

Abstract
Purpose of review
Turner syndrome (TS) is a relatively common disorder of female development with cardinal features of short stature and congenital cardiovascular defects (CHD). TS is the most common established cause of aortic dissection in young women, but has received little attention outside of pediatric literature. This review focuses on emerging knowledge of the characteristics of aortic disease in TS in comparison with Marfan-like syndromes and isolated aortic valve disease.
Recent findings
The incidence of aortic dissection is significantly increased in individuals with TS at all ages, highest during young adult years and in pregnancy. Pediatric patients with dissection have known CHD, but adults often have aortic valve and arch abnormalities detected only by screening cardiac MR (CMR). Thoracic aortic dilation in TS must be evaluated in relation to body surface area (BSA). Dilation is most prominent at the ascending aorta similar to the pattern seen in non-syndromic bicuspid aortic valve (BAV), is equally prevalent (20-30%) in children and adults, and does not seem to be rapidly progressive. Cardiovascular anomalies and risk for aortic dissection in TS are strongly linked to a history of fetal lymphedema, evidenced by the presence of neck webbing and shield chest.
Summary
Risk for acute aortic dissection is increased by more than 100-fold in young and middle-aged women with TS. Monitoring frequency and treatment modalities are decided on an individual basis until more information on outcomes becomes available.
Keywords: aorta, X chromosome, adult congenital heart disease, bicuspid aortic valve, coarctation
Introduction
Turner syndrome (TS) is caused by complete or partial monosomy for the X chromosome during embryonic development. Analysis of cytogenetic screening studies indicate that TS occurs in ∼1/200 conceptions but only 1/2000 live female births[1] with congenital cardiovascular defects leading to a high rate of fetal demise[2,3]. Survival is better in gestations that are mosaic for normal cell lines resulting from sex chromosome loss in early embryonic mitotic cell divisions [2]. The phenotype in such cases depends upon the temporal and anatomical distribution of 45,X cell populations in the developing embryo. Clinical features of TS are also seen in individuals with major deletions of the X chromosome short arm (Xp). The most consistent features are short stature and premature ovarian failure. Full scale intelligence is usually normal with verbal scores typically greater than performance IQ scores[4]. With a timely diagnosis and current medical treatment, girls with TS may be fully feminized and reach a near average adult height [5]. Their educational attainments are often outstanding, and quality of life and independence as adults are steadily improving[6]. However, prospects for a normal lifespan are limited by premature mortality due largely to cardiovascular disease. Epidemiological data from European National Registries indicate a large excess of deaths due to complications from congenital heart disease and also increased ischemic heart disease[1]. This article focuses on several recent papers that have provided new information about the prevalence and risk factors for aortic disease in TS [7-11].
Congenital Heart Disease (CHD) in TS
As mentioned above, most 45,X fetuses die from cardiovascular defects mainly involving the left ventricular outflow tract. A significant number of infants diagnosed at birth with TS succumb to left heart and/or aortic hypoplasia [12]. Common congenital defects in surviving girls and adults with TS [13] include bicuspid aortic valve (∼30%) and aortic coarctation (∼12%). Including anatomic anomalies such as partial anomalous pulmonary connection, persistent left SVC or elongation of the transverse arch of the aorta (Fig. 1), approximately 50% of karyotypically-proven, asymptomatic women with TS have evidence of abnormal cardiovascular development [14]. The cause of CHD in TS remains unknown. The observation of statistically significant association between neck webbing and the presence of congenital cardiovascular defects supported Clark's hypothesis that jugular lymphatic sac obstruction in 45,X fetuses led to distended thoracic ducts, compression of the ascending aorta and reduced intracardiac blood flow[15]. Clark proposed that reduced blood flow caused impaired development of aortic valve and arch, thus invoking an extra-cardiac teratogenic event, i.e., jugular lymphatic obstruction, in altered cardiovascular morphology[15]. Subsequent studies have confirmed the association between neck webbing, the residua of fetal jugular lymphatic distention (Fig. 2), and bicuspid aortic valve (BAV) and aortic coarctation in TS[9,16]. However, it now seems likely that haploinsufficiency for a pseudoautosomal, X-chromosome gene may cause fetal lymphedema and/or cardiovascular defects, independent of each other. In support of this view, cardiovascular defects and lymphedema appear to be independent effects of haploinsufficiency for the autosomal gene FOXC2, which encodes a forkhead winged-helix transcription factor. Thus, by analogy with the key role of the pseudoautosomal gene SHOX in skeletal development and stature, it seems likely that bi-allelic expression of an X- and Y-linked gene could be essential for normal cardiovascular development. At least 20 genes are expressed from both sex chromosomes and escape X-inactivation. Most are located on Xp, consistent with observations that the TS CHD phenotype is specifically associated with haploinsufficiency for Xp[8,9].
