

Abstract
Mutations in the presenilin 1 gene cause most early-onset familial Alzheimer’s disease (FAD). Here we report that a defect in the cell cycle—improper chromosome segregation—can be caused by abnormal presenilin function and therefore may contribute to AD pathogenesis. Specifically we find that either over-expression or FAD mutation in presenilin 1 (M146L and M146V) leads to chromosome missegregation and aneuploidy in vivo and in vitro: 1) Up to 20% of lymphocytes and neurons of FAD-PS-1 transgenic and knockin mice are aneuploid by metaphase chromosome analysis and in situ hybridization, 2) Transiently transfected human cells over-expressing normal or mutant PS-1 develop similar aneuploidy within 48 hours, including trisomy 21. 3) Mitotic spindles in the PS-1 transfected cells contain abnormal microtubule arrays and lagging chromosomes. Several mechanisms by which chromosome missegregation induced by presenilin may contribute to Alzheimer’s disease are discussed.
Keywords: Alzheimer’s disease, mitosis, chromosome segregation, microtubules, presenilin, trisomy 21, Down syndrome
1. Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder that arises when neurons in certain regions of the brain, particularly those involved in memory and cognition, are damaged and ultimately killed, probably as a consequence of the abnormal production of amyloidogenic Aβ peptides. Our molecular understanding of this pathogenic pathway has been greatly aided by the analysis of mutant genes and proteins that cause rare cases of familial AD, which appear to mimic closely the more common, sporadic form of the disease.
The majority of FAD is caused by mutant forms of the two presenilin genes, PS-1 and PS-2, most commonly PