate the BDNF-induced increase in CREB phosphorylation in these same neurons. In sharp contrast, however, KN62 had no effect on the ability of db-cAMP to activate CREB. Figure 1A shows that CREB is phosphorylated in response to db-cAMP as effectively with or without KN62. Consistent with this, KN62 also has no effect on the ability of db-cAMP to overcome inhibition by MAG; neurons on MAG-expressing cells in the presence of db-cAMP, either with or without KN62, extend neurites as long as on the control cells not expressing MAG (Fig. 1B). Hence, we find that blocking the activity of CaMKIV does indeed interfere with the BDNF-induced priming effect, via a mechanism which works either upstream or in parallel to the cAMP-elevation step.
Figure 1. Calcium-calmodulin kinase activity is necessary for the BDNF-induced priming effect.
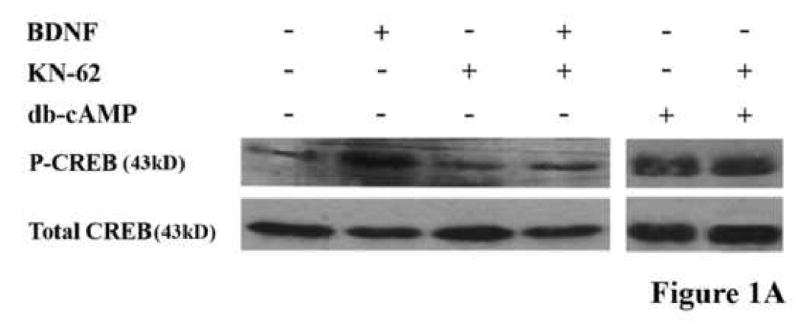
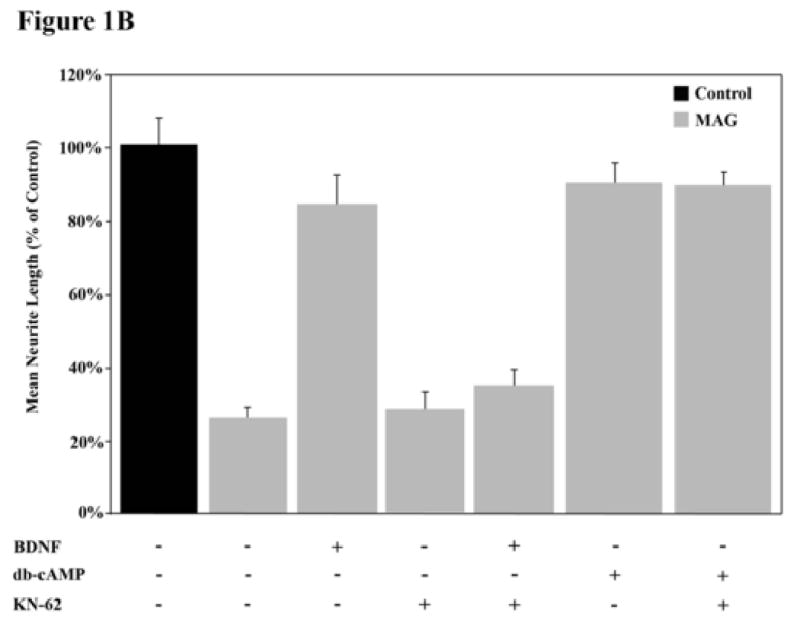
(A) Cerebellar granule neurons (CGNs) were treated with or without BDNF, db-cAMP and/or KN-62 and phosphorylation of CREB was assessed by western blot analysis. (B) CGNs were primed with or without BDNF and grown in the presence (grey bars) or absence (black bars) of MAG and with or without db-cAMP. Data shows the mean length (as a percent of control) for the longest neurite per neuron (±SEM) for 175-200 neurons and represents a composite of 3 separate experiments.