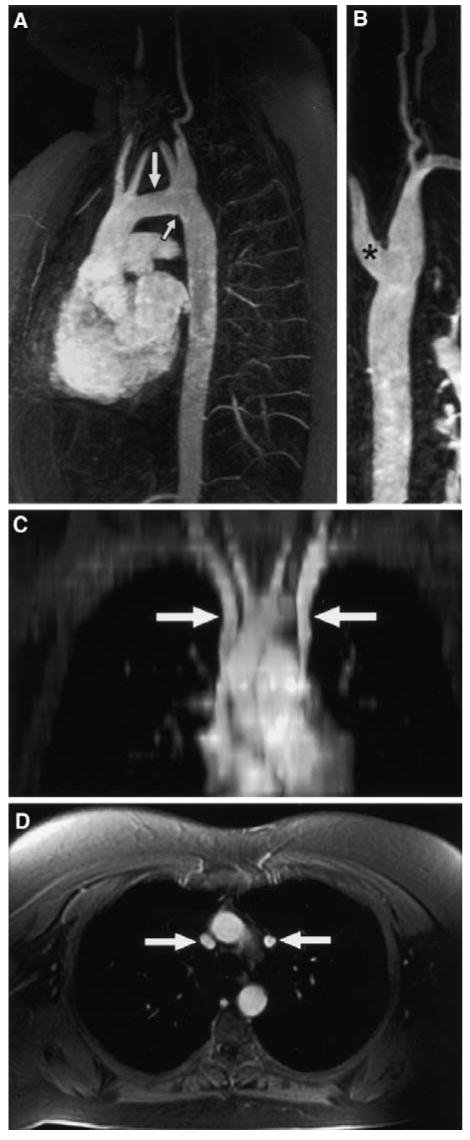
Fig. 1. Elongated transverse arch of the aorta (ETA) and associated anomalies in a woman with TS revealed by Gd-enhanced 3D MRA.
(A) demonstrates flat and elongated transverse arch (large arrow) with characteristic kink in lesser curvature (small arrow). This same woman had aberrant origins of both right and left carotids at the greater curvature (A) and both subclavian arteries off a common, dilated vessel at the lesser curvature (A&B). This type of kinking at the usual coarctation site has been termed “pseudocoarctation” in other studies. Subject also had persistent LSVC as seen on reformatted coronal (C) and axial (D) postcontrast fat-suppressed spoiled gradient echo images. From Ho et al.[14].
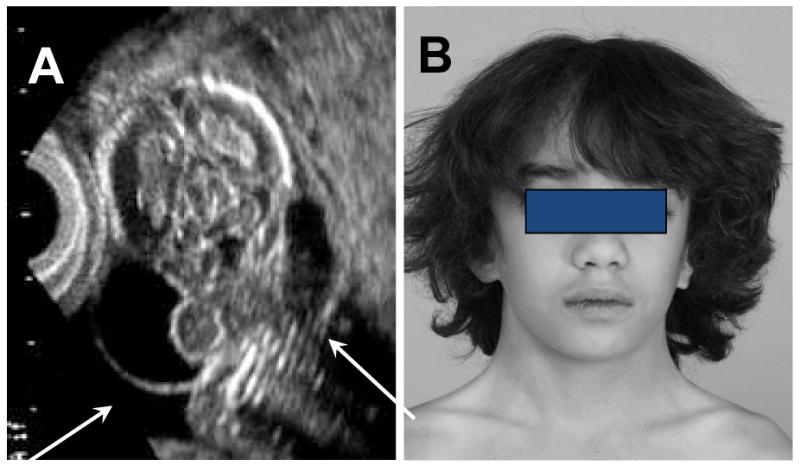
Fig. 2.
Fetal nuchal cystic hygroma result in neck webbing. A) Fetal ultrasound showing massively dilated jugular lymphatic sacs (arrows). B) Symmetric neck webbing in a 12 yo girl with TS, who has bicuspid aortic valve and aortic coarctation. Neck webbing or pterygium colli, defined as redundant skin folds extending from mastoid to acromion, result from tenting of the fetal skin over the dilated lymphatics, and is usually associated with low posterior hairline and shield chest.
An increased rate of aortic dissection in TS was initially attributed to the presence of aortic coarctation, but an influential early study suggested aortic disease in TS was similar to that in Marfan syndrome, characterized by “cystic medial degeneration”[17]. This view has been very persistent, despite lack of actual evidence for similar pathophysiology in Marfan and Turner syndromes over the years[17-19]. Recent studies have provided new information addressing the issue whether the increased risk for aortic aneurysm and dissection in TS is confined to those with aortic valve disease and coarctation, or whether there is, in addition, a Marfan-like aortopathy that predisposes to aortic complications, even in those without identifiable macrovascular defects[7-11]. This is a critically important question with direct impact on our ability to identify individuals at risk and determine safe and effective interventions, especially now as new medical treatments that may prevent aortic deterioration in Marfan-like syndrome are emerging[20].
