

Summary
Atomic level structural information on αB-Crystallin (αB), a prominent member of the small Heat Shock Protein (sHSP) family has been a challenge to obtain due its polydisperse, oligomeric nature. We show that magic-angle spinning solid-state NMR can be used to obtain high-resolution information on ∼ 580 kDa human αB assembled from 175-residue, 20 kDa subunits. An ∼100-residue α-crystallin domain is common to all sHSPs and solution-state NMR was performed on two different α-crystallin domain constructs isolated from αB. In vitro, the chaperone-like activities of full-length αB and the isolated α-crystallin domain are identical. Chemical shifts of the backbone and the Cβ resonances have been obtained for residues 64-162 (α-crystallin domain plus part of the C-terminus) in αB and the isolated α-crystallin domain by solid- and solution-state NMR, respectively. Both sets of data strongly predict six β-strands in the α-crystallin domain. A majority of residues in the α-crystallin domain have similar chemical shifts in both solid- and solution-state indicating a similar structure for the domain in its isolated and oligomeric forms. Sites of inter-subunit interaction are identified from chemical shift differences that cluster to specific regions of the α-crystallin domain. Multiple signals are observed for the resonances of M68 in the oligomer, identifying the region containing this residue as existing in heterogeneous environments within αB. Evidence for a novel dimerization motif in the human α-crystallin domain is obtained by a comparison of (i) solid- and solution-state chemical shift data and (ii) 1H-15N HSQC spectra as a function of pH. The isolated α-crystallin domain undergoes a dimer-monomer transition over the pH range of 7.5 to 6.8. This steep pH-dependent switch may be important for αB to function optimally, e.g., to preserve the filament integrity of cardiac muscle proteins such as actin and desmin during cardiac ischemia which is accompanied by acidosis.
Keywords: αB-Crystallin, chaperone, sHSP, solid-state NMR, solution-state NMR
Introduction
αB-Crystallin (αB) forms a polydisperse oligomer of ∼580 kDa assembled from 175-residue, 20 kDa subunits and has diverse biological roles. αB was originally identified as the basic B subunit of α-crystallin, a protein that is essential for maintaining the transparency of the eye-lens. As a stress chaperone belonging to the small Heat Shock Protein family (sHSP), αB has been shown to prevent aggregation of denatured proteins in vitro.1 αB is also expressed in other tissues, most predominantly in cardiac and skeletal muscle and in the brain. A major constituent of cardiac muscle cells (3-5%), αB has been shown to associate with myofibrillar proteins such as titin, desmin, and actin in vivo.2,3 αB translocates from the cytosol of the muscle cells to the myofibrils as a result of cardiac ischemia, implicating αB in the preservation of filament integrity of the myofibrillar proteins. A missense mutation in αB, Arg120 to Gly, is associated with desmin-related cardiomyopathy, a disease characterized by aggregates of desmin and R120G-αB in heart tissue.4 C-terminal truncations in αB are also associated with myofibrillar myopathies.5 R120G-αB forms larger oligomers than αB6 and C-terminally truncated mutations have decreased solubility.7 In vitro, R120G-αB was shown to be a less effective chaperone than αB. The structural basis for the altered properties associated with the mutations is unknown.
More recently, several new biological roles of αB have been reported. The SCF (Skp1/Cul1/F-box) E3 ligase regulates cell cycle progression at the G1/S phase transition by poly-ubiquitinating phosphorylated Cyclin D1, uncontrolled expression of which is associated with lymphomas, breast, and esophageal cancers. αB was found to be an essential cofactor for FBX4, a substrate-specific subunit of SCF.8 Another report showed that αB reverses the effects of Multiple Sclerosis in a mouse model of the disease, a finding that could potentially lead to its therapeutic use.9 In a more recent study, αB was identified as the cytosolic interaction partner of Ksp (kidney-specific)-cadherin implying that this interaction is important for maintaining tissue integrity in the human kidney.10
The functional diversity of αB is mirrored in its structural plasticity, the very nature of which has proven refractory for structure determination by x-ray crystallography. The structure of αB has therefore remained an enigma for decades. Lack of structural information characterizes the small heat shock protein field in general: structures of only three small heat shock proteins have been solved to date despite more than 100 sHSPs identified in the genomes of organisms from bacteria to human. The primary structures of sHSPs have a common organization comprised of a variable N-terminal region, a conserved ∼100-residue α-crystallin domain, and a variable C-terminal region. In the solved structures, a dimer comprised of the α-crystallin domain forms a fundamental building block of the oligomer while interactions involving the N- and C-terminal regions with the α-crystallin domain lead to higher order oligomeric structures. Variations in the length and sequence of the N- and C-terminal regions are thought to give rise to different oligomeric structures. MjHSP16.5 (from Methanococcus jannaschii) forms a 24-subunit sphere11 and wHSP16.9 (from wheat, Triticum aestivum) forms a double disk dodecameric oligomer.12 In contrast, Tsp36 (from beef tapeworm, Taenia saginata) forms only a dimer.13 Tsp36 also has an unusual domain organization in which the N-terminal region is followed by two α-crystallin domains (ACD1 and ACD2) and there is no C-terminal region.
The α-crystallin domain is fairly conserved across species: the sequence similarity ranges from 19% to 60%. In structures determined thus far, the α-crystallin domain adopts a 7-stranded β-sandwich fold. The α-crystallin domains from MjHSP16.5 and wHSP16.9 both form domain-swapped dimers. On the other hand, ACD1 and ACD2 in Tsp36 are stacked end-to-end to form a single subunit of the dimer.
The requirement for long range order in crystals posed by x-ray crystallography and the size limitation posed by solution-state NMR make structure determination of heterogeneous, oligomeric supramolecules a challenge. Solid-state NMR is rapidly emerging as a technique capable of providing high-resolution structural information on proteins and has the potential to be applicable to heterogeneous oligomers. Here we report a hybrid solution- and solid-state NMR study aimed at generating structural information on human αB. Solution-state NMR was applied to an 89-residue α-crystallin domain construct that forms a well-behaved dimer in solution; full-length αB was investigated by solid-state NMR. Backbone resonance assignments obtained from both solution- and solid-state NMR data demonstrate that the secondary structure of the α-crystallin domain in αB is maintained in the dimeric construct. Comparison of solution- and solid-state NMR data suggests a novel dimerization interface for αB and identifies inter-subunit interaction sites within the oligomer. The isolated α-crystallin domain exhibits a steep pH-dependent dimer-monomer transition that provides additional evidence for the dimerization interface and may be critically linked to the optimal function of the oligomer. The known heterogeneity of αB oligomers is reflected in solid-state NMR data but does not undermine the ability to extract structural information.
Results
Optimization and characterization of the α-crystallin domain constructs
Optimization of the human α-crystallin domain construct and the conditions used for acquiring NMR spectra such as pH, temperature, and salt was critical for successful solution NMR studies. Two different α-crystallin domain constructs were used in solution NMR studies as shown in Figure 1. We began our studies with αB11.4 (residues 57-158, molecular weight =11.4 kDa) that had previously been identified as a minimally stable proteolysis product of human αB.14 The 1H-15N HSQC spectra showed a steep pH- and temperature-dependent behavior. Temperatures below 35 °C gave rise to extremely broad peaks in the spectrum. Considerable peak doubling was present in the spectrum at pH 6.5 which was abrogated by increasing the pH to 7.5. Hence we proceeded to characterize the molecule at pH 7.5 and 35 °C. The correlation time, τc, determined from the average 15N R2/R1 ratio gives a reasonable estimate of the multimeric state of a protein. The value of τc for αB11.4 at pH 7.5 and 35 °C was found to be 13.5 ± 0.5 ns at a protein concentration of 1 mM which corresponds more closely to a dimeric species (2 × 11.4). In comparison, the reported values of τc at 35 °C for some proteins are as follows: (i) human thioredoxin, 4.7 ns (11.8 kDa), 15 (ii) staphylococcal nuclease, 9.1 ns (18 kDa), 16 and Escherichia coli (E. coli) adenylate kinase, 10.3 ns (corrected for temperature, 23.6 kDa). 17 Sedimentation velocity experiments confirmed that αB11.4 exists predominantly as a dimer at pH 7.5 and behaves as a monomer at pH 6.5 (data not shown). The presence of higher order aggregates at pH 7.5 was also observed in the sedimentation velocity experiments, consistent with the higher τc value. TROSY-based triple resonance spectra of perdeuterated αB11.4 contained only 40-50% of the expected peaks, greatly hampering efforts to assign the spectrum. We therefore searched for a construct that would be more amenable to solution NMR studies.
Figure 1.
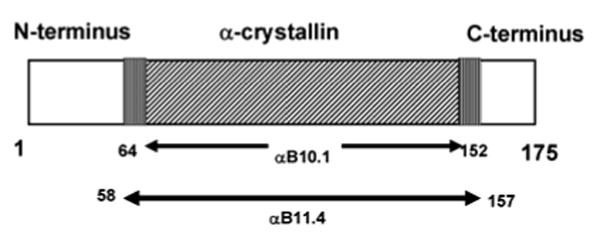
“Domain” organization of αB and the α-crystallin domain constructs used in solution-state NMR studies.
Previous work on the related protein, αA-Crystallin (αA) that shares 54% sequence identity with αB has shown that 36- and ∼10-residues N- and C-terminal to the α-crystallin domain, respectively, are responsible for forming polydisperse oligomers. 18 We found that removal of residues on both ends of the αB11.4 construct to make αB10.1 (residues 64-152, molecular weight = 10.1 kDa) gave greatly improved NMR spectra. Gel filtration profiles showed similar pH-dependent monomer-dimer transition for αB10.1 to that observed for αB11.4. However, for the shorter construct, the 1H- 15N spectra at pH 7.5 were further improved by lowering the temperature to 19 °C. At this temperature, the value of τc was determined to be 17.5 ± 1 ns for αB10.1. Correcting for temperature and viscosity of water for the three proteins listed above (τc increases with decreasing temperature and increasing viscosity), a τc value of 14-15 ns is expected for dimeric αB10.1 (2 × 10.1 kDa). Hence the measured value of τc obtained for αB10.1 still indicates the presence of some higher molecular weight species. A variant, N146D-αB10.1, with a τc value of 15 ns gives spectra that are highly similar to αB10.1. The structure of the variant is thus virtually identical to the wild-type (wt) construct but has a lower propensity to form aggregates under NMR conditions. We therefore chose N146D-αB10.1 for solution-state NMR experiments. To further confirm the multimeric state of N146D-αB10.1, sedimentation velocity experiments were performed. Over a concentration range of 8 μM to 170 μM, N146D-αB10.1 exists as a dimeric species at 20 °C in 50 mM sodium phosphate buffer (pH 7.5) containing 100 mM Na2SO4.
sHSPs are thought to sequester proteins partially denatured by cellular stress until refolding by ATP-dependent chaperones is initiated. The ability of sHSPs to serve as chaperones is usually assessed in vitro by their ability to inhibit aggregate formation, often measured by light scattering. A chemically- or heat-denatured protein is used as the substrate and light scattering is monitored at either 340 or 360 nm. We used DTT-denatured α-Lactalbumin (α-Lac) as the substrate and followed light scattering at 340 nm as a function of time (Figure 2). In the absence of either αB or αB10.1, solutions of α-Lac began to scatter light after about 1 hr. The increase in scattering is fairly linear until it begins to plateau after ∼4 hrs. In stark contrast, the presence of an equimolar amount (protein concentrations of 0.1 mM were used) of either αB or αB10.1 completely abrogates the appearance of light scattering over the time course of the assay (∼5 hrs). The simplest interpretation of the observations is that α-Lac does not form large aggregates when either αB or αB10.1 is present. αB10.1 is dimeric under the assay conditions, indicating that an α-crystallin domain dimer is sufficient for in vitro chaperone activity at protein concentrations used in this assay, viz., 0.1 mM. N146D-αB10.1 was indistinguishable from wt-αB10.1 in the in vitro chaperone assay (data not shown).