Inhibition of CaMK activity has no effect on young neurons' ability to grow on MAG
Previously, we showed that axon growth by a variety of young neurons, either embryonic or neonatal, depending on the neuron type, are not inhibited by MAG or myelin (Cai et al., 2001; DeBellard et al., 1996). With development, however, each neuronal type switches their response to MAG and become strongly inhibited. Furthermore, the ability of these young neurons to grow in an inhibitory environment and indeed to spontaneously regenerate in vivo, is due to higher endogenous cAMP levels than in their older post-natal counterparts (Cai et al., 2001). For most neurons, this switch has occurred by birth, however, dorsal root ganglion neurons (DRG) switch their responsiveness and show a decrease in endogenous cAMP levels postnatally, at about day 3 or 4 after birth. Therefore, we wanted to assess whether CaMK plays any role in the ability of these young neurons to grow in an inhibitory environment, a situation where cAMP levels are already endogenously elevated. Figure 2 shows that post-natal day 1 (PND1) DRG neurons are not inhibited by MAG; they grow as well on the MAG-expressing CHO cells as they do on the control cells not expressing MAG. When CaMK is inhibited by the addition of KN62, there is no change and the young neurons are still not inhibited by MAG. These results are consistent with what we suggest above, namely, that when cAMP is elevated, activation of CaMK is not necessary for the induction of axonal growth in an inhibitory environment. Previous work from our lab has shown that blocking either PKA or ERK also blocks both the neurotrophin priming-induced and dbcAMP-induced CREB phosphorylation resulting in a block of axonal growth inhibition in older neurons (Gao et al., 2003). This suggests that their effects are downstream from the signaling step that induces the elevation of endogenous cAMP levels. However, unlike these enzymes, CaMK inhibition has no effect on these phenomena once cAMP levels have been elevated via application of db-cAMP or as shown here, in young DRG neurons (Fig.2). These findings concur with the work of Finkbeiner and colleagues who found that CREB activation and transcription can be induced in cortical neurons by application of BDNF and that this effect is CaMKIV-dependent (Finkbeiner et al., 1997). Furthermore, this previously published work also suggests that the BDNF-induced signaling may bifurcate and regulate both CaMKIV and ERK-dependent signaling, as we find is also the case in the neurotrophin-induced priming effect. These findings, while surprising, provide some insight into the precise “location” of CaMKIV in this regeneration-promoting signaling pathway and opens the door for further investigation of this pathway: it is not, as is the case with PKA and ERK, downstream of cAMP, so therefore, it must be either upstream or working in parallel.
Figure 2. CaMK activation is not necessary for the improved axonal outgrowth from uninhibited neurons.
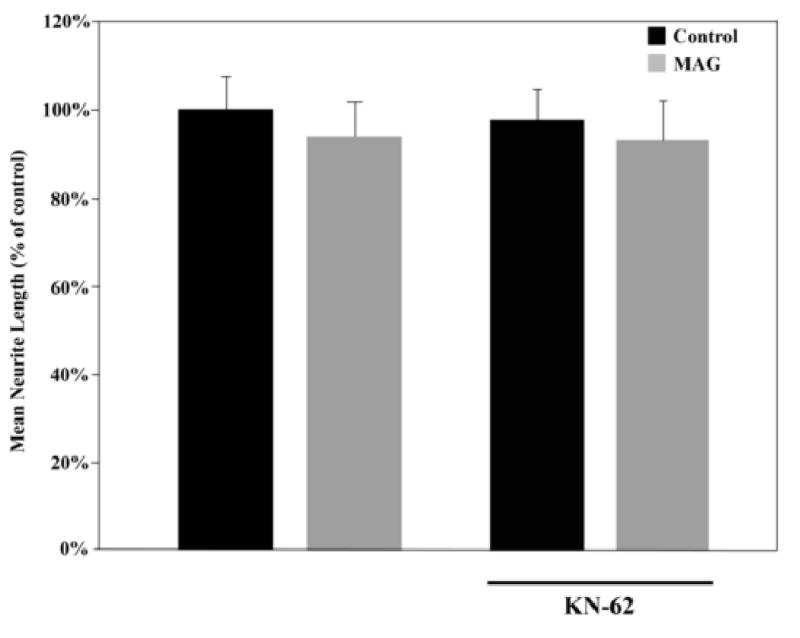
DRG neurons from post-natal day 1 (PND1) rat pups were grown on monolayers of control (black bars) or MAG-expressing (grey bars) CHO cells with or without KN-62. Results indicate the mean length (as a percent of control) for the longest neurite per neuron (±SEM) for 175-200 neurons and represent a composite of 3 separate experiments.
The activation of CaMK and cAMP by BDNF is in parallel, not sequential:
Previously, we showed that the elevation of cAMP and consequential activation of PKA by BDNF is brought about by the inhibition of phosphodiesterase 4 (PDE4) by activated Erk (Nikulina et al., 2004). To test if CaMK is upstream from cAMP, we measured the activation of Erk and the levels of cAMP in response to BDNF, with and without KN62. As reported before by us and others, BDNF activates ERK, and the presence of KN62 has no effect on this activation (Fig.3A). Consistent with this and with our working hypothesis that BDNF must activate Erk to bring about the increase in cAMP, the CaMK inhibitor has no effect on the increase in cAMP levels in neurons in response to BDNF; with or without KN62, BDNF induces approximately a 2-fold increase in cAMP levels (Fig.3B). These results strongly suggest that BDNF activates CaMK and elevates cAMP in parallel but both events are necessary for priming with BDNF to block inhibition by MAG. Furthermore, we also reported that induction of all three enzymes (PKA, ERK and CaMK) is necessary for CREB activation and the resulting improvement in axonal growth, for if any one of these enzymes is inhibited, the BDNF-induced priming effect is lost (Gao et al., 2004); unpublished results, Spencer and Filbin). Thus, we hypothesize that treatment of primary neurons with BDNF induces a multi-faceted signaling cascade which converges on CREB phosphorylation and activation. In this scenario, a “threshold level” of CREB activation is necessary for the induction of transcription required to overcome inhibition by MAG.