Characteristics of aortic dissection in TS
About 2/3 of dissections begin in the ascending aorta (Stanford type A) and one third originate in descending aorta [7,21]. Dissections are usually acute, associated with chest, neck and/or back pain and often involve proximal rupture, hemopericardium and tamponade [7]. Aortic dissection in TS is commonly fatal, and many times this diagnosis is simply not considered in young women with chest pain. Regrettably, TS is usually not listed as a risk factor for aortic dissection in texts and handbooks designed for emergency care providers. The histopathology of the aorta involved in TS dissection has been described as cystic medial necrosis (CMN) in about half the cases where any pathology report was available[7], but specific molecular studies of elastin fibrils, smooth muscle cell number and vitality have not been reported.
Risk factors for Aortic dissection in TS: Age
A recent epidemiological study of Danish and Swedish records [7] found the median age of onset of aortic dissection or rupture in TS to be 35 yrs, and projected an annualized incidence of approximately 15 cases/100,000 TS years for individuals <20 years of age, 73-78 cases/100,000 for women 20-40 yrs old and ∼50/100,000 for older women with TS (Fig. 3). This is against a background rate of 6/100,000 in the general Danish population, with average age of 71 years and male: female ratio of 2:1[7]. A review of 85 cases of aortic dissection in TS reported in the medical literature found the median age to be 30 yrs [21]. Twenty cases (24%) occurred in girls less than 20 yrs old, and all of these had aortic valve disease or coarctation. As found in the Danish population based study, the highest risk for aortic dissection was among young adults aged 20-40 yrs (56% of cases).
Fig. 3.
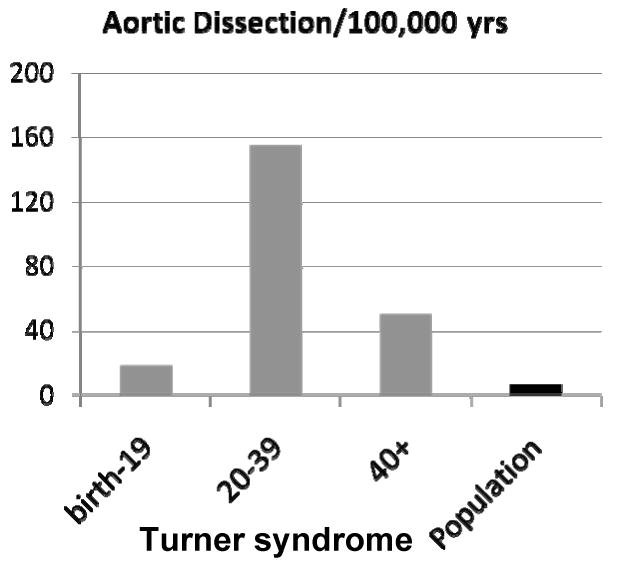
Incidence of aortic dissection in Turner syndrome by age group. This is a summary of epidemiological data reported by Gravholt et al[7]. The frequency of aortic dissection is greater in females with TS at all ages compared to the general population, with the highest incidence in young adults.
Karyotype and Phenotype
Girls with a ‘pure’ 45,X karyotype are more consistently affected with developmental anomalies including fetal lymphedema, BAV and aortic coarctation. Not unexpectedly, most reported cases of aortic dissection have had this karyotype as well[7,21]. Clinically, neck webbing and other signs of fetal lymphedema such as “shield chest” are more common in those with CHD and aortic dilation and dissection [8,10].
Hypertension
The prevalence of hypertension is clearly increased in both girls and adults with TS [22], however there has not been a convincing correlation between the blood pressure elevation and dissection in this syndrome. In the Danish study, only 4/16 had hypertension [7] and in the literature survey, about half the cases had hypertension and half did not[21], while the prevalence of hypertension in adults with TS is 40-50%. Where specific blood pressure parameters were described, hypertension in individuals with dissection was often relatively mild (e.g., 142/82 mm Hg in Case #2 of Carlson and Silberbach). The relation between blood pressure and ascending aorta diameters in TS is discussed below.
Pregnancy
In recent years, increasing numbers of women with TS have become pregnant via assisted reproduction. While the pregnancy and live birth rate have been good, there has been a high rate of maternal complications such as eclampsia, with an estimated 2% of mothers dying from aortic dissection or rupture[23]. A few women had known CHD, but most had no specific cardiac screening prior to beginning fertility treatment so cardiovascular status was unknown [23]. Pregnancy was implicated in 7/85 cases reviewed by Carlson & Silberbach, and 2/16 cases in Gravholt et al.
Underlying CHD
Most TS girls and women that develop aortic dissection have aortic valve disease and/or coarctation[7,21]. The average age of onset is, however, much younger in TS (20's and 30's) vs non-syndromic BAV and/or coarctation, which generally occurs in the 50's [24]. While apparently 100% of pediatric cases (<20 yrs) have some structural CHD [21], about 10% of adults with TS that have died from aortic dissection have no evidence of major structural defects on autopsy[7,21].