Figure 2.

Chaperone-like activity of αB10.1 and αB towards DTT-denatured α-Lac. Relative light scattering intensity curves measured at 340 nm (A340 nm) at 42 °C as a function of time for α-Lac (◆), 1:1 α-Lac:αB (∗), and 1:1 α-Lac:αB10.1 are shown. 0.1 mM protein samples in 50 mM sodium phosphate buffer (pH 7.5) containing 100 mM NaCl, 1 mM EDTA, and 15 mM DTT were used. No light scattering from denatured α-Lac is observed in the presence of either αB10.1 or αB indicating that both the α-crystallin domain and the oligomer confer equal protection from aggregation under the conditions of the experiment.
Solution-State NMR Resonance Assignments of αB10.1
In spite of the relatively small number of resonances, assignment of N146D-αB10.1 proved challenging and required perdeuteration in conjunction with the application of TROSY-based triple resonance experiments. An overlay of 1H-15N HSQC spectra of the variant and wt α-crystallin domains is shown in Figure 3a. The spectra are well-dispersed, indicative of stably folded domains. Figure 3b shows the 1H-15N TROSY-HSQC spectrum of [2H,13C,15N]-N146D-αB10.1; 80 out of 84 non-proline backbone amide groups are observed. Complete backbone assignments were obtained from triple resonance TROSY-based spectra except for residues Gly64, Leu65, His83, Phe84, His119, R120, and S139. The assignments are indicated with residue numbers in Figure 3b. Transfer of assignments from the variant to the wt protein was easily achieved by comparing 3D TROSY-HNCACB spectra of the two proteins. Only a few amide resonances exhibit chemical shift changes between the variant and wt and these are labeled in Figure 3a.
Figure 3.
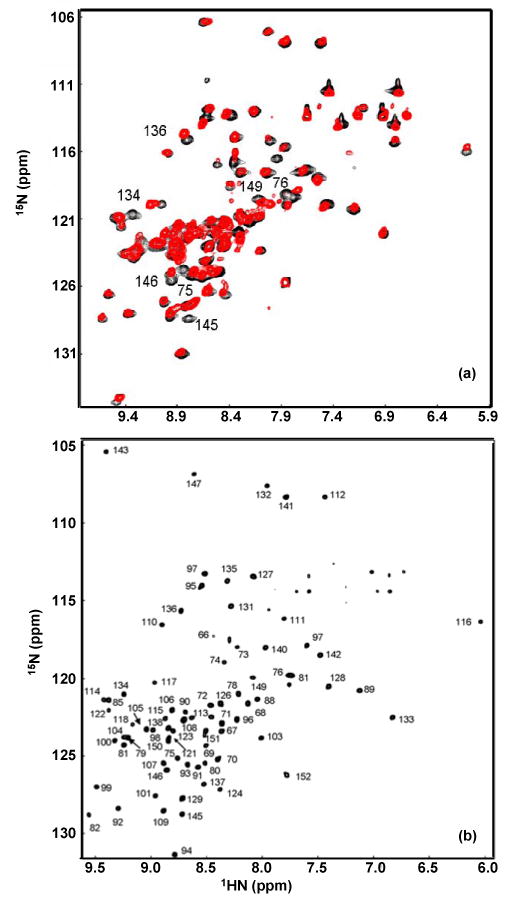
(a). 1H-15N HSQC overlay of 15N-αB10.1 (red) and 15N-N146D-αB10.1 (black). The spectra were acquired on Bruker DMX500 at 22 °C on 0.5 mM samples in 50 mM sodium phosphate buffer (pH 7.5) containing 100 mM Na2SO4. 32 transients per t1 value were acquired. (b). 1H-15N TROSY-HSQC spectrum of [2H, 13C, 15N]-N146D-αB10.1 acquired on Varian Inova 800 on a 1 mM sample. 8 transients per t1 value were acquired. Other conditions are similar to that described in (a). The spectral width in (b) is smaller compared to (a) and so the most downfield peak in (a) corresponding to L143 is aliased in (b). Peaks in (b) are labeled with assignments. Resonances exhibiting chemical shift changes between the wt and the variant spectra are labeled in (a). These resonances belong to residues in β3, β8, and β9 as expected for the topology β3-β9-β8.
Solid-State NMR Resonance Assignments of αB
Full-length αB was precipitated by different methods to obtain spectra suitable for solid-state NMR studies. In Figure 4, 1D 13C spectra of U-αB precipitated from a solution with PEG 8000 (a) and lyophilized U-αB (b) are shown. Resolved Cβ resonances of threonines at ∼ 70 ppm and methyl resonances of isoleucines at ∼ 15 ppm are visible in Figure 4a.
Figure 4.
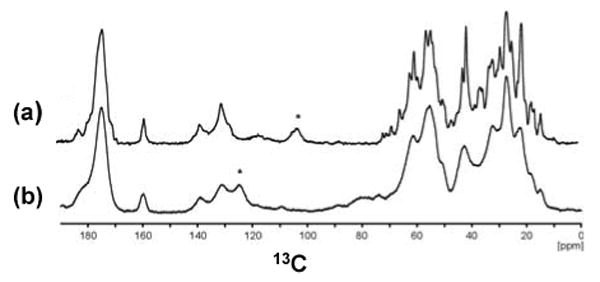
1D 13C CPMAS spectra of U-αB obtained from different sample preparations. (a). αB precipitated from a solution with PEG8000. (b). Lyophilized αB. Both spectra were recorded with ∼85 kHz 1H decoupling and 15 ms acquisition time at 12 °C. During CP, a field of ∼60 kHz and ∼50 kHz was applied on 13C and 1H, respectively. The spectrum in (a) was recorded at a field of 16.4 Tesla and a MAS frequency of ωr=10 kHz. The spectrum in (b) was recorded at a field of 18.8 Tesla and a MAS frequency of ωr=8 kHz. Spinning sidebands are marked with an asterisk.
3D NCACX, NCACB, and NCOCX triple resonance experiments and 2D 13C-13C correlation experiments were acquired on three samples: (1) U-αB, (2) 1,3-αB, and (3) 2-αB (see Materials and Methods). The spectra of the spin-diluted samples, 2-αB and 1,3-αB, show reduced complexity with different labelling patterns for the various amino acid types19 which was exploited for the assignment strategy.
The sequential assignment strategy is illustrated for the segment L131-T132-I133-T134-S135-S136-L137 in Figure 5. The sequential walk starts at the 15N frequency of L137 in an NCACX spectrum of U-αB (Figure 5a). The amino-acid type is identified from characteristic chemical shifts, which is confirmed by the following assignment procedure. At the same 15N frequency in the NCOCX spectrum of 1,3-αB (Figure 5b), there is a cross peak at 66.0 ppm (labeled 1), characteristic for serine Cβ, and a signal at 52.9 ppm (labeled 2). Since only the carbons indicated by green letters in Figure 5 are labeled in 1,3-αB, the signal at 52.9 ppm is attributed to a sequential transfer from S136 C′ to L137 Cα. The Cα resonance of S136 is found at 56.7 ppm in the NCOCX spectrum of U-αB at the 15N frequency of L137 (Figure 5c). These assignments are further confirmed by the cross peaks observed in the NCACB spectrum of U-αB: at the 15N frequency of S136, the intra-residue Cα-Cβ correlation is observed (Figure 5d). In the NCOCX spectrum of U-αB at the S136 15N frequency (Figure 5e), the C′(173.3 ppm), Cα(56.9 ppm) and Cβ (66.9 ppm) resonances of S135 are identified. The Cα and Cβ shifts of S135 are further confirmed by the cross peaks observed in the NCACB spectrum at the 15N frequency of S135 (Figure 5f). In the NCACX spectrum of 2-αB at the 15N frequency of S135 (Figure 5g), correlations between S135 Cα and a Cα peak at 60.1 ppm (labeled 3) and a C′ cross peak at 173.0 ppm (labeled 4) are observed. In the NCOCX spectrum of U-αB at the S135 15N frequency (Figure 5h), a cross peak between a Cα peak at 60.1 ppm, and a C′ peak at 173.0 ppm is observed. In addition, we observe a cross peak involving a Cβ resonance at 72.5 ppm, characteristic for a Thr residue. Thus, the sequential Cα-Cα (labeled 4) and Cα-C′ (labeled 3) cross peaks in Figure 5g connect S135 and T134 unambiguously. The assignments for T134 Cα, Cβ and C′ are further confirmed by the NCACX spectrum of U-αB (Figure 5i) at the 15N frequency of T134. A sequential Cα-C′ cross peak (labeled 5) is observed in an NCACX spectrum recorded on 2-αB at the 15N frequency of T134, which we assigned to C′ of I133 (Figure 5j). This assignment is further confirmed by the NCOCX spectrum of U-αB, where at the T134 15N frequency (Figure 5k), correlations characteristic of isoleucine involving the C′ resonance of I133 and the Cα(60.5 ppm), Cβ(37.7 ppm), Cγ1(27.5 ppm) and Cγ2(18.2 ppm) resonances are observed. The 15N frequency of I133 can be found by searching for matching Cα and Cβ frequencies in an NCACB spectrum of U-αB (Figure 5l). Correlations are observed between the 15N resonances of I133 and the 13C resonances of T132 in the NCOCX spectrum (Figure 5m). As in the case of I133, the 15N frequency of T132 is found by searching for a plane that contains matching 13C resonances (NCACX spectrum in Figure 5n). Following a similar procedure, L131 can be sequentially connected with T132. Figure 5o shows cross-peaks between the 15N resonance of T132 and the Cα(56.7 ppm), Cβ (41.0 ppm), Cγ (27.6 ppm) and C′(178.6 ppm) resonances of L131. At the 15N frequency of L131, the same L131 Cα, Cβ, Cγ and C′ cross peaks are observed in the NCACX spectrum recorded on U-αB (Figure 5p). The assignment of L131 is further corroborated by sequential correlations between L131 Cα and T132 Cα (labeled 6) and T132 Cβ (labeled 7) in the NCACX spectrum of 1,3-αB at the 15N frequency of L131 (Figure 5q).