Figure 3. CaMK activation does not regulate ERK signaling or cAMP elevation following BDNF treatment.
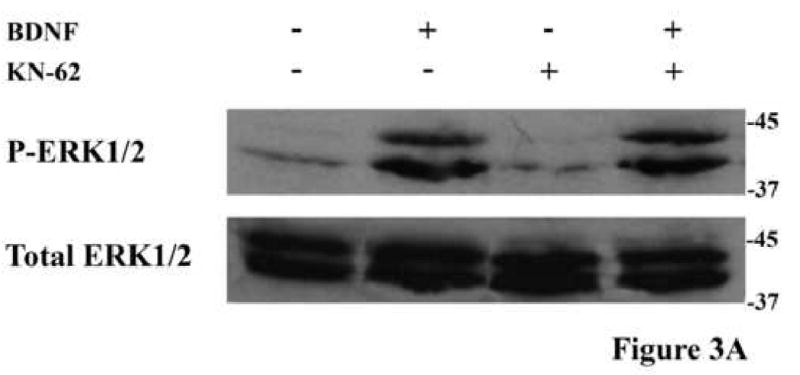
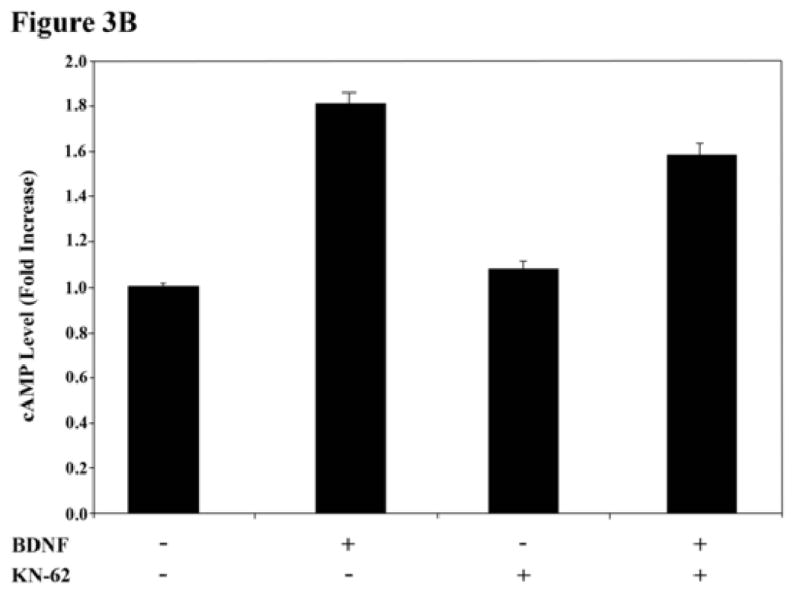
(A) Phosphorylation of ERK1/2 in rat PND5 CGNs was assessed by western blot analysis in response to treatment with BDNF and/or KN-62. Blocking CaMK activation with KN-62 has no effect on the increase in ERK1/2 phosphorylation in response to BDNF. (B) Primary CGNs were treated with or without BDNF and/or KN-62 and cAMP levels (±SEM) were assessed by ELISA. Data represents results from 3 independent experiments.
CaMK signaling is dependent on calcium release from intracellular stores
As the name implies, CaMK requires an influx of calcium to the cytoplasm and induction of the calcium/calmodulin complex to be activated. Therefore, if BDNF is activating this enzyme, it must induce a calcium flux. Increased cytosolic calcium can be achieved by either influx from the extracellular space, through voltage-sensitive channels or certain glutamate receptor channels, or from internal stores via inositol-1,4,5-trisphosphate (IP3)-sensitive channels. Since the mode of calcium flux can often determine the intracellular signaling response which results from CaMK activation (Berridge, 1998), we wanted to know which source of calcium was responsible for mediating the BDNF effect and activating CaMK. To address this issue, inhibitors of glutamate NMDA channels (2-amino-5-phosphonovaleric acid, APV), of voltage-gated channels (nimodipine and nifedipine) or of IP3 channels (2-Aminoethoxydiphenyl borate, 2-APB) were each included during priming of DRG neurons with BDNF and the effect on neurite outgrowth and activation of CREB was assessed. Of the three types of inhibitors, only the IP3 channel inhibitor, 2-APB, blocked the ability of BDNF to both overcome inhibition by MAG and to activate CREB (Fig. 4A and B). From these results we conclude that BDNF induces a flux of calcium from internal stores via IP3-sensitive channels. This mode of flux is in agreement with previous work which reported that treatment of cortical neurons with BDNF can induce CREB activation in a PLCγ-dependent manner (Finkbeiner et al., 1997). If indeed PLCγ is activated and necessary in the priming paradigm, we would then expect calcium flux to be via IP3 channel-mediated release from intracellular stores. This is particularly interesting given that this is the first time that calcium flux of any sort— particularly from intracellular stores—has been implicated in the block of MAG-mediated inhibition of axonal growth.
Figure 4. Identification of the mode of calcium flux which mediates the CaMK effect.