Aortopathy in TS
Because of the young age of onset of aortic pathology, the high rate of occurrence in pregnancy and reports of CMN, aortic disease in TS has been likened to a “connective tissue” disorder similar to that seen in Marfan syndrome[19]. There is some evidence for a generalized vasculopathy in TS. The internal diameters of the ascending aorta and internal carotid and brachial arteries were significantly increased in women with TS compared to age-matched control women by 10, 17 and 17%, respectively[25]. Arterial wall compliance, measured by pulse wave analysis and augmentation index, was not different in TS and control groups[25]. However, another smaller study found similar carotid diameters and increased pulse velocity in women with TS[26], although differences in height and estrogen exposure may have somewhat confounded the latter results. Girls and women with TS do not have any non-vascular signs of connective tissue defects such as joint laxity or lens dislocation. However, recent progress in elucidating the molecular pathways leading to aortic aneurysm formation in Marfan and related syndromes suggest that the concept of “connective tissue” disorder may have outlived its usefulness. It appears that disorders of diverse genetic etiologies may disrupt arterial TGF-beta signaling resulting in aneurysm formation and risk for dissection or rupture (Braverman, this issue). This is a very exciting development raising the possibility that TGF-beta antagonism by losartan or other new medications may prevent or reverse aortic pathology in these syndromes. This prospect makes the clarification of aortic pathophysiology in TS all the more urgent.
Aortic dilation
Aortic dilation has been reported on occasion in adults with TS that later experienced dissection, but the site of dilation, actual diameter, diameter in relation to body size, and rate of dilation has never been defined. For example, in the Danish study, one patient that dissected reportedly had “aorta root dilation” as a predisposing condition, but no further information was available[7]. The presence of progressive aortic root dilation is the hallmark of the TGF-beta related aortopathies, both in the human and in murine models. For clinical purposes, in Loewys-Dietiz and Marfan syndromes, a diameter of 4.5-5 cm at the aortic root (sinuses of Valsalva) or rate of increase > 0.5 cm/yr, is taken as indication for surgical intervention to prevent further deterioration It remains to be determined whether there is a similar process in TS. Patients with Marfan have a relatively focal pathology involving mainly the aortic root with aortic valve dysfunction arising secondary to root dilation hence incompetency. In contrast, 30% of individuals with TS have a congenitally abnormal valve characterized by complete or partial fusion of the right and left coronary leaflets [9]. This anomaly results in typical bicuspid aortic valve problems with development of aortic stenosis and/or regurgitation in many patients with TS. The anatomic pattern of aortic dilation in non-syndromic BAV involves the root and ascending aorta generally, with most marked effect on the ascending aorta[24,27]. It is not entirely clear whether the extent of dilation predicts dissection in isolated BAV or other etiologies of aortic disease.
Several outstanding issues are important in considering the incidence of ascending aortic dilation in TS. Firstly, most TS individuals are very small (current average adult height = 4 ft 7 in; this will increase in the future as girls are achieving greater adult height due to treatment with growth hormone). Pediatricians usually adjust aortic diameters to body surface area using Roman nomograms or more recent normative data [11], but this is rarely considered for the adult patient. However when the adult is the size of a child, it is clearly not correct to use absolute values, i.e., 5-6 cm, determined in the general population. Secondly, the prevalence of aortic dilation varies depending on the method of size normalization used and the site measured. For example, normalizing to height and measuring at the annulus, Ostberg et al reported 16% of women with TS had dilation, while measured at the ascending aorta on cardiac MR and compared with descending aortic diameter at the same level, they determined that 33% of TS women had significant dilation [28]. Recent, large studies that have compared dimensions at the annulus, sinuses, STJ and ascending aorta have found (1) BSA normalization is the most appropriate approach for determining aortic dilation in TS and (2) the ascending aorta is the most affected and informative site[8,9,11]. For example, Lopez et al. using BSA normalization in a pediatric population[11] found dilation at the annulus (12%), sinuses (20%), sinotubular junction (STJ, 18%) and ascending aorta (30%). At the NIH we found a similar anatomical profile in 250 girls and women with TS[9]. It is critically important to further progress in this area that investigators and clinicians reporting on aortic disease specify exactly where they are measuring aortic diameter, using specific terms not including “aortic root” and provide body size measurements associated with the aortic diameters to allow for size normalization.