Figure 5.
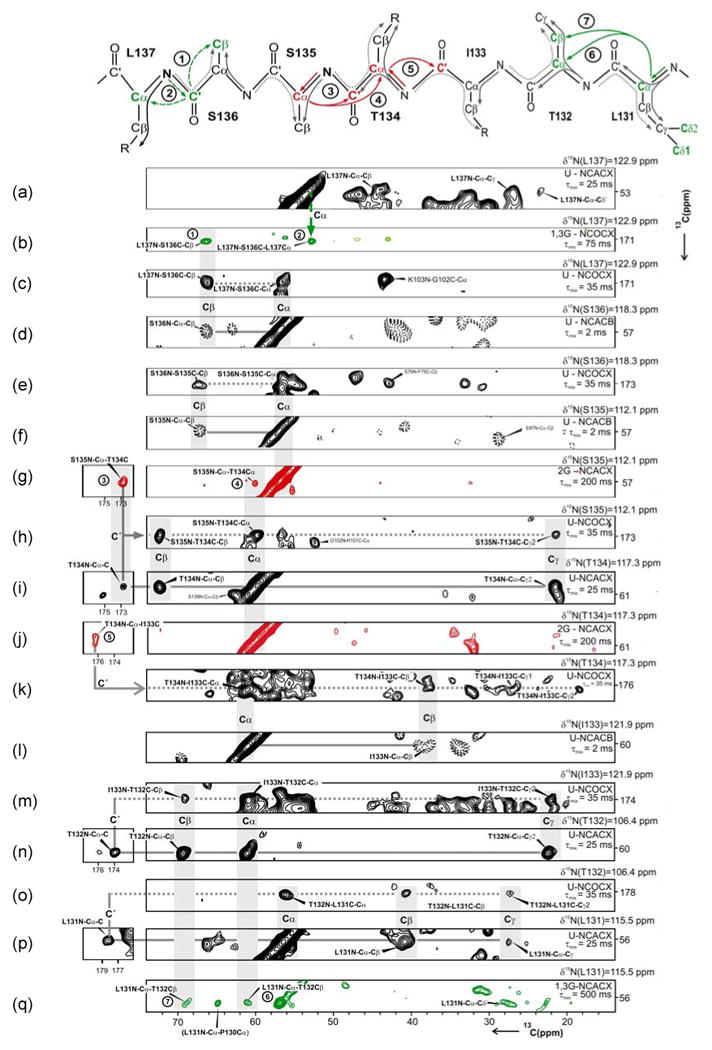
Strips extracted from various solid-state 3D triple resonance spectra are shown. The assignment of the fragment, L137 to L131 using strips from 3D NCACX, NCACB and NCOCX spectra and the analysis of sequential cross peaks derived from 1,3-αB and 2-αB samples are illustrated. The labeling scheme, experiment type, and the mixing time are indicated on the right hand side. The experiment type is indicated with NCOCX, NCACX or NCACB with the prefixes U, 1,3G, and 2G, indicating spectra acquired on U-, 1,3-, and 2-αB samples, respectively. In NCACX spectra, the 15N and Cα resonances of residue i are correlated with the side chain of residue i. In NCACB, the 15N and Cα resonances of residue i are correlated with the Cβ of residue i. NCOCX spectra provide the corresponding sequential information and correlate 15N of residue i and C′ of residue i-1 with the side chain of residue i-1. (See main text for details).
As illustrated above, the differential labeling patterns in the spectra acquired on 1,3-αB and 2-αB samples lead to unambiguous assignments. The improved resolution in spectra obtained by the partial removal of J-couplings as described in Materials and Methods facilitated the assignment process further. Use of these procedures resulted in almost complete assignments for the α-crystallin domain within the full-length αB from solid-state NMR data. The confidence level of the assignments from solid state reported here (Supplementary Data, Table S1) is close to 100% since no other assignments were possible. Some side-chain carbon resonances such as Arginine Cδ, Lysine Cε, and some aromatic side-chains could not be assigned due either to overlap of signals or mobility.
Analysis of Solution- and Solid-State NMR Chemical Shifts
Backbone chemical shifts measured in solution are a rich source of information. Secondary structure can be predicted with high confidence from 1H, 15N, and 13C chemical shifts, and 15N chemical shift changes are routinely used to detect sites of intermolecular contacts. Backbone chemical shifts have also provided accurate secondary structure predictions in cases where resonance assignments have been made from solid-state measurements.20,21 Both solution- and solid-state chemical shifts of the 13Cα, 13Cβ, 13C′, and 15N resonances predict six β-strands in the α-crystallin domain. Since 1Hα chemical shifts are routinely used in solution-state data to predict secondary structure, the prediction program, PECAN was run two separate times, once with and once without 1Hα shifts. Both datasets predict identical β-strands except for the end strands, β3 and β9, where inclusion of the 1Hα shifts produced a decrease in the probability of the extended conformation.
As expected, the data from solution- and solid-state begin to diverge at the N-terminal end of the α-crystallin domain where residues in full-length αB can influence the chemical shifts and/or the secondary structure of the regions at boundary of the domain. These results are summarized in Figure 6 where the sequence alignment and secondary structures of the α-crystallin domains from human αB, wHSP16.9, Tsp36 (ACD1 and ACD2), and MjHSP16.5 are shown. The β-strands of the α-crystallin domains in solution- (αB10.1) and in the solid-state (αB) are shown above the sequence and as shaded regions, respectively, in Figure 6. The β-strands in αB are numbered β3-β9, following the convention established for other sHSPs.11 Most importantly, the positions and the lengths of the strands are similar in the isolated domain and in the oligomer indicating that the secondary structure is preserved in both forms/states. The sequence and secondary structure elements of the α-crystallin domains from the three sHSPs whose crystal structures have been solved are shown below that of αB in Figure 6 for comparison. β-strands 3, 4, 5, 8, and 9 are essentially the same in all the sequences while there is variability in two distinct regions of the α-crystallin domains. First, the region between β5 and β7 is the most divergent in length and in sequence amongst all known sHSP sequences. The 21-residue β5-β7 region is similar in MjHSP16.5 and wHSP16.9 with a 5-residue β6-strand that forms part of the domain swapped dimer interface.11,12 On the other hand, the shorter β5-b7 region in Tsp36 is either a 13- or a 17-residue loop (ACD1 or ACD2) that does not contain a β-strand. In a third variation, our data for the human α-crystallin domain indicate that the β5-β7 region contains a 10-residue loop followed by one contiguous 11-residue β-strand that we call β6+7. A second region that differs between the human α-crystallin domain and the other sHSPs is the segment labeled, HR1 (Heterogeneous Region 1) in Figure 6, at the extreme N-terminus of the α-crystallin domain. The corresponding region in Tsp36, wHSP16.9, and MjHSP16.5 forms a β-strand labeled as β2. As our analysis of solid-state NMR 3D spectra did not yield unambiguous assignment for the HR1 region, we prepared an αB sample in which only the amino acids, Pro, Phe, Met, and Trp were uniformly labelled with 13C and 15N (called PFMW-αB). There are only two methionines in the sequence of αB, M1 and M68, and our expectation was that the unambiguous identification of Met68 resonances in the HR1 region using PFMW-αB would provide a starting point for the sequential assignment. The average chemical shifts for the 13Cα, 13Cβ, and 13Cγ resonances of methionines in proteins are 56.1 ppm, 33.0 ppm, and 32.0 ppm, respectively (Biological Magnetic Resonance Data Bank, BMRB). A 2D 13C-13C correlation spectrum acquired with 15 ms spin diffusion mixing time on PFMW-αB (Figure 7) reveals more than five Cα- Cβ cross peaks due to methionines in αB. 1HN and Hα resonances that arise from flexible residues in αB, including the first five N-terminal residues, have been assigned in full-length αB by solution-state 1H NMR by Carver et al.22 It is normally difficult to observe flexible residues by solid-state NMR due to inefficient cross polarization (CP) transfer, so it is likely that all observed cross peaks in Figure 7 arise from M68. The 13Cα shifts vary from 54.5 to 56.0 ppm and the 13Cβ shifts vary from 34.0 to 37.5 ppm. The spread and multiplicity of chemical shifts in the solid-state data indicate that M68 is found in multiple distinct environments within the oligomer. We propose that the HR1 region is involved in the formation of higher order multimers and that several types of inter-subunit interactions can exist, leading to the observed heterogeneity of the signals. This is in contrast to a monodisperse sHSP such as wHSP16.9 where defined inter-molecular interactions of the N- and C-termini with the dimeric α-crystallin domain stabilize the oligomeric structure. In αB, the HR1 region may exist in different environments by interacting with different sites in the N-terminus for example, or exist in multiple conformations contributing to the polydispersity of the molecule. Hence we name the region in αB that corresponds to β2 in other sHSPs as HR1: Heterogeneous Region 1.
Figure 6.
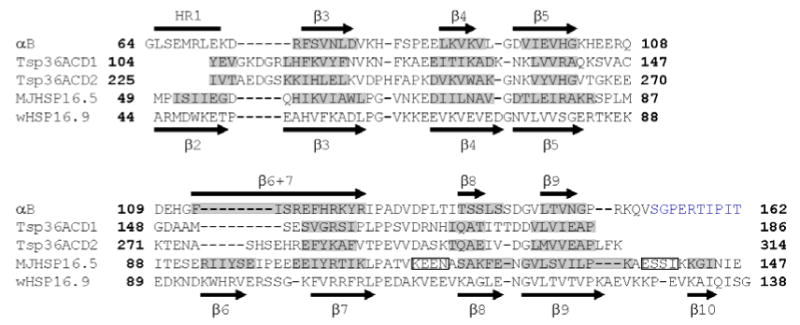
Secondary structures and sequences of the α-crystallin domains from αB, Tsp36, MjHSP16.5, and wHSP16.9 are shown. Tsp36 has two α-crystallin domains, ACD1 and ACD2. The sequence of the α-crystallin domain with an additional 10 C-terminal residues in αB and the corresponding region from wHSP16.9 are shown. The β-strands predicted from solution-state chemical shifts are shown as black arrows above the sequence of αB, those predicted by solid-state data are highlighted in grey along the sequence. The C-terminal residues in αB assigned by solid-state NMR are colored blue. The region corresponding to β2 in other sHSPs is labeled HR1 in αB. Residues M68-K72 have not been assigned by solid-state NMR. Residues G64-E67 and D73 have been assigned and solid-state chemical shifts predict random coil structure similar to solution-state. Residues G64, L65, His83, Phe84, H119, R120, and S139 have not been assigned in solution-state. The secondary structure elements of the α-crystallin domain in wHSP16.9 are shown below the sequence. Segments shaded in grey indicate β-strands in the α-crystallin domains of Tsp36 and MjHSP16.5. Rectangles indicate α-helices in MjHSP16.5.