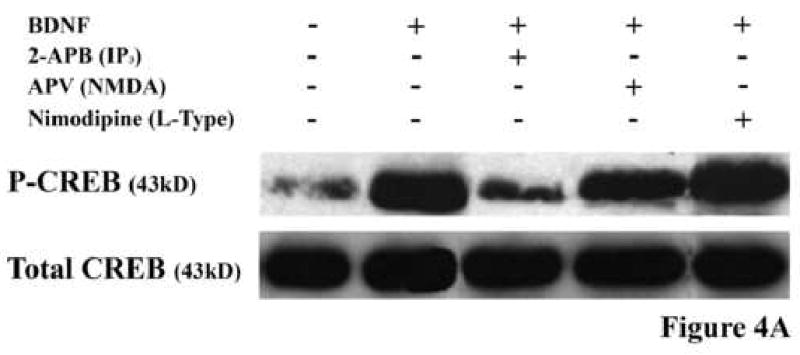
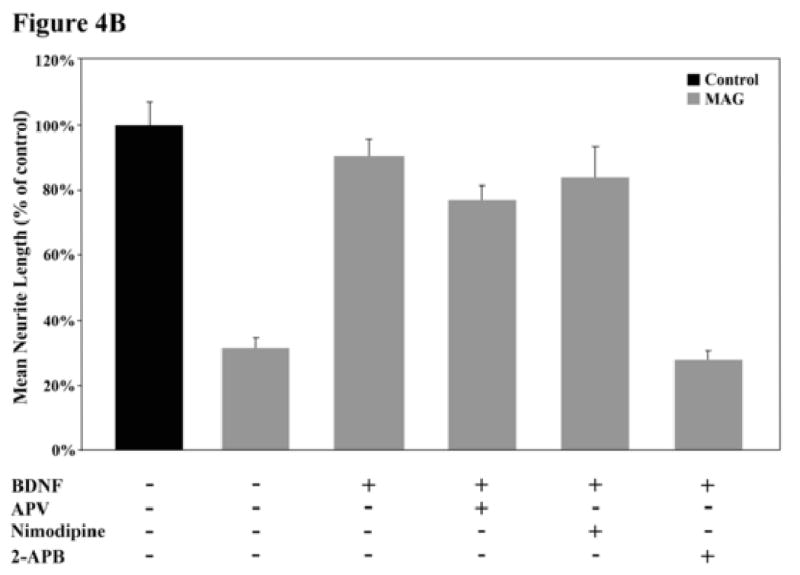
(A) Rat PND5-8 CGNs were treated with or without BDNF and one of the following calcium flux inhibitors: 2-APB, APV or nimodipine. Only the IP3-channel inhibitor, 2-APB, inhibited the BDNF-induced CREB phosphorylation. (B) CGNs were primed with BDNF in the presence or absence of these inhibitors and grown on MAG-expressing (grey bars) or control (black bars) CHO cells. The only inhibitor that blocked the BDNF-induced priming effect was the IP3-channel inhibitor, 2-APB. Histograms show the mean length (as a percent of control) for the longest neurite per neuron (±SEM) for 175-200 neurons and represent a composite of 3 separate experiments.
Dominant-negative CaMKIV blocks the neurotrophin-induced priming effect while a constitutively-active mutant can mimic it
While these experiments provide evidence to suggest that a calcium/calmodulin-dependent kinase is indeed involved in the neurotrophin-induced priming of primary neurons, they do not distinguish between the effects of CaMKII versus CaMKIV, as KN62 blocks both. To address this question directly and to complement the pharmacological data, a dominant-negative (DN) protein specific for CaMKIV (“kinase activity dead”) was expressed in neurons. In addition, a constitutively-active (CA) form of CaMKIV, consisting of a truncated version of the protein which does not require Ca2+/calmodulin binding, was also used. The cDNAs for these proteins were placed in adenoviral vectors, along with green fluorescent protein (GFP) as a reporter (Fig.5A-C). As a control, vectors with only GFP were used. DRG neurons were infected with the various viruses and left for 24 hours to allow expression of the transgenes. The neurons infected with the DN-CaMKIV virus were then primed with BDNF before being transferred to MAG-expressing or control cells for the neurite outgrowth assay. For neurons expressing CA-CaMKIV, neurons were transferred immediately to the MAG or controls cells and the neurite outgrowth assay was carried out.
Figure 5. Adenoviral vectors encoding mutant forms of CaMKIV.