Interestingly, most recent studies have found little correlation between blood pressure and aortic diameter or dilation in TS [9-11,28]. These studies all had more than 80 participants in contrast to an earlier small study of 38 adults reporting that hypertension was a major risk factor for aortic dilation in TS[29]. I think the reason for the discrepancy is that the earlier study had a substantial number of subjects with long-standing, poorly controlled hypertension, while subjects involved in more recent studies generally have well-controlled hypertension. A more important factor in determining ascending aortic diameter in TS is the presence or absence of BAV. Isolated BAV is associated with larger proximal aortic diameters, especially at the ascending aorta, even with normal valve function[24]. The same is true in TS, with aortic diameters significantly greater in those with BAV vs. normal tricuspid AV[9]. However, there is measureable aortic dilation in a significant number of girls and women with TS that have apparently normal aortic valves[9-11]. It is not yet clear whether aortic dilation is associated with the various other anomalies commonly found in TS (Fig. 1)[14].
Finally, there is the important question as to whether aortic dilation is progressive in individuals with TS. To my knowledge there has been only one study addressing this issue[10]. Lanzarini et al. found little change in aortic diameters measured by echocardiography at the sinuses, STJ and ascending aorta over a median 3 years of follow-up. Similar to other recent studies, they found a ∼20% prevalence of aortic dilation that was not associated with hypertension or increasing age. Nor was there evidence of progressive dilation in those with dilated aorta at baseline. Further studies are needed, but these initial data[10], coupled with the similar prevalence of aortic dilation in pediatric and adult groups[9,11], suggests that rapidly progressive aortic dilation is not a common feature of aortic disease in TS. The salient characteristics of aortic dissection in Marfan and related syndromes compared with isolated or non-syndromic BAV and TS are compared in Table 1.
Table 1. Characteristics of Aortic Dissection in Different Populations.
| Marfan et al. | Isolated BAV | TS | |
|---|---|---|---|
| Age onset (yrs) | 20-40 | 50's | 20-40 |
| Site of dilation | Sinuses V | Asc. aorta | Asc. aorta |
| Progressive dilation | Yes | +/- | ? |
| Histology | CMN | CMN | CMN |
BAV – bicuspid aortic valve; Sinuses V – sinuses of Valsalva. Asc. – ascending aorta; CMN – cystic medial necrosis.
NIH experience
In the course of a TS genotype phenotype study in the intramural clinical research program at the National Institutes of Health (NIH), 4 of our first 60 enrolled adult subjects experienced acute aortic dissection, fatal in three women. One died just before her scheduled admission but we had extensive cardiovascular data on the others and therefore have made a concerted effort to determine prospective risk factors for aortic complications in our study population in a longitudinal, natural history study. We compared girls and women with TS to age-matched control women using cardiac MR to measure ascending and descending aortic diameters at the level of the right pulmonary artery[8]. The ascending aortic diameter is normalized to BSA and termed the aortic size index (ASI). The average ASI is greater in women with TS as a group compared to healthy females (Fig. 4) and nine of the first 158 women (∼6%) had ASI greater than or equal to the 99th percentile for controls (2.5 cm/m2). The three women all had abnormal aortic valves (leaflet thickening and partial to complete fusion of coronary cusps without significant stenosis and with mild to moderate regurgitation). They also had elongated transverse arches (ETA) and other anomalies of the great vessels. They all had clear signs of fetal lymphedema with neck webbing and shield chests. Ages ranged from 44-57 yrs and absolute ascending aortic diameters from 3.7 to 4.8 cm. All three had ASI greater than the 99th percentile for controls (Fig. 5). The ascending to descending aortic diameter ratio was also higher in TS but was not a useful predictor of complications in our population [8].
Fig. 4.
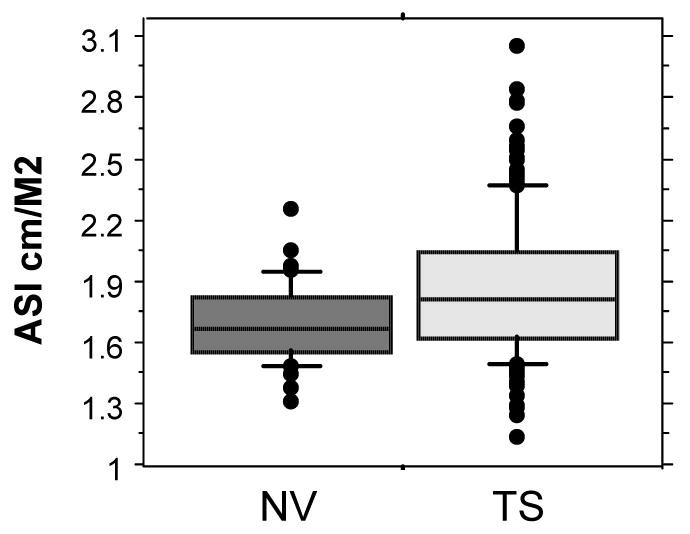
Ascending aortic diameter normalized for BSA in 160 adults with TS and age-matched control women. This normalization termed aortic size index or ASI, showed that mean ascending aortic diameter is significantly increased in TS, and that a group of women with TS have extremely elevated values. Outcomes for these cases are illustrated in Fig. 5 Data extracted from Matura et al. [8].