Figure 7.
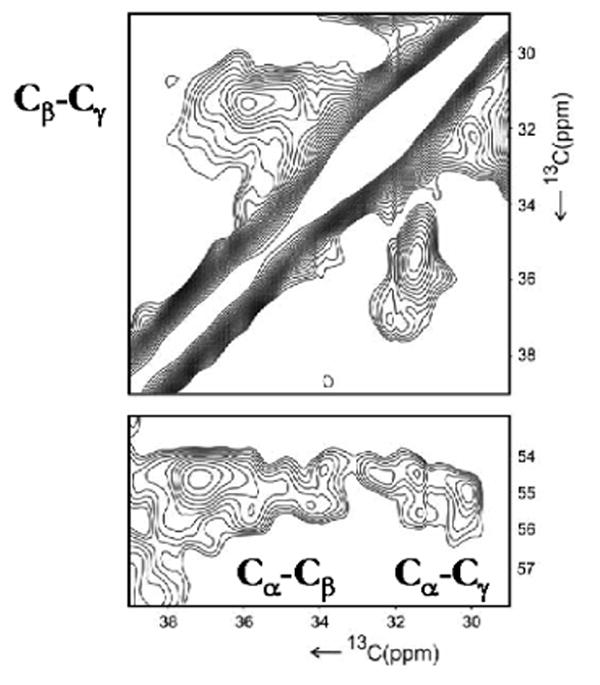
2D 13C/13C PDSD spectrum acquired with 15 ms mixing time on PFMW-αB is shown. The plot shows the Cγ-Cβ, Cα-Cβ and Cα-Cγ cross peaks from M68. The spectrum was recorded at a field of 14.1 T and 86 kHz 1H decoupling.
Solution-state chemical shifts predict a random coil structure for HR1 in αB10.1 which is further corroborated by 15N backbone dynamics data. HR1 is characterized by negative to low steady-state {1H}-15N heteronuclear NOE values ranging from -1.0 to 0.6 (Supplementary Data, Figure S1). Model-free analysis yields low order parameters (S2 = 0.4-0.6) for these residues (data not shown). Residues from 74 through 150 have strong positive {1H}-15N NOEs (>0.7) and S2 values of 0.9. The dynamics data indicate a molecule that is conformationally rigid on the ps-ns timescale with an N-terminal region that is highly flexible in solution.
Residues 153-162 that are C-terminal to the α-crystallin domain and absent in αB10.1, have been assigned in αB by solid state NMR (colored blue in Figure 6). This region contains the conserved IXI/V motif found in all sHSPs. No evidence for regular secondary structure elements was found in this region of αB. On the other hand, a short β-strand, β10, is found in both wHSP16.9 and MjHSP16.5.
Comparison of Solution- and Solid-State NMR Data
Chemical shifts observed in solution (δ solution) and in the solid state (δ solid) were compared residue by residue. Chemical shift values of 15N, 13Cα, 13Cβ, and 13C′ resonances for the α-crystallin domains in both solid- and solution-state are listed in Supplementary data, Table S1. The chemical shift differences (δ solid-δ solution) for 15N, 13Cα, 13Cβ, and 13C′ resonances are shown as histograms in Supplementary Data, Figure S2 A, B, C, and D, respectively. The mean ± standard deviation chemical shift differences for 15N, 13Cα, 13Cβ, and 13C′ resonances are 1.3 ± 1.4, 0.6 ± 0.7, 0.5 ± 0.4, and 0.6 ± 0.6 ppm, respectively. These values are within the range observed in other comparative studies of solid- and solution-state data.20,23 To facilitate interpretation of chemical shift differences, the sum of the absolute values of 15N, 13Cα, 13Cβ and 13C′ chemical shift differences (ΣN,Cα,Cβ,C′ (|δsolid-δsolution|)) for each residue was computed and plotted as a histogram versus residue number in Figure 8a. A majority of residues have differences below the mean value of 2.8 ppm, indicating that the structure of the α-crystallin domain in the oligomer is preserved in its isolated form in solution. Notably, chemical shift differences that are greater than the mean values are clustered in specific regions, viz., β3, β4, and β8. The chemical shift differences are shown mapped onto the topology of αB10.1 in Figure 8b. The topology of an αB10.1 protomer was determined by qualitative analysis of dN-N NOEs observed in the 15N-edited NOESY spectrum (data not shown). As shown in Figure 8b, the topology consists of two β-sheets with β3-β9-β8 strands forming one sheet and β4-β5-β6+7 forming a second sheet. Chemical shift differences between solid- and solution-state chemical shift data in the range, 3-4 ppm are colored orange and differences >4 ppm are colored red in Figure 8b. Strands β3, β4, and β8 where a majority of chemical shift differences are observed correspond to edge strands in the two sheets. Thus these edge strands experience different environments in the oligomer (αB) as compared to the isolated dimer (αB10.1). A dissimilar environment could be due to (i) the proximity of either the intra- or inter-subunit N- and C-terminal regions of αB, or (ii) the proximity of another dimer within the oligomer. The crystal structure of wHSP16.9 can be used as a model to rationalize the chemical shift differences between solid- and solution-state NMR data. We chose the structure of wHSP16.9 as our model because the N-terminus of MjHSP16.9 is not observed in the crystal structure and because TSP36 lacks a C-terminal region. In wHSP16.9, residues in the N-terminus are involved in intra- and inter-molecular interactions with residues in β2 and β7. The conserved IXI/V motifs (found in sHSPs) in the C-termini of one dimer strap across a hydrophobic groove formed by the edge strands β4 and β8 in different dimers. Such intermolecular interactions between neighboring subunits lead to the higher order oligomeric structure of wHSP16.9.12 Similar inter-molecular interactions in αB between the C-terminal region and the strands, β4, and β8 can potentially give rise to the observed chemical shift differences. The observed chemical shift differences in β3 could arise from interactions with either the N-terminal region and/or the HR1 region. Significant perturbations (>4 ppm) are also observed in the loops between HR1 and β3 (residues D73 and R74) and between β3 and β4 (residues V81 and K82). Some assignments are missing in these segments so a more thorough comparison between the solid- and solution-chemical shift data cannot be made at this time.
Figure 8.
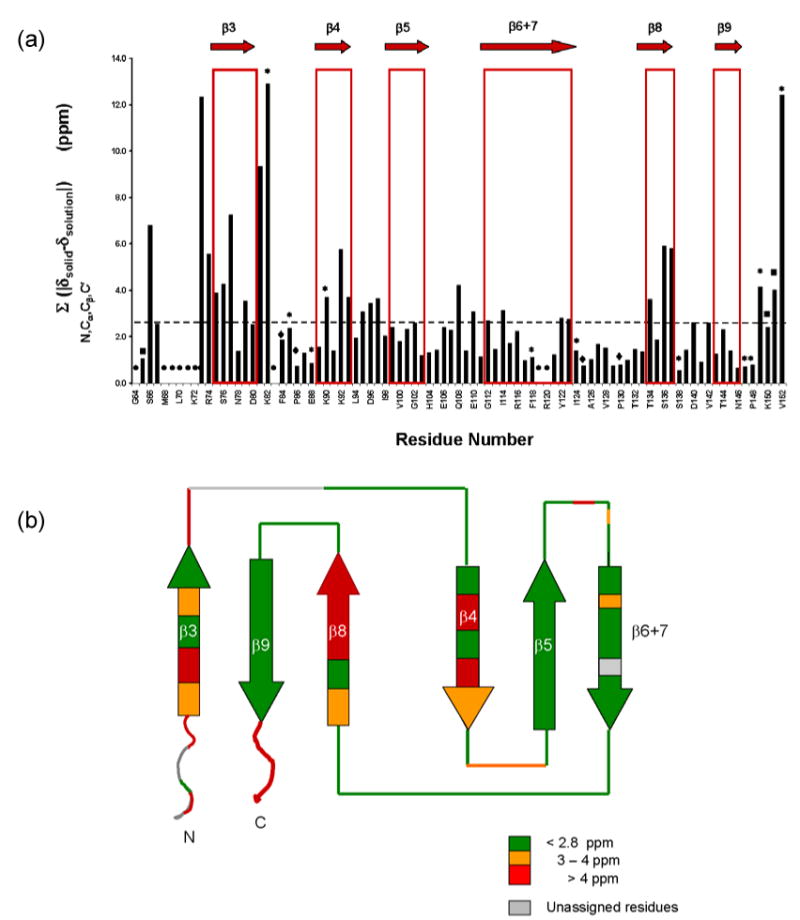
(a). Sum of the absolute values of the chemical shift differences of 15N, 13Cα, 13Cβ, and 13C′ resonances (ΣN,Cα,Cβ,C′|δsolid-δsolution|) plotted versus residue number. The secondary structure is shown at the top of the histogram. Dashed horizontal line represents the mean value. Residues that have either some or all resonance assignments missing are indicated as follows:
• : 15N, 13Cα, 13Cβ, and 13C′; ◆ :15N; ∗ :13C′; ¦ :13C′ and 13Cβ
(b). Chemical shift differences between solid- and solution-state NMR data shown mapped on the topology of αB10.1. Color coding is as follows: < 2.8 ppm: green (no perturbation); 3-4 ppm: orange; > 4 ppm: red. Edge strands, β3, β4, and β8, some loop regions as well as the N and C-termini experience significant chemical shift perturbations. No information is available for regions colored gray due to missing assignments.
Chemical shift similarities between the solution- and solid-state NMR data are clustered in β5, β7, and β9 and in the loops β5-β6+7, β6+7-β8 and β8-β9. Lack of shift differences in β5 and β9 is not unexpected, as they are each the middle strands of a sheet (Figure 8b). The similarity of the chemical environment for residues in β6+7, an edge strand, in the solution- and solid-states is notable and leads us to propose that strand β6+7 comprises the dimer interface. We favor a dimer model in which the β6+7 strands from the two protomers align antiparallel to each other. The residues in β6+7 would then be buried in the dimer interface and involved in similar contacts in αB and αB10.1, resulting in the observed similarities of chemical shifts between solution- and solid-state data.