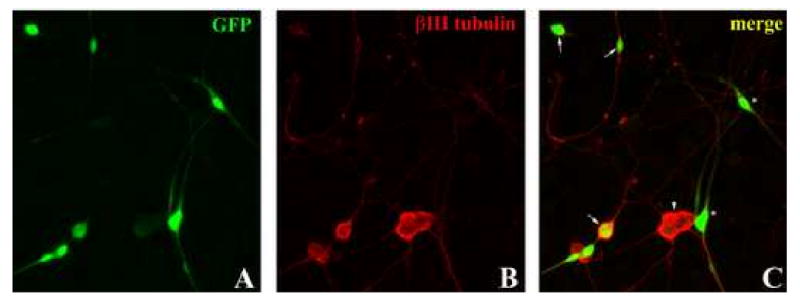

(A-B) PND5-8 rat DRG neurons were infected with mutant CaMKIV-containing adenovirus (including a GFP reporter) prior to priming and neurite outgrowth. (C) Infected DRG neurons were identified as cells which were positive for both GFP and βIII tubulin (arrows). DRG neurons which were not infected (arrowheads) and infected non-neuronal cells (asterisks) were not counted. (D) Following infection, DRG neurons were primed with or without BDNF and grown on MAG-expressing (grey bars) or control (black bars) CHO cells. DRG neurons infected with a dominant-negative CaMKIV (DN) were unable to be primed with BDNF. Cells infected with constitutively-active CaMKIV (CA) extended axonal processes as long as that on control cells even in the absence of BDNF. Results show the mean length (as a percent of control) for the longest neurite per neuron (±SEM) for 175-200 neurons and represent a composite of 3 separate experiments.
Neurons infected with the DN-CaMKIV-containing viruses were still inhibited by MAG even after priming with BDNF (Fig.5D). In sharp contrast, neurons infected with the CA CaMKIV-containing viruses were not inhibited by MAG even without priming with BDNF; the neurons extended neurites of equivalent length on the MAG-cells as on the control cells (Fig.5D). These results indicate that it is indeed CaMKIV that is involved in the ability of priming with BDNF to overcome inhibition by MAG.
The finding that a constitutively-active mutant CaMKIV is sufficient to mimic the priming effect, even in the absence of exogenous treatment, was particularly surprising given that we believe that in response to BDNF CaMK, as well as PKA and ERK, all need to be activated in order to reach the threshold of CREB phosphorylation required to overcome inhibition. One hypothesis to explain this phenomenon may lie in the unique nature of the CaMKIV enzyme. CaMKIV is activated by both Ca2+/calmodulin binding and a CaMK kinase (CaMKK)-mediated phosphorylation event (reviewed in (Soderling and Stull, 2001)). Activation by CaMKK-mediated phosphorylation can lead to prolonged CaMKIV activation, even in the absence of elevated calcium levels and may be necessary to induce gene transcription (Enslen et al., 1994; Enslen et al., 1995). In addition, it has been proposed that autophosphorylation of an autoinhibitory domain may mediate the availability of CaMKIV as a substrate for CaMKK (Chatila et al., 1996; Okuno et al., 1995). Thus, overexpression of a constitutively-active CaMKIV moiety may mimic this calcium-independent activation and signaling which may facilitate further activation of endogenous CaMKIV by CaMKK due to relief of inhibition by autophosphorylation. Therefore, continuous activation of CaMKIV by the constitutively-active mutant may, itself, be sufficient to reach the “threshold” CREB activation, even in the absence of additional signaling.
The functional relevance of CaMKIV activity
The induction of the improved axonal growth on the inhibitory substrate observed in neurons which have been primed with BDNF appears to occur in two stages. The first is a rapid induction of the primary signaling components such as cAMP, PKA and ERK. To this list, we can now add the calcium/calmodulin-dependent kinase. However, while we believe we know precisely how and where these other components fit in the signaling cascade, the role of CaMKIV appears to be a bit more enigmatic. It does not seem to mediate the BDNF-induced elevation of cAMP levels. In addition, CaMKIV activation in response to BDNF is not necessary for the phosphorylation and activation of the extracellular signal regulated kinase (ERK), itself a necessary step in the activation of CREB, cAMP elevation and the block of MAG-mediated inhibition of axonal regeneration. Thus, while we have not yet pinpointed the precise position and role of CaMKIV in the neurotrophin-induced priming effect, we can say that it is not upstream of cAMP elevation and nor is it downstream of cAMP elevation, but rather, must exist in a separate and parallel pathway which leads from BDNF binding to the Trk B receptor to the phosphorylation and activation of CREB and the subsequent induction of transcription. We can, however, say that the elevation of intracellular calcium levels in response to treatment of neurons with BDNF is dependent on flux from intracellular stores and that this pathway is necessary for both CREB phosphorylation and the improved axonal growth on an inhibitory substrate.
It has been reported by others that MAG can also induce spatially-localized calcium flux from intracellular stores and provoke axonal growth cone repulsion (Henley et al., 2004), an effect often considered analogous to axonal growth inhibition. While this may suggest a theoretical contradiction, there are several key differences in the mode of activity of these two phenomena. First, Henley et al. find that this flux occurs exclusively in the growth cone and is limited to the side of the cone nearest the MAG gradient and, thus, appears to be a transient and localized effect more likely to regulate cytoskeletal rearrangement than gene transcription. Furthermore, growth cone guidance effects occur in the order of minutes rather than hours, as is the case with axonal regeneration. Hence, it is highly probable that the immediate effects of intracellular calcium flux are transient and independent of novel gene transcription. Therefore, the difference in the growth cone repulsion and axonal regeneration inhibition effects of MAG appears to be that of acute versus chronic responses to the same signaling agent. But while both axonal repulsion by MAG and the BDNF-induced block of axonal inhibition depend on fluctuations in intracellular calcium levels and cAMP concentrations (Henley et al., 2004; Hong et al., 2000; Ming et al., 1997; Ming et al., 2002; Song et al., 1997), we believe that they differ dramatically in the signaling cascades which they activate and while the former effect occurs quickly and appears to be transient, the latter requires some hours for complete induction (Spencer and Filbin, unpublished observations) and requires new gene transcription.