Fig. 5.
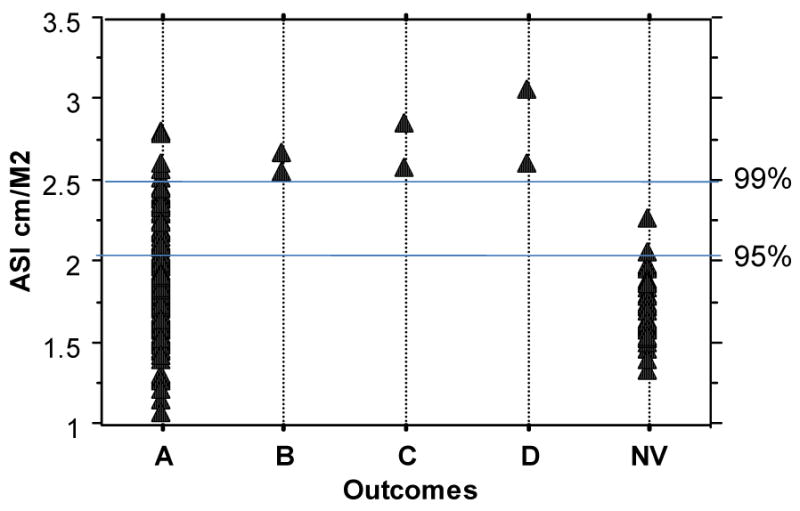
Outcomes among 158 women followed at the NIH for an average of 3 years according to baseline ASI.
A – All undergoing regular monitoring for aortic diameters; those with ASI>2 are informed about the risk, aware of potential symptoms and need for urgent care, and wear medic alert ids
B – Aortic valve replacement for valvular dysfunction was accompanied by proximal aortic graft
C – Acute aortic dissection treated with emergent surgical intervention (an additional patient since recent Matura et al paper)
D – Died from acute aortic dissection/rupture
NV – ASI data for 37 age-matched female volunteers
The stories of these three individuals are representative of the background of most of the TS adults seen in the NIH study. They all had postgraduate degrees, were productively employed and in good general health. Only one had previously known CHD; she had had surgical repair of aortic coarctation at age 7 yrs and was discharged from the pediatric cardiologist after high school. The others had echocardiography screening prior to NIH evaluation, but no abnormalities were reported. Review of their prior echocardiography reports, however, revealed no information on aortic valve structure or aortic diameters. At the NIH, transthoracic echocardiographic visualization of the aortic valve and thoracic aorta was inadequate in these three patients, but cardiac MR visualization was excellent. The difficulty with echocardiographic visualization in females with TS may be due to unusual thoracic cage architecture associated with fetal lymphedema, and it is possible that those most at risk may be the most difficult to image with routine echocardiography. We suggested the use of beta-adrenergic blocker and angiotensin converting enzyme inhibitors because of high heart rates and mild hypertension, and possibly to prevent further aortic dilation. These medications were prescribed for all three women at the time of the acute dissections. The women were referred to cardiologists in their home areas for follow-up of aortic valve function and aortic diameters. Outpatient follow-up at home indicated that blood pressures were in the normal range and aortic diameters seemed to be stable based on imaging done 9-12 mos prior to dissection. In summary, 33% (3/9) of adults with TS in the NIH study that had an ASI>2.5 cm/m2 experienced acute aortic dissection within 3 years, and all had abnormal aortic valve and arch structure detected on cardiac MR but not on transthoracic echocardiography. The overall prevalence of aortic dissection in TS based on NIH data is ∼620/100,000 TS years observation, for a relative risk of ∼200-fold compared to the general female population. Among TS women with BAV, the incidence is ∼ 1600/100,000 TS years.
At present it is not clear if the TS aortopathy is more similar to that in Marfan syndrome or to that associated with BAV and coarctation. To wit, we don't know if girls and women with TS would benefit from treatment recommendations that seem effective in Marfan syndrome such as activity limitations, treatment with beta adrenergic and/or angiotensin receptor blockers, and surgical intervention at a pre-specified aortic diameter. Additional research on the outcomes for prospectively evaluated patients and elucidation of the genetic etiology of the TS cardiovascular phenotype will provide insight into these questions. In the meantime, it is recommended that
TS girls with known CHD should be transferred to adult CHD clinics as they mature
All young adults with TS need a comprehensive CMR evaluation
-
Those with aortic anomalies and/or dilation
need close cardiological follow-up
education of patient and family about signs & symptoms of aortic dissection
medical alert ids
caution re pregnancy
control of BP
trial of angiotensin antagonism vs. beta adrenergic blockade
For those with significant aortic valve disease and aortic dilation, consider the replacement of ascending aorta at the time of aortic valve replacement
Conclusion
The incidence of aortic dissection is significantly increased in individuals with TS at all ages, but highest during young adult years and in pregnancy. Pediatric patients with dissection all had known CHD. Adults prospectively evaluated by CMR prior to dissection all have abnormal aortic valve and arch structure and dilated ascending aorta. Thoracic aortic dilation in TS must be evaluated in relation to BSA. It is most prominent at the ascending aorta similar to the pattern seen in non-syndromic BAV, is equally prevalent in children and adults, and does not seem to be rapidly progressive. Monitoring frequency and treatment modalities are decided on an individual basis until more information on outcomes becomes available.