The pH-dependent monomer-dimer transition properties of the human α-crystallin domain constructs provides further corroboration of the proposed dimer interface. The longer αB11.4 construct has pH-dependent behavior more amenable to NMR analysis than that of αB10.1: considerable broadening is observed in the 1H-15N HSQC spectrum of αB10.1 at lower pH. An overlay of 1H-15N HSQC spectra of αB11.4 at pH 6.8 (black) and pH 7.5 (blue) is shown in Figure 9. The spectra are remarkably different given the less than 1 pH unit difference in conditions. A comparison of the 1H-15N HSQC spectra at pH 7.5 and 6.8 should then reveal the identity of residues that experience chemical shift changes due to dimer-monomer transition. Though de novo resonance assignments of αB11.4 proved to be a challenge as discussed earlier, 1HN and 15N assignments could be transferred from the HSQC spectrum of αB10.1 to that of αB11.4 at pH 7.5. Triple resonance spectra acquired on perdeuterated samples of αB11.4 at pH 6.8 contained only ∼50% of the expected resonances, but these could be assigned. A majority of resonances have identical chemical shifts in both spectra and include regions comprising β3, β4, β5, β8, β9, and the loops between β4-β5, β7-β8, and β8-β9. In general, lowering of the pH inhibits exchange of labile backbone amide resonances with the solvent resulting in the appearance of these peaks in the 1H-15N HSQC spectrum. As expected, new peaks appear in the spectrum at pH 6.8 (indicated by circles in Figure 9). Remarkably, several peaks disappear or move from their original positions, indicated by arrows in Figure 9. Of the sixteen resonances that move or disappear as a function of pH, seven are in β6+7 (residues 114-118, 122, and 123), i.e., essentially all of the observable residues in β6+7 other than K121 which could not be clearly identified due to overlap. The remaining perturbed residues are located in (i) the β5-β6+7 loop (residues 106, 108, 110-112), (ii) the β3-β4 loop (residues 82, 85, 86), and (iii) the β4-β5 loop (residue 103). Hence, a majority of the resonances that are perturbed on lowering the pH to a value where the monomer is favored are in the putative dimerization interface, β6+7. In conclusion, a comparison of the solution- and solid-state chemical shift data and an analysis of the pH-dependent 1H-15N HSQC spectra both predict an identical dimer interface for the human α-crystallin domain.
Figure 9.
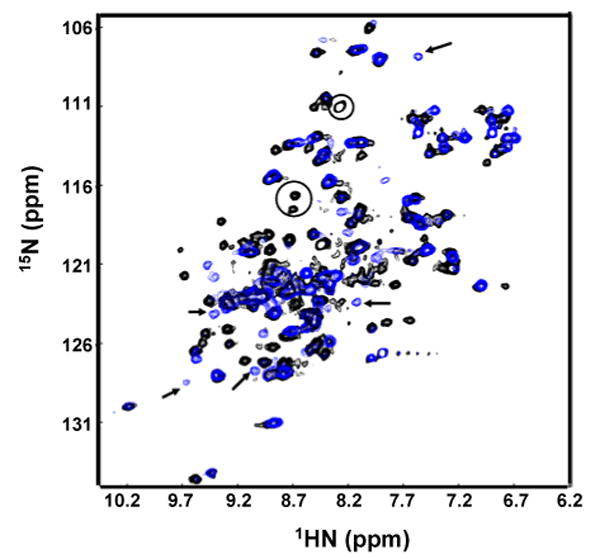
Overlay of 1H-15N HSQC spectra of αB11.4 at pH6.8 (black) and pH7.5 (blue). The spectra were acquired at 500 MHz at 37 °C on a 1 mM sample in 50 mM sodium phosphate buffer containing 100 mM NaCl. 32 transients per t1 value were acquired. Arrows indicate peaks in the spectrum at pH 7.5 that move/disappear on lowering the pH to 6.5. Circles indicate peaks that appear at pH 6.5.
Discussion
Solid-State NMR and polydisperse oligomers
The increasing number of entries in the BMRB database for solid-state NMR is a testament to the rapid growth of this technique as a high-resolution structural tool for proteins. Chemical shift assignments and structures have been obtained for fibrils and membrane proteins that are otherwise difficult to obtain by either solution-state NMR or x-ray crystallography. Polydisperse oligomeric proteins such as αB pose a huge challenge for structure determination. It has been shown that secondary structures predicted from solid state chemical shifts (15N, 13Cα, 13Cβ, and 13C′) have a very close correspondence to structures calculated previously by either x-ray crystallography24,25 or solution-state NMR.20 Structures of several proteins ranging in molecular weight from ∼4 kDa to 20 kDa have been determined by solid-state NMR, e.g., Kaliotoxin,26 GB1,27 SH3 domain,28 Ubiquitin,29 and Crh (dimer, 2 × 10.1 kDa).30 We have shown that it is possible to obtain structural information at atomic level for the α-crystallin domain and part of the C-terminal region in αB with solid-state NMR. Furthermore, we have begun to identify sites contributing to polydispersity of the molecule. We observe a multitude of signals for M68 indicating structural heterogeneity in full-length αB using a sample with a few selectively labeled amino acids (PFMW-αB). There are only two Met residues in αB, M1 and M68. M68 is in the middle of the HR1 region (the region that corresponds to β2 strand in other sHSPs) and this heterogeneity could arise from different conformations and/or chemical environments. EPR studies performed on αA using several spin-labeled Cys mutants to determine the location of β-strands in the α-crystallin domain do not provide rigorous evidence for the existence of β2 in this highly related protein (there is 54% sequence identity between αB and αA).31,32 The characteristic periodicity observed for β-strands in EPR parameters such as the linewidth of the central EPR signal and the accessibility values was not observed in the putative β2 (HR1 in αB) strand. The lack of periodicity, especially in the accessibility parameter was attributed to quaternary interactions involving the putative β2 strand. On the other hand, absence of a discrete structure in the HR1 region as shown by NMR data on αB could also explain the EPR data on αA. The conformational flexibility of residues in HR1 may contribute to different types of inter-subunit interactions that give rise to the known polydispersity of αB.
The human α-crystallin Domain
The design of the human α-crystallin domain construct proved to be extremely crucial for solution NMR studies. A few N-terminal-deleted αA constructs varying in the start of the α-crystallin domain have been described in literature. 18 They are reported to form smaller multimers with molecular weights in the range 40 – 60 kDa. An N-terminal deletion construct, αB(64-175), made in our lab had a very broad gel filtration profile indicative of a heterogeneous species (data not shown). Thus it was important to optimize both ends of the α-crystallin domain to generate a monodisperse species. Careful design of α-crystallin domain constructs from other sHSPs can yield structurally viable molecules which should help to increase the limited structural database for sHSPs (Rajagopal and Klevit, unpublished results).
A few resonances (G64, L65, H83, F84, H119, R120, and S139) could not be assigned from solution-state NMR data on αB10.1 because they were not observed in spectra under any conditions. Labile amide resonances at the extreme N-terminus (G64 and L65) or in exposed loop regions (H83 and F84 in β3-β4 loop; S139 in β8-β9 loop) are not observed probably due to rapid exchange with H2O especially at the relatively high pH value of 7.5 used in these studies. Residues in dynamic protein-protein interfaces may not be observed due to exchange broadening from motions in the micro- to milli-second timescale. Line broadening as well as reduced intensities are observed for most of the residues in the β6+7 strand that forms the dimer interface in αB10.1 and in the extreme case, residues H119 and R120 are broadened beyond detection. These resonances are observed in solid-state NMR spectra probably due to the much lower temperature (-3 °C) used for data acquisition. In fact, solution-state 1H-15N HSQC spectra on αB10.1 show that increased exchange broadening of resonances is observed with elevated temperatures of 35 °C and above (data not shown).
The topologies of the α-crystallin domains of sHSPs ranging from bacterial to human are similar: six or seven β-strands constitute these domains. But are there differences in the structure beyond the topological level? Initial glimpses into the protomeric structure and the dimer interface in αA have been afforded by EPR investigations. Structural models of the monomeric unit of the α-crystallin domain from αA have been determined from both EPR spin labeling studies32 and the program, Rosetta, using EPR distance restraints.33 In the models, the α-crystallin domain from αA forms a β-sandwich structure similar to the α-crystallin domains in sHSP structures solved by x-ray crystallography. However, modeling studies can overlook differences such as the lengths and positions of strands/or loops that are important for defining higher order structures. Our NMR data show that there are differences in certain segments of αB especially in the region comprising β6 that have important ramifications for higher order architecture. In our constructs and in full-length αB, β6 is found to be contiguous with β7, forming one single strand. Chemical shift data indicate that the dimer interface is comprised mainly of the long β6+7 strand (residues F113-R123) in αB unlike in wHSP16.9 and MjHSP16.5 where the dimer interface is formed from β2- and β6-strands from two subunits. A modeling study in conjunction with X-ray scattering data on αB11.4 proposed a V-shaped dimer with a subset of residues in the β6+7 strand (residues G112-S115) constituting the dimer interface.14 This model, however, is not consistent with our NMR data in terms of secondary structure elements. EPR studies on αA have identified β-strands in positions identical to those found in αB.32 The dimer interface investigated in another EPR study using spin-labeled Cys mutants in the region corresponding to β6+7 strand proposed that residues I110-E113 (αB residues I114-E117) form the dimer interface.34,35 EPR is an elegant approach to obtain structural information on proteins that are otherwise intractable by either x-ray crystallography or solution-state NMR. However, the need to introduce non-native cysteines for spin-labeling may yield data that is misleading, especially if the Cys is in a buried position in the folded protein. The critical importance of the β6+7 strand is underscored by disease-associated mutations in both αB and αA: R120G in αB is associated with cardiomyopathy and R116C in αA is associated with cataract. It was shown in a recent study that introduction of the R120C mutation in αB causes aggregation of this extremely soluble protein.6 Hence perturbations caused by the introduction of cysteine residues at certain sites in β6+7 strand may explain the lack of EPR spin-spin interactions expected for residues in the dimer interface. Comparison of (i) chemical shift data from solid- and solution-state NMR and (ii) 1H-15N HSQC spectra at pH 7.5 and 6.8 provide strong evidence that the dimer interface is formed by the β6+7 strand. Further experimentation will provide additional structural details such as the orientation of the two protomeric units and side-chain packing. Our NMR data show that a de novo, rigorous structure determination without recourse to modeling is now feasible.
Functional Implications
It is thought that sHSPs either dissociate to form smaller multimers or undergo a conformational change under stress such as changes in pH or elevation in temperature.36 Preservation of structure and function of the isolated human α-crystallin domain constructs as compared to full-length αB makes them useful functional mimics to test this hypothesis. A steep pH-dependent dimer-monomer transition is observed for both the human α-crystallin domains, αB11.4 and αB10.1. It has been shown that the affinity of αB for cardiac muscle proteins such as desmin and actin increases at pH 6.5,37 presumably to preserve the filament integrity of these proteins under stress such as cardiac ischemia which is accompanied by acidosis. There are a few other known examples of steep pH-dependent monomer-dimer transitions that are linked to function. For example, Glycinamide Ribonucleotide Transformylase (GART) is a dimer at pH 6.8 and dissociates into a monomer at pH 7.5 where its catalytic efficiency is increased.38 Similarly, dissociation of the α-crystallin domain dimer may be necessary for triggering a conformational switch in αB that is tuned to changes in the environment. Residues in strands β3 and β4 have been shown to be important for chaperone activity in αB.39,40 Comparison of solid- and solution-state NMR chemical shift data show differences in β3 and β4 (Figure 8) implying their involvement in inter-subunit interactions. Taken together, the observations suggest that a change in αB is necessary for exposure of the chaperone binding sites masked by quaternary contacts.