In summary, we propose a model in which CaMKIV is a necessary component of the BDNF-induced priming effect and that activation of this kinase is mediated by the release of calcium from intracellular stores. The activation of CaMKIV is simultaneous and in parallel to that of PKA and ERK, with all three activities combining to produce sufficient CREB phosphorylation/activation such that the transcription of further downstream effectors, such as arginase I (Cai et al., 2002), can produce a block of the MAG-mediated inhibition of neurite outgrowth. These signaling pathways are similar to that observed in synaptic plasticity and long term potentiation and may suggest a similar mechanism which governs both neuronal paradigms as well as revealing a new potential target for therapeutic intervention for encouraging axonal regeneration following injury.
Materials and Methods
Priming with BDNF and the Neurite Outgrowth Assay
Cerebellar and DRG neurons were isolated and 5 × 105 neurons/per well in a poly-L-lysine-coated 24-well dish were primed overnight with 200ng/ml BDNF with 10μM KN62, 100μM 2-APB, 10μM Nimodipine, 10μM Nifedipine or 100μM APV, included where indicated, as previously described (Cai et al., 1999). Confluent monolayers of MAG-expressing or control CHO cells were established over a 24-hour period in individual chambers of an eight-well tissue culture slide (Lab-Tek) and co-cultures were established as described previously (Doherty et al., 1990; Mukhopadhyay et al., 1994) by tryspinizing the primed neurons and transferring them in Sato to the CHO monolayers. After 16–18 hours at 37°C, the co-cultures were fixed for 30 minutes with 4% paraformaldehyde, and permeabilized with ice-cold methanol for 2 minutes. Blocking was performed with DMEM containing 10% goat serum for 30 minutes and the neurons were then incubated overnight with a monoclonal antibody against βIII tubulin (1:1000; Covance). The cultures were washed with PBS and incubated with biotinylated goat anti-mouse IgG antibody (1:500; Amersham) for 30 minutes and finally, with streptavidin-conjugated Texas Red (1:500; Amersham). The length of the longest neurite per neuron was measured for at least 200 neurons per well using the Compix image analysis software. All animal protocols were approved and supervised by the Hunter College IACUC board.
CREB Activation/Phosphorylation
5 × 106 neurons were starved in DMEM for 2 hours in 6-well poly-L-lysine-coated plates and then treated for 30 minutes at 37°C with 1mM db-cAMP or 200ng/ml BDNF with 10μM KN-62, 10μM nimodipine, 100μM APV or 100μM 2-APB, as indicated. Neurons were lysed in 2% SDS sample buffer 10-20μg protein were loaded per well onto a 10% polyacrylamide gel. Separated proteins were then transferred onto a PVDF membrane (Millipore), incubated with rabbit anti-phosphorylated CREB (Ser133) antibody (1:1000; Cell Signaling) overnight at 4°C. Blots were washed 3 times with PBS-Tween20 and incubated for 1hour at room temperature with shaking in HRP-linked goat anti-rabbit IgG antibody (Cell Signaling) at a dilution of 1:2000. Visualization of the proteins was performed using the ECL chemiluminescence kit (Amersham).
Quantification of cAMP Levels
Cellular cAMP levels were quantified using the BIOMOL Format A cAMP “PLUS” EIA kit (BIOMOL). Neurons were plated onto 6-well poly-L-lysine-coated plates and allowed to adhere overnight at 37°C. The neurons were starved for 2 hours in DMEM and then treated for 30 minutes with 200ng/ml BDNF and/or 10μM KN-62. Cells were lysed with 0.1M HCl and cAMP quantitated as described by the manufacturer.
Recombinant Adenovirus and neuron Infection
Recombinant adenovirus containing CaMKIV mutant constructs were constructed by sub-cloning the cDNAs (kindly provided by Dr. Talal Chatila of the Mattel Children's Hospital, UCLA) into pTRACK CMV and then inserting it by homologous recombination into pAdeasy-1 and the viral preparation and purification was carried out as described (He et al., 1998). DRG neurons were plated onto poly-l-lysine coated 24-well plates and infected with adenovirus at 100 Fluorescent units (Fu) / cell. After overnight culture neurons were either primed with neurotrophins or transferred directly to monolayers of CHO cells and fixed and stained as described above. Neurite length was measured only from those neurons that were both βIII tubulin and GFP positive.