Acknowledgments
This work was supported by the intramural research program of the NICHD, NIH
Footnotes
The author has no disclosures.
References
- 1.Stochholm K, Juul S, Juel K, et al. Prevalence, Incidence, Diagnostic Delay, and Mortality in Turner Syndrome. J Clin Endocrinol Metab. 2006;91:3897–3902. doi: 10.1210/jc.2006-0558. [DOI] [PubMed] [Google Scholar]
- 2.Hook EB, Warburton D. The distribution of chromosomal genotypes associated with Turner's syndrome: livebirth prevalence rates and evidence for diminished fetal mortality and severity in genotypes associated with structural X abnormalities or mosaicism. Hum Genet. 1983;64:24–27. doi: 10.1007/BF00289473. [DOI] [PubMed] [Google Scholar]
- 3.Surerus E, Huggon IC, Allan LD. Turner's syndrome in fetal life. Ultrasound Obstet Gynecol. 2003;22:264–267. doi: 10.1002/uog.151. [DOI] [PubMed] [Google Scholar]
- 4.Ross J, Roeltgen D, Zinn A. Cognition and the sex chromosomes: studies in Turner syndrome. Horm Res. 2006;65:47–56. doi: 10.1159/000090698. [DOI] [PubMed] [Google Scholar]
- 5.*Bondy CA. Care of Girls and Women with Turner Syndrome: A Guideline of the Turner Syndrome Study Group. J Clin Endocrinol Metab. 2007;92:10–25. doi: 10.1210/jc.2006-1374. [DOI] [PubMed] [Google Scholar]; Consensus recommendations for multidisciplinary care.
- 6.Lagrou K, Froidecoeur C, Verlinde F, et al. Psychosocial functioning, self-perception and body image and their auxologic correlates in growth hormone and oestrogen-treated young adult women with Turner syndrome. Horm Res. 2006;66:277–284. doi: 10.1159/000095547. [DOI] [PubMed] [Google Scholar]
- 7.Gravholt CH, Landin-Wilhelmsen K, Stochholm K, et al. Clinical and epidemiological description of aortic dissection in Turner's syndrome. Cardiol Young. 2006;16:430–436. doi: 10.1017/S1047951106000928. [DOI] [PubMed] [Google Scholar]
- 8.**Matura LA, Ho VB, Rosing DR, et al. Aortic dilatation and dissection in Turner syndrome. Circulation. 2007;116:1663–1670. doi: 10.1161/CIRCULATIONAHA.106.685487. [DOI] [PubMed] [Google Scholar]; Prospective study of aortic anatomy in 158 women followed for an average of 3 yrs. Three of the nine with greatest ascending aortic dilation (all less than 5 cm) had acute dissection.
- 9.**Sachdev V, Matura LA, Sidenko S, et al. Aortic Valve Disease in Turner Syndrome. Journal of the American College of Cardiology. 2008;51:1904–1909. doi: 10.1016/j.jacc.2008.02.035. [DOI] [PubMed] [Google Scholar]; Transthoracic echocardiography and cardiac MR delineate aortic valve and proximal aortic architecture in 250 females with TS.
- 10.**Lanzarini L, Larizza D, Prete G, et al. Prospective Evaluation of Aortic Dimensions in Turner Syndrome: A 2-Dimensional Echocardiographic Study. Journal of the American Society of Echocardiography. 2007;20:307–313. doi: 10.1016/j.echo.2006.08.028. [DOI] [PubMed] [Google Scholar]; Longitudinal study found 20% of a group of 78 girls and women with TS had dilated proximal aorta at baseline with little progression over median 3 yr follow-up.
- 11.**Lopez L, Arheart KL, Colan SD, et al. Turner Syndrome Is an Independent Risk Factor for Aortic Dilation in the Young. Pediatrics. 2008;121:e1622–1627. [Google Scholar]; Cross sectional echocardiographic study describes aortic dilation in girls using Z-scores for BSA-adjusted diameters. Highest incidence was 30% at the level of the ascending aorta.
- 12.Sybert VP. Cardiovascular Malformations and Complications in Turner Syndrome. Pediatrics. 1998;101:e11. doi: 10.1542/peds.101.1.e11. [DOI] [PubMed] [Google Scholar]
- 13.*Bondy CA. Congenital cardiovascular disease in Turner syndrome. Congenit Heart Dis. 2008;3:2–15. doi: 10.1111/j.1747-0803.2007.00163.x. [DOI] [PubMed] [Google Scholar]; Reviews the spectrum of CHD, ECG and autonomic abnormalities and premature coronary artery disease in TS.