Arginines and aromatic residues are often found in protein-protein interfaces.41 The site of R120 in αB is conserved in all sHSPs known to date. However, the interaction of this conserved residue varies from molecule to molecule. For example, in MjHSP16.5, the equivalent R107 forms an intramolecular bifurcated hydrogen bond and R108 in wHSP16.9 forms an intermolecular salt bridge. The unique dimer interface in αB may provide yet another interaction for R120. The demonstrated ability to obtain structural information using a combination of solution- and solid-state NMR on the previously intractable αB system will allow investigation of the pH-dependent properties, the structural consequences of the R120G mutation, and other regulatory mechanisms including phosphorylation in the future. In this context, the solid-state NMR approach has the potential of resolving structural details of the different protomers constituting the variety of oligomers. Furthermore, the approaches presented here will be applicable to a wide variety of dynamical complexes with similar properties, opening new avenues for structural biology.
Materials and Methods
Protein Expression and Purification
αB was expressed and purified as described elsewhere42 with some modifications to the growth protocol. BL21 Star (DE3) cells (Invitrogen) were used instead of BL21 (DE3) cells. Uniform [13C,15N]-labeled αB was grown in M9 minimal media supplemented with 15NH4Cl (1 g/L) and 13C6-glucose (2 g/L). Dilutely labeled 13C samples were grown on M9 media supplemented with either 1,3-13C-glycerol or 2-13C-glycerol (2 g/L). Cells were incubated at 37 °C until growth to a cell density of 0.6 at 600 nm. Cultures were transferred to a 22 °C shaker and protein expression was induced with 1 mM IPTG. Cells were harvested after 12-16 hrs. 1,3-13C-glycerol and 2-13C-glycerol were purchased from Cambridge Isotope Laboratories (Andover, USA).
The α-crystallin domains were cloned into the pET-24a vector (Novagen, EMD Chemicals Inc., San Diego) and over-expressed in E.coli BL21 Star (DE3) cells. Two α-crystallin domain constructs were used in this study, (i) αB11.4 (residues 58-157) and (ii) αB10.1 (residues 64-152). N146D-αB10.1 was constructed with Quick-Change site-directed mutagenesis Kit from Stratagene (La Jolla, CA). [13C,15N]- and [2H,13C,15N]-labeled samples were grown with 4 g/L of 13C-glucose in M9 minimal media, induced with 0.5 mM IPTG and the cells were harvested after 18 hrs and 36 hrs, respectively. Cell lysis and subsequent centrifugation steps were performed as described in literature.14 Since the α-crystallin domains were extremely susceptible to proteolysis, the purification protocol was modified from that described before to generate long-lived samples required for NMR experiments. The pellet from the 2.5 M NH2SO4 cut was dissolved in 20 mM Tris (pH 8.0) buffer (Buffer A) and desalted on a G25 Sephadex column. Fractions containing protein were pooled and loaded onto a DEAE-Sepharose anion-exchange column. The sample was eluted with a series of step gradients between Buffer A and Buffer B (Buffer A plus 1 M NaCl). Pooled protein fractions were desalted with a G25-Sephadex column and applied to MonoQ HR 10/10 anion-exchange column (Pharmacia). Step gradients between Buffer A and B were employed to elute > 90% pure protein as judged by SDS PAGE. Finally, G75-Sephadex gel filtration yielded protein with > 98% purity. Fractions were pooled and dialyzed against three changes of sodium phosphate buffer (pH 7.0), lyophilized, and stored at -20 °C for future use. 15N NH4Cl and 13C-glucose were obtained from CIL (Cambridge Isotope Laboratories). Protease inhibitor cocktail for non His-tagged proteins and protease inhibitors such as Pefabloc and PMSF (Phenylmethylsulfonyl Fluoride) were purchased from SIGMA.
Analytical Ultracentrifugation
Analytical ultracentrifugation was performed at the University of Connecticut with a Beckman-Coulter XL-I analytical ultracentrifuge at 20 °C and 55,000 rpm. Sedimentation velocity experiments were performed in double sector cells with sapphire windows using the interference optics system. Interference scans were acquired at 1 min intervals for 7 hrs. The sample concentrations used were: 8 μM, 22 μM, 90 μM, and 170 μM.
Assays for Chaperone-Like Activity
DTT-denatured α-Lac was used as the substrate. DTT and α-Lac were purchased from SIGMA. Equimolar amounts of chaperone and substrate (0.1 mM) in sodium phosphate buffer (pH 7.5) containing 150 mM NaCl and 5 mM EDTA were used to generate the mixtures: (i) αB10.1 + α-Lac + DTT, (ii) αB + α-Lac + DTT, and (iii) α-Lac + DTT. The samples were incubated at 37 °C. Light scattering was measured simultaneously for multiple samples with a Multiskan MCC/340 plate reader using a 96-well micro-titre plate, allowing several time courses to be collected in parallel.
Solution-State NMR
All NMR experiments were performed on 0.5 – 1.0 mM samples in 90% H20/10% D2O solution containing 50 mM sodium phosphate buffer (pH 7.5), 100 mM Na2SO4, 100 μM EDTA, 1 mM PMSF, 1 mM Pefabloc, and 1 μg/ml Leupeptin. The standard suite of triple resonance43 experiments required for the assignment of the backbone resonances were acquired on [85% 2H, U-13C, 15N]-N146D-αB10.1 at 15 °C. TROSY-HNCACB and TROSY-HNCA experiments were acquired at Pacific Northwest National Laboratories (PNNL, Richland, WA) on a Varian INOVA800. TROSY-HN(CO)CA, TROSY-HN(CO)CACB, and TROSY-HNCO were acquired in-house on Bruker DMX500. Assignments were transferred to αB10.1 (wt α-crystallin domain) with a TROSY-HNCACB experiment acquired on perdeuterated αB10.1. TROSY-HNCO and a 15N-edited TOCSY-HSQC with 25-ms mixing time were acquired to obtain C′ and Hα assignments, respectively. Triple resonance spectra were acquired as data matrix of 512 × 32 × 48 complex points with acquisition times of 46 ms, 13 ms, and 3 ms in the 1H, 15N, and 13C dimensions, respectively; 16-32 transients were acquired per t1 value. The 15N-edited TOCSY spectrum was acquired with 512 × 32 × 80 complex points with acquisition times of 73 ms, 23 ms, and 11.4 ms in the HN, 15N, and 1H dimensions, respectively, with 32 transients per t1 value.
All spectra were processed with NMRPipe software.44 Raw data matrices were transformed into 1024 ×256 ×256 real matrices. Mirror-image linear prediction in the 15N-dimension and forward linear prediction to double the number of points in the 13C and 1H dimensions were used. In each dimension, data were apodized with a squared sine bell shifted by 60°. 1H chemical shifts were referenced to DSS (2,2-dimethyl-2-silapentane-5-sulfonate sodium salt) at 0 ppm. 15N and 13C chemical shifts were referenced indirectly using the 15N/1H and 13C/1H gyromagnetic ratios, respectively.
Peak picking and data analysis were performed with NMRView.45 Assignments were obtained with AutoAssign46 and confirmed manually. Secondary structure was determined from PECAN (Protein Energetic Conformational Analysis from NMR chemical shifts)47 using 15N, 13Cα, 13Cβ, 13C′, and 1Hα chemical shifts. The chemical shifts of 13Cα and 13Cβ resonances were corrected for 2H isotope shift before being used as input for PECAN.
Solution-State 15N relaxation measurements
HSQC-based experiments were used to collect 15N R1, R2, and {1H}-15N steady-state heteronuclear NOE data on Bruker DMX500 and DMX600 spectrometers at 22 °C. 48 The 15N R1 and R2 data required to determine the values of τc were acquired on the α-crystallin domains on Bruker DMX500. 32 transients and 64 transients per t1 value were acquired for 15N R1, R2 and {1H}-15N NOE experiments, respectively. 512 × 160 complex data matrices in 1H and 15N dimensions, respectively, were transformed into 2048 × 2048 real matrices and data were apodized with a squared sine-bell shifted by 60°. 15N R1 values were determined from spectra acquired with 10, 50,100, 200, 400, 600, 800, and 1000 ms relaxation delays, with a duplicate acquired at a relaxation delay of 200 ms. 15N R2 values were determined from spectra acquired with relaxation delays: 8, 24, 40, 56, 72, 88, and 112 ms, with duplicates acquired for relaxation delays of 8 and 40 ms. Estimates of error were determined from duplicate spectra as described previously.48 {1H}-15N heteronuclear NOE spectra were recorded using a saturation time of 3 s with a 2 s relaxation delay prior to saturation. The reference spectrum was acquired with a relaxation delay of 5 s. The program Modelfree49 was used to calculate the order parameters (S2) and correlation times.
Sample Preparation for Solid-State NMR
Lyophilized full-length αB was dissolved in 50mM sodium phosphate buffer (pH 7.6) containing 50 mM NaCl and 0.002% NaN3 to a final concentration of 40mg/ml. 0.5 ml (20 mg) of the αB solution was mixed with ∼80-100 μl of a 50% PEG8000 solution until the start of precipitation. The precipitation was complete after 3 days in a vapor diffusion chamber over a reservoir solution with 50% PEG8000. A ZrO2 rotor was filled with the precipitant and further used for solid-state NMR experiments. Samples grown in media containing 13C-glucose are referred to as U-αB, 1,3-[13C]-glycerol as 1,3-αB, and 2-[13C] -glycerol as 2-αB. All protein samples contain uniform 15N-labelling.
Solid-State NMR
The NCACX and NCOCX spectra50 of U-αB were recorded with 25 ms and 35 ms spin diffusion mixing, respectively. The NCACX and NCOCX experiments on 1,3-αB were recorded with 500ms and 75 ms spin diffusion mixing,51 respectively. The NCACX on 2-αB was recorded with 200 ms and 60 ms spin diffusion mixing. The NCACB spectrum52 was recorded with 2ms DREAM mixing. DREAM mixing was established with a weak 13C B1 field that was linearly amplitude-modulated from 6 to 3 kHz. The carrier frequency was centered at 45 ppm between Cα and Cβ.