Acknowledgments
We thank Marie Handler and Arkadiy Palvanov for their outstanding technical support, Dr. Talal Chatila for the CaMKIV mutant cDNA constructs, Dr. Lloyd Williams for his help with the image analysis and Dr. Christine Cain for assistance with the manuscript. This work was supported by grants from the New York State Spinal Cord fund, the NIH NS 37060, the National Multiple Sclerosis Society, and core facility grants from the Research Centers for Minorities Institute-NIH and Specialized Neuroscience Research Programs-NIH (NS41073).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersen PL, Webber CA, Kimura KA, Schreyer DJ. Cyclic AMP prevents an increase in GAP-43 but promotes neurite growth in cultured adult rat dorsal root ganglion neurons. Exp Neurol. 2000;166:153–65. doi: 10.1006/exnr.2000.7485. [DOI] [PubMed] [Google Scholar]
- Bach ME, Hawkins RD, Osman M, Kandel ER, Mayford M. Impairment of spatial but not contextual memory in CaMKII mutant mice with a selective loss of hippocampal LTP in the range of the theta frequency. Cell. 1995;81:905–15. doi: 10.1016/0092-8674(95)90010-1. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–14. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- Cai D, Deng K, Mellado W, Lee J, Ratan RR, Filbin MT. Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron. 2002;35:711–9. doi: 10.1016/s0896-6273(02)00826-7. [DOI] [PubMed] [Google Scholar]
- Cai D, Qiu J, Cao Z, McAtee M, Bregman BS, Filbin MT. Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci. 2001;21:4731–9. doi: 10.1523/JNEUROSCI.21-13-04731.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Shen Y, De Bellard M, Tang S, Filbin MT. Prior exposure to neurotrophins blocks inhibition of axonal regeneration by MAG and myelin via a cAMP-dependent mechanism. Neuron. 1999;22:89–101. doi: 10.1016/s0896-6273(00)80681-9. [DOI] [PubMed] [Google Scholar]
- Chatila T, Anderson KA, Ho N, Means AR. A unique phosphorylation-dependent mechanism for the activation of Ca2+/calmodulin-dependent protein kinase type IV/GR. J Biol Chem. 1996;271:21542–8. doi: 10.1074/jbc.271.35.21542. [DOI] [PubMed] [Google Scholar]
- Chen MS, Huber AB, van der Haar ME, Frank M, Schnell L, Spillmann AA, Christ F, Schwab ME. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature. 2000;403:434–9. doi: 10.1038/35000219. [DOI] [PubMed] [Google Scholar]
- DeBellard ME, Tang S, Mukhopadhyay G, Shen YJ, Filbin MT. Myelin-associated glycoprotein inhibits axonal regeneration from a variety of neurons via interaction with a sialoglycoprotein. Mol Cell Neurosci. 1996;7:89–101. doi: 10.1006/mcne.1996.0007. [DOI] [PubMed] [Google Scholar]
- Doherty P, Cohen J, Walsh FS. Neurite outgrowth in response to transfected N-CAM changes during development and is modulated by polysialic acid. Neuron. 1990;5:209–19. doi: 10.1016/0896-6273(90)90310-c. [DOI] [PubMed] [Google Scholar]
- Enslen H, Sun P, Brickey D, Soderling SH, Klamo E, Soderling TR. Characterization of Ca2+/calmodulin-dependent protein kinase IV. Role in transcriptional regulation. J Biol Chem. 1994;269:15520–7. [PubMed] [Google Scholar]
- Enslen H, Tokumitsu H, Soderling TR. Phosphorylation of CREB by CaM-kinase IV activated by CaM-kinase IV kinase. Biochem Biophys Res Commun. 1995;207:1038–43. doi: 10.1006/bbrc.1995.1289. [DOI] [PubMed] [Google Scholar]
- Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci. 2003;4:703–13. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–47. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- Gao Y, Deng K, Hou J, Bryson JB, Barco A, Nikulina E, Spencer T, Mellado W, Kandel ER, Filbin MT. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron. 2004;44:609–21. doi: 10.1016/j.neuron.2004.10.030. [DOI] [PubMed] [Google Scholar]
- Gao Y, Nikulina E, Mellado W, Filbin MT. Neurotrophins elevate cAMP to reach a threshold required to overcome inhibition by MAG through extracellular signal-regulated kinase-dependent inhibition of phosphodiesterase. J Neurosci. 2003;23:11770–7. doi: 10.1523/JNEUROSCI.23-37-11770.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–3. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- GrandPre T, Nakamura F, Vartanian T, Strittmatter SM. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature. 2000;403:439–44. doi: 10.1038/35000226. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–14. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley JR, Huang KH, Wang D, Poo MM. Calcium mediates bidirectional growth cone turning induced by myelin-associated glycoprotein. Neuron. 2004;44:909–16. doi: 10.1016/j.neuron.2004.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong K, Nishiyama M, Henley J, Tessier-Lavigne M, Poo M. Calcium signalling in the guidance of nerve growth by netrin-1. Nature. 2000;403:93–8. doi: 10.1038/47507. [DOI] [PubMed] [Google Scholar]