- 14.Ho VB, Bakalov VK, Cooley M, et al. Major Vascular Anomalies in Turner Syndrome: Prevalence and Magnetic Resonance Angiographic Features. Circulation. 2004;110:1694–1700. doi: 10.1161/01.CIR.0000142290.35842.B0. [DOI] [PubMed] [Google Scholar]
- 15.Clark EB. Neck Web and Congenital Heart Defects: A Pathogenic Association in 45 X-O Turner Syndrome. Teratology. 1984;29:355–361. doi: 10.1002/tera.1420290305. [DOI] [PubMed] [Google Scholar]
- 16.Mazzanti L, Cacciari E. Congenital heart disease in patients with Turner's syndrome. Italian Study Group for Turner Syndrome (ISGTS) J Pediatr. 1998;133:688–692. doi: 10.1016/s0022-3476(98)70119-2. [DOI] [PubMed] [Google Scholar]
- 17.Kostich ND, Opitz JM. Ullrich-Turner Syndrome Associated with Cystic Medial Necrosis of the Aorta and Great Vessels: Case Report and Review of the Literature. Am J Med. 1965;38:943–950. doi: 10.1016/0002-9343(65)90014-8. [DOI] [PubMed] [Google Scholar]
- 18.Price WH, Wilson J. Dissection of the aorta in Turner's syndrome. J Med Genet. 1983;20:61–63. doi: 10.1136/jmg.20.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin AE, Lippe BM, Geffner ME, et al. Aortic dilation, dissection, and rupture in patients with Turner syndrome. J Pediatr. 1986;109:820–826. doi: 10.1016/s0022-3476(86)80700-4. [DOI] [PubMed] [Google Scholar]
- 20.Brooke BS, Habashi JP, Judge DP, et al. Angiotensin II Blockade and Aortic-Root Dilation in Marfan's Syndrome. N Engl J Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.*Carlson M, Silberbach M. Dissection of the aorta in Turner syndrome: two cases and review of 85 cases in the literature. J Med Genet. 2007;44:745–749. doi: 10.1136/jmg.2007.052019. [DOI] [PMC free article] [PubMed] [Google Scholar]; Reports two new cases of young Turner women with known CHD who died from ascending aortic dissection that was not diagnosed in time.
- 22.Nathwani NC, Unwin R, Brook CG, et al. Blood pressure and Turner syndrome. Clin Endocrinol (Oxf) 2000;52:363–370. doi: 10.1046/j.1365-2265.2000.00960.x. [DOI] [PubMed] [Google Scholar]
- 23.Karnis MF, Zimon AE, Lalwani SI, et al. Risk of death in pregnancy achieved through oocyte donation in patients with Turner syndrome: a national survey. Fertility and Sterility. 2003;80:498–501. doi: 10.1016/s0015-0282(03)00974-9. [DOI] [PubMed] [Google Scholar]
- 24.Braverman AC, Guven H, Beardslee MA, et al. The Bicuspid Aortic Valve. Current Problems in Cardiology. 2005;30:470–522. doi: 10.1016/j.cpcardiol.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Ostberg JE, Donald AE, Halcox JPJ, et al. Vasculopathy in Turner Syndrome: Arterial Dilatation and Intimal Thickening without Endothelial Dysfunction. J Clin Endocrinol Metab. 2005;90:5161–5166. doi: 10.1210/jc.2005-0677. [DOI] [PubMed] [Google Scholar]
- 26.Baguet JP, Douchin S, Pierre H, Rossignol AM, Bost M, Mallion JM. Structural and functional abnormalities of large arteries in Turner syndrome. Heart. 2005;91:1442–1446. doi: 10.1136/hrt.2004.048371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beroukhim RS, Roosevelt G, Yetman AT. Comparison of the Pattern of Aortic Dilation in Children With the Marfan's Syndrome Versus Children With a Bicuspid Aortic Valve. The American Journal of Cardiology. 2006;98:1094–1095. doi: 10.1016/j.amjcard.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 28.Ostberg JE, Brookes JAS, McCarthy C, et al. A Comparison of Echocardiography and Magnetic Resonance Imaging in Cardiovascular Screening of Adults with Turner Syndrome. J Clin Endocrinol Metab. 2004;89:5966–5971. doi: 10.1210/jc.2004-1090. [DOI] [PubMed] [Google Scholar]
- 29.Elsheikh M, Casadei B, Conway GS, et al. Hypertension is a major risk factor for aortic root dilatation in women with Turner's syndrome. Clin Endocrinol (Oxf) 2001;54:69–73. doi: 10.1046/j.1365-2265.2001.01154.x. [DOI] [PubMed] [Google Scholar]