All solid-state NMR spectra were recorded at -3 °C and a MAS frequency of ωR/2π = 10.0 kHz except the methyl-filtered DARR53 experiment, which was recorded at ωR/2π = 12.5 kHz. 2D and 3D experiments on αB were acquired on a wide-bore Bruker Avance DMX700 spectrometer. 3D experiments on 2- and 1,3-αB were acquired on a wide-bore Bruker Avance DMX600 spectrometer. To facilitate methyl assignments, a methyl-filtered DARR experiment54 was recorded on a narrow-bore Bruker Avance DMX900 spectrometer. The wide-bore DMX 600 and 700 spectrometers were equipped with a 4 mm triple-resonance CP/MAS probe (Bruker, Karlsruhe, Germany). The DMX900 spectrometer was equipped with a 3.2 mm triple-resonance CP/MAS probe (Bruker, Karlsruhe, Germany).
To establish sequential assignments, triple resonance 3D NCACX, 3D NCOCX and 3D NCACB spectra were recorded on U-, 2-, and 1,3-αB. At relatively low mixing times of 25-35 ms, NCACX and NCOCX spectra contain correlations from 15N (i) to the C′ and the aliphatic carbons of the i and i-1 residues, respectively. At spin-diffusion mixing times longer than 50 ms, e.g., 200-500 ms, additional inter-residue cross peaks appear: correlations of the type, N(i)-Cα(i)-Cα (i-1), N(i)-Cα(i)-Cα(i+1) and N(i)-Cα(i)-C′(i-1) are observed in NCACX spectra and N(i)-C′(i-1)-C′(i), N(i)-C′(i-1)-C′(i-2) and N(i)-C′(i-1)-Cα (i) correlations are observed in NCOCX spectra. NCACX and NCACB spectra were acquired with 48 scans as a data matrix of 1024 × 64 × 40 points with acquisition times of 12 ms, 5.6 ms, and 6 ms in F3 (13C), F2 (13Cα) and F1 (15N), respectively. NCOCX spectra were acquired with 48 scans as a data matrix of 1024 × 32 × 40 points with acquisition times of 12 ms, 5.6 ms, and 6 ms in F3 (13C), F2 (13C′) and F1 (15N), respectively. To establish 1H/15N CP, a 100-75% ramp on 1H was used. The B1 fields during CP were ∼50 kHz and ∼60 kHz for 1H and 15N, respectively. Magnetization transfer between low-γ nuclei 15N and 13C was established with a 3.5 ms adiabatic CP using a field of 25 kHz and 35 kHz, respectively. 2D PDSD spectra were recorded with a 15 ms mixing time for U-αB and 50 ms for 2- and 1,3-αB. For the methyl-filtered 13C/13C experiment, CP with 50 ms phase inversion and 60 μs dipolar dephasing was used prior to t1-evolution and 75 ms DARR mixing. During dipolar dephasing, the 13C chemical shifts were refocused with a π-pulse.
A t1-J-decoupled spectrum55 was recorded on U-αB with 40 ms PDSD mixing. During t1-evolution a 350 μs gaussian π-pulse on Cα and a hard pulse on 13C were applied to remove J-couplings from the indirect dimension. The spectrum was further processed with the Software Rowland NMR toolkit to remove J-couplings from the direct dimension for the selected cross peak region (Cα-Cβ) using the Maximum Entropy Algorithm.56 The B1 fields during 1H/13C CP (1.5ms) in 2D experiments were ∼55 kHz and ∼65 kHz for 1H and 13C, respectively. A 50-100% ramp on 13C channel was used for CP. 2D 13C/13C spectra were recorded as a data matrix of 1024 × 768 points with acquisition times of 15 ms and 10 ms in the direct and indirect dimension, respectively. The J-decoupled PDSD was recorded using States-TPPI for phase-sensitive detection in the indirect dimension; all other spectra were recorded using TPPI. During DARR mixing, a B1 field on protons equal to ωR was applied. Typical π/2-pulse length on 13C and 1H were 3.5-4.3 μs and 3.8 μs, respectively. SPINAL64 decoupling of 1H spins with 87 kHz was applied in all 2D and 3D experiments.
2D spectra were zero filled to 2048 points in each dimension. All 3D spectra were zero filled to a data matrix of 2048 × 256 × 256 points. Linear prediction in the indirect dimensions was used to increase the number of points by a factor of 1.3 for all spectra. Gaussian and square cosine filters were used in the direct and indirect dimensions, respectively. Data were processed with Topspin 1.3 (Bruker, Karlsruhe, Germany) if not mentioned otherwise and further analyzed using Sparky 3.110 (T.D. Goddard & D.G. Kneller, University of California, San Francisco).
Supplementary Material
Acknowledgments
We thank Dr. Ingeborg Feil and Dr. John Clark for providing us with the plasmids for αB11.4 and αB. We thank Dr. Borris Demmler for performing sedimentation velocity experiments on αB11.4 at CAUMA in San Antonio, Texas. A portion of the research was performed at EMSL (Environmental and Molecular Sciences Lab), a national scientific user facility sponsored by the Department of Energy's office of Biological and Environmental Research located at Pacific Northwest National Laboratory. This work was supported by the National Eye Institute (1R01 EY017370) and Deutsche Forshungsgemeinshaft (SSB449).
Abbreviations
- αB
αB-Crystallin
- HSQC
heteronuclear single quantum coherence
- αA
αA-Crystallin
- wt
wild type
- TROSY
transverse relaxation optimized spectroscopy
- CP
cross polarization
- τc
correlation time
- PEG
polyethylene glycol
- α-Lac
α-Lactalbumin
- NOE
Nuclear Overhauser Effect
- MAS
magic-angle spinning
- PDSD
Proton Driven Spin Diffusion
- sHSP
small Heat Shock Protein
- E.coli
Escherichia coli
- DTT
dithiothreitol, MjHSP16.5, Methanoccocus jannaschii HSP16.9
- wHSP16.9
wheat HSP16.9
- Tsp36
Taenia saginata HSP36
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Horwitz J. Alpha-crystallin can function as a molecular chaperone. Proc Natl Acad Sci U S A. 1992;89:10449–53. doi: 10.1073/pnas.89.21.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bullard B, Ferguson C, Minajeva A, Leake MC, Gautel M, Labeit D, Ding L, Labeit S, Horwitz J, Leonard KR, Linke WA. Association of the chaperone alphaB-crystallin with titin in heart muscle. J Biol Chem. 2004;279:7917–24. doi: 10.1074/jbc.M307473200. [DOI] [PubMed] [Google Scholar]
- 3.Singh BN, Rao KS, Ramakrishna T, Rangaraj N, Rao Ch M. Association of alphaB-crystallin, a small heat shock protein, with actin: role in modulating actin filament dynamics in vivo. J Mol Biol. 2007;366:756–67. doi: 10.1016/j.jmb.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Bova MP, Yaron O, Huang Q, Ding L, Haley DA, Stewart PL, Horwitz J. Mutation R120G in alphaB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function. Proc Natl Acad Sci U S A. 1999;96:6137–42. doi: 10.1073/pnas.96.11.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun Y, MacRae TH. The small heat shock proteins and their role in human disease. Febs J. 2005;272:2613–27. doi: 10.1111/j.1742-4658.2005.04708.x. [DOI] [PubMed] [Google Scholar]
- 6.Simon S, Michiel M, Skouri-Panet F, Lechaire JP, Vicart P, Tardieu A. Residue R120 is essential for the quaternary structure and functional integrity of human alphaB-crystallin. Biochemistry. 2007;46:9605–14. doi: 10.1021/bi7003125. [DOI] [PubMed] [Google Scholar]
- 7.Hayes VH, Devlin GL, Quinlan RA. Truncation of alphaB-crystallin by the myopathy-causing Q151X mutation significantly destabilizes the protein leading to aggregate formation in transfected cells. J Biol Chem. 2008 doi: 10.1074/jbc.M706453200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ, Rustgi A, Fuchs SY, Diehl JA. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol Cell. 2006;24:355–66. doi: 10.1016/j.molcel.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ousman SS, Tomooka BH, van Noort JM, Wawrousek EF, O'Connor KC, Hafler DA, Sobel RA, Robinson WH, Steinman L. Protective and therapeutic role for alphaB-crystallin in autoimmune demyelination. Nature. 2007;448:474–9. doi: 10.1038/nature05935. [DOI] [PubMed] [Google Scholar]
- 10.Thedieck C, Kalbacher H, Kratzer U, Lammers R, Stevanovic S, Klein G. alphaB-Crystallin is a Cytoplasmic Interaction Partner of the Kidney-Specific Cadherin-16. J Mol Biol. 2008 doi: 10.1016/j.jmb.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 11.Kim Y, Valentine K, Opella SJ, Schendel SL, Cramer WA. Solid-state NMR studies of the membrane-bound closed state of the colicin E1 channel domain in lipid bilayers. Protein Sci. 1998;7:342–8. doi: 10.1002/pro.5560070214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Montfort RL, Basha E, Friedrich KL, Slingsby C, Vierling E. Crystal structure and assembly of a eukaryotic small heat shock protein. Nat Struct Biol. 2001;8:1025–30. doi: 10.1038/nsb722. [DOI] [PubMed] [Google Scholar]
- 13.Stamler R, Kappe G, Boelens W, Slingsby C. Wrapping the alpha-crystallin domain fold in a chaperone assembly. J Mol Biol. 2005;353:68–79. doi: 10.1016/j.jmb.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 14.Feil IK, Malfois M, Hendle J, van Der Zandt H, Svergun DI. A novel quaternary structure of the dimeric alpha-crystallin domain with chaperone-like activity. J Biol Chem. 2001;276:12024–9. doi: 10.1074/jbc.M010856200. [DOI] [PubMed] [Google Scholar]
- 15.Campos-Olivas R, Horr I, Bormann C, Jung G, Gronenborn AM. Solution structure, backbone dynamics and chitin binding of the anti-fungal protein from Streptomyces tendae TU901. J Mol Biol. 2001;308:765–82. doi: 10.1006/jmbi.2001.4622. [DOI] [PubMed] [Google Scholar]
- 16.Grasberger BL, Gronenborn AM, Clore GM. Analysis of the backbone dynamics of interleukin-8 by 15N relaxation measurements. J Mol Biol. 1993;230:364–72. doi: 10.1006/jmbi.1993.1152. [DOI] [PubMed] [Google Scholar]
- 17.Shapiro YE, Kahana E, Tugarinov V, Liang Z, Freed JH, Meirovitch E. Domain flexibility in ligand-free and inhibitor-bound Escherichia coli adenylate kinase based on a mode-coupling analysis of 15N spin relaxation. Biochemistry. 2002;41:6271–81. doi: 10.1021/bi012132q. [DOI] [PubMed] [Google Scholar]