- Kottis V, Thibault P, Mikol D, Xiao ZC, Zhang R, Dergham P, Braun PE. Oligodendrocyte-myelin glycoprotein (OMgp) is an inhibitor of neurite outgrowth. J Neurochem. 2002;82:1566–9. doi: 10.1046/j.1471-4159.2002.01146.x. [DOI] [PubMed] [Google Scholar]
- Matthews RP, Guthrie CR, Wailes LM, Zhao X, Means AR, McKnight GS. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol Cell Biol. 1994;14:6107–16. doi: 10.1128/mcb.14.9.6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–83. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- McKerracher L, David S, Jackson DL, Kottis V, Dunn RJ, Braun PE. Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron. 1994;13:805–11. doi: 10.1016/0896-6273(94)90247-x. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song HJ, Berninger B, Holt CE, Tessier-Lavigne M, Poo MM. cAMP-dependent growth cone guidance by netrin-1. Neuron. 1997;19:1225–35. doi: 10.1016/s0896-6273(00)80414-6. [DOI] [PubMed] [Google Scholar]
- Ming GL, Wong ST, Henley J, Yuan XB, Song HJ, Spitzer NC, Poo MM. Adaptation in the chemotactic guidance of nerve growth cones. Nature. 2002;417:411–8. doi: 10.1038/nature745. [DOI] [PubMed] [Google Scholar]
- Monsul NT, Geisendorfer AR, Han PJ, Banik R, Pease ME, Skolasky RL, Jr, Hoffman PN. Intraocular injection of dibutyryl cyclic AMP promotes axon regeneration in rat optic nerve. Exp Neurol. 2004;186:124–33. doi: 10.1016/S0014-4886(03)00311-X. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT. A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron. 1994;13:757–67. doi: 10.1016/0896-6273(94)90042-6. [DOI] [PubMed] [Google Scholar]
- Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI. Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron. 2002;34:885–93. doi: 10.1016/s0896-6273(02)00702-x. [DOI] [PubMed] [Google Scholar]
- Nikulina E, Tidwell JL, Dai HN, Bregman BS, Filbin MT. The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc Natl Acad Sci U S A. 2004;101:8786–90. doi: 10.1073/pnas.0402595101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno S, Kitani T, Fujisawa H. Full activation of brain calmodulin-dependent protein kinase IV requires phosphorylation of the amino-terminal serine-rich region by calmodulin-dependent protein kinase IV kinase. J Biochem. 1995;117:686–90. doi: 10.1093/oxfordjournals.jbchem.a124764. [DOI] [PubMed] [Google Scholar]
- Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT, Bunge MB. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med. 2004;10:610–6. doi: 10.1038/nm1056. [DOI] [PubMed] [Google Scholar]
- Prinjha R, Moore SE, Vinson M, Blake S, Morrow R, Christie G, Michalovich D, Simmons DL, Walsh FS. Inhibitor of neurite outgrowth in humans. Nature. 2000;403:383–4. doi: 10.1038/35000287. [DOI] [PubMed] [Google Scholar]
- Qiu J, Cai D, Dai H, McAtee M, Hoffman PN, Bregman BS, Filbin MT. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron. 2002;34:895–903. doi: 10.1016/s0896-6273(02)00730-4. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–61. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992a;257:206–11. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992b;257:201–6. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- Soderling TR, Stull JT. Structure and regulation of calcium/calmodulin-dependent protein kinases. Chem Rev. 2001;101:2341–52. doi: 10.1021/cr0002386. [DOI] [PubMed] [Google Scholar]
- Song HJ, Ming GL, Poo MM. cAMP-induced switching in turning direction of nerve growth cones. Nature. 1997;388:275–9. doi: 10.1038/40864. [DOI] [PubMed] [Google Scholar]
- Sun P, Enslen H, Myung PS, Maurer RA. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994;8:2527–39. doi: 10.1101/gad.8.21.2527. [DOI] [PubMed] [Google Scholar]
- Wang KC, Koprivica V, Kim JA, Sivasankaran R, Guo Y, Neve RL, He Z. Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature. 2002;417:941–4. doi: 10.1038/nature00867. [DOI] [PubMed] [Google Scholar]
- Wei F, Qiu CS, Liauw J, Robinson DA, Ho N, Chatila T, Zhuo M. Calcium calmodulin-dependent protein kinase IV is required for fear memory. Nat Neurosci. 2002;5:573–9. doi: 10.1038/nn0602-855. [DOI] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR. Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron. 1999;23:787–98. doi: 10.1016/s0896-6273(01)80036-2. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Fujitani M, Yamagishi S, Hata K, Mimura F. Multiple signals regulate axon regeneration through the nogo receptor complex. Mol Neurobiol. 2005;32:105–11. doi: 10.1385/MN:32:2:105. [DOI] [PubMed] [Google Scholar]