- 18.Bova MP, McHaourab HS, Han Y, Fung BK. Subunit exchange of small heat shock proteins. Analysis of oligomer formation of alphaA-crystallin and Hsp27 by fluorescence resonance energy transfer and site-directed truncations. J Biol Chem. 2000;275:1035–42. doi: 10.1074/jbc.275.2.1035. [DOI] [PubMed] [Google Scholar]
- 19.Fossi M, Castellani F, Nilges M, Oschkinat H, van Rossum BJ. SOLARIA: a protocol for automated cross-peak assignment and structure calculation for solid-state magic-angle spinning NMR spectroscopy. Angew Chem Int Ed Engl. 2005;44:6151–4. doi: 10.1002/anie.200501884. [DOI] [PubMed] [Google Scholar]
- 20.Franks WT, Zhou DH, Wylie BJ, Money BG, Graesser DT, Frericks HL, Sahota G, Rienstra CM. Magic-angle spinning solid-state NMR spectroscopy of the beta1 immunoglobulin binding domain of protein G (GB1): 15N and 13C chemical shift assignments and conformational analysis. J Am Chem Soc. 2005;127:12291–305. doi: 10.1021/ja044497e. [DOI] [PubMed] [Google Scholar]
- 21.Igumenova TI, Wand AJ, McDermott AE. Assignment of the backbone resonances for microcrystalline ubiquitin. J Am Chem Soc. 2004;126:5323–31. doi: 10.1021/ja030546w. [DOI] [PubMed] [Google Scholar]
- 22.Carver JA, Aquilina JA, Truscott RJ, Ralston GB. Identification by 1H NMR spectroscopy of flexible C-terminal extensions in bovine lens alpha-crystallin. FEBS Lett. 1992;311:143–9. doi: 10.1016/0014-5793(92)81386-z. [DOI] [PubMed] [Google Scholar]
- 23.Balayssac S, Bertini I, Falber K, Fragai M, Jehle S, Lelli M, Luchinat C, Oschkinat H, Yeo KJ. Solid-state NMR of matrix metalloproteinase 12: an approach complementary to solution NMR. Chembiochem. 2007;8:486–9. doi: 10.1002/cbic.200600408. [DOI] [PubMed] [Google Scholar]
- 24.Luca S, Filippov DV, van Boom JH, Oschkinat H, de Groot HJ, Baldus M. Secondary chemical shifts in immobilized peptides and proteins: a qualitative basis for structure refinement under magic angle spinning. J Biomol NMR. 2001;20:325–31. doi: 10.1023/a:1011278317489. [DOI] [PubMed] [Google Scholar]
- 25.Seidel K, Etzkorn M, Heise H, Becker S, Baldus M. High-resolution solid-state NMR studies on uniformly [13C,15N]-labeled ubiquitin. Chembiochem. 2005;6:1638–47. doi: 10.1002/cbic.200500085. [DOI] [PubMed] [Google Scholar]
- 26.Lange A, Becker S, Seidel K, Giller K, Pongs O, Baldus M. A concept for rapid protein-structure determination by solid-state NMR spectroscopy. Angew Chem Int Ed Engl. 2005;44:2089–92. doi: 10.1002/anie.200462516. [DOI] [PubMed] [Google Scholar]
- 27.Franks WT, Wylie BJ, Schmidt HL, Nieuwkoop AJ, Mayrhofer RM, Shah GJ, Graesser DT, Rienstra CM. Dipole tensor-based atomic-resolution structure determination of a nanocrystalline protein by solid-state NMR. Proc Natl Acad Sci U S A. 2008;105:4621–6. doi: 10.1073/pnas.0712393105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature. 2002;420:98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- 29.Zech SG, Wand AJ, McDermott AE. Protein structure determination by high-resolution solid-state NMR spectroscopy: application to microcrystalline ubiquitin. J Am Chem Soc. 2005;127:8618–26. doi: 10.1021/ja0503128. [DOI] [PubMed] [Google Scholar]
- 30.Loquet A, Bardiaux B, Gardiennet C, Blanchet C, Baldus M, Nilges M, Malliavin T, Bockmann A. 3D structure determination of the Crh protein from highly ambiguous solid-state NMR restraints. J Am Chem Soc. 2008;130:3579–89. doi: 10.1021/ja078014t. [DOI] [PubMed] [Google Scholar]
- 31.Koteiche HA, Berengian AR, McHaourab HS. Identification of protein folding patterns using site-directed spin labeling. Structural characterization of a beta-sheet and putative substrate binding regions in the conserved domain of alpha A-crystallin. Biochemistry. 1998;37:12681–8. doi: 10.1021/bi9814078. [DOI] [PubMed] [Google Scholar]
- 32.Koteiche HA, McHaourab HS. Folding pattern of the alpha-crystallin domain in alphaA-crystallin determined by site-directed spin labeling. J Mol Biol. 1999;294:561–77. doi: 10.1006/jmbi.1999.3242. [DOI] [PubMed] [Google Scholar]
- 33.Alexander N, Bortolus M, Al-Mestarihi A, McHaourab H, Meiler J. De novo high-resolution protein structure determination from sparse spin-labeling EPR data. Structure. 2008;16:181–95. doi: 10.1016/j.str.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berengian AR, Bova MP, McHaourab HS. Structure and function of the conserved domain in alphaA-crystallin. Site-directed spin labeling identifies a beta-strand located near a subunit interface. Biochemistry. 1997;36:9951–7. doi: 10.1021/bi9712347. [DOI] [PubMed] [Google Scholar]
- 35.Berengian AR, Parfenova M, McHaourab HS. Site-directed spin labeling study of subunit interactions in the alpha-crystallin domain of small heat-shock proteins. Comparison of the oligomer symmetry in alphaA-crystallin, HSP 27, and HSP 16.3. J Biol Chem. 1999;274:6305–14. doi: 10.1074/jbc.274.10.6305. [DOI] [PubMed] [Google Scholar]
- 36.Haslbeck M, Franzmann T, Weinfurtner D, Buchner J. Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol. 2005;12:842–6. doi: 10.1038/nsmb993. [DOI] [PubMed] [Google Scholar]
- 37.Bennardini F, Wrzosek A, Chiesi M. Alpha B-crystallin in cardiac tissue. Association with actin and desmin filaments. Circ Res. 1992;71:288–94. doi: 10.1161/01.res.71.2.288. [DOI] [PubMed] [Google Scholar]
- 38.Moormann RJ, den Dunnen JT, Mulleners L, Andreoli P, Bloemendal H, Schoenmakers JG. Strict co-linearity of genetic and protein folding domains in an intragenically duplicated rat lens gamma-crystallin gene. J Mol Biol. 1983;171:353–68. doi: 10.1016/0022-2836(83)90034-7. [DOI] [PubMed] [Google Scholar]
- 39.Bhattacharyya J, Padmanabha Udupa EG, Wang J, Sharma KK. Mini-alphaB-crystallin: a functional element of alphaB-crystallin with chaperone-like activity. Biochemistry. 2006;45:3069–76. doi: 10.1021/bi0518141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghosh JG, Estrada MR, Houck SA, Clark JI. The function of the beta3 interactive domain in the small heat shock protein and molecular chaperone, human alphaB crystallin. Cell Stress Chaperones. 2006;11:187–97. doi: 10.1379/CSC-186.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guharoy M, Chakrabarti P. Secondary structure based analysis and classification of biological interfaces: identification of binding motifs in protein-protein interactions. Bioinformatics. 2007;23:1909–18. doi: 10.1093/bioinformatics/btm274. [DOI] [PubMed] [Google Scholar]
- 42.Muchowski PJ, Valdez MM, Clark JI. AlphaB-crystallin selectively targets intermediate filament proteins during thermal stress. Invest Ophthalmol Vis Sci. 1999;40:951–8. [PubMed] [Google Scholar]
- 43.Yamazaki T, Lee W, Arrowsmith CH, Muhandiram DR, Kay LE. A suite of triple resonance NMR experiments for the backbone assignment of 15N, 13C, 2H labeled proteins with high senstivity. J Am Chem Soc. 1994;116:11655–11666. [Google Scholar]
- 44.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer's beta -amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci U S A. 2002;99:16742–7. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang H, Johnson PM, Ko KC, Kamio M, Germann MW, Derby CD, Tai PC. Cloning, characterization and expression of escapin, a broadly antimicrobial FAD-containing L-amino acid oxidase from ink of the sea hare Aplysia californica. J Exp Biol. 2005;208:3609–22. doi: 10.1242/jeb.01795. [DOI] [PubMed] [Google Scholar]
- 46.Moseley A, Graw J, Delamere NA. Altered Na,K-ATPase pattern in gamma-crystallin mutant mice. Invest Ophthalmol Vis Sci. 2002;43:1517–9. [PubMed] [Google Scholar]
- 47.Eghbalnia HR, Wang L, Bahrami A, Assadi A, Markley JL. Protein energetic conformational analysis from NMR chemical shifts (PECAN) and its use in determining secondary structural elements. J Biomol NMR. 2005;32:71–81. doi: 10.1007/s10858-005-5705-1. [DOI] [PubMed] [Google Scholar]
- 48.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 49.Mandel AM, Akke M, Palmer AG., 3rd Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J Mol Biol. 1995;246:144–63. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- 50.Castellani F, van Rossum BJ, Diehl A, Rehbein K, Oschkinat H. Determination of solid-state NMR structures of proteins by means of three-dimensional 15N-13C-13C dipolar correlation spectroscopy and chemical shift analysis. Biochemistry. 2003;42:11476–83. doi: 10.1021/bi034903r. [DOI] [PubMed] [Google Scholar]
- 51.Suter D, Ernst RR. Spin diffusion in resolved solid-state NMR spectra. Phys Rev B Condens Matter. 1985;32:5608–5627. doi: 10.1103/physrevb.32.5608. [DOI] [PubMed] [Google Scholar]
- 52.Pauli J, Baldus M, van Rossum B, de Groot H, Oschkinat H. Backbone and side-chain 13C and 15N signal assignments of the alpha-spectrin SH3 domain by magic angle spinning solid-state NMR at 17.6 Tesla. Chembiochem. 2001;2:272–81. doi: 10.1002/1439-7633(20010401)2:4<272::AID-CBIC272>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 53.Jehle S, Hiller M, Rehbein K, Diehl A, Oschkinat H, van Rossum BJ. Spectral editing: selection of methyl groups in multidimensional solid-state magic-angle spinning NMR. J Biomol NMR. 2006;36:169–77. doi: 10.1007/s10858-006-9078-x. [DOI] [PubMed] [Google Scholar]
- 54.Takegoshi K, Nakamura S, Terao T. 13C-1H dipolar assisted rotational resonance in magic-angle spinning NMR. Chemical Physics Letters. 2001;344:631–637. [Google Scholar]
- 55.Straus SK, Bremi T, Ernst RR. Resolution Enhancement by homonuclear J decoupling in solid-state MAS NMR. Chemical Physics Letters. 1996;262:709–715. [Google Scholar]
- 56.Scholz I, Jehle S, Schmieder P, Hiller M, Eisenmenger F, Oschkinat H, van Rossum BJ. J-deconvolution using maximum entropy reconstruction applied to 13C-13C solid-state cross-polarization magic-angle-spinning NMR of proteins. J Am Chem Soc. 2007;129:6682–3. doi: 10.1021/ja070849g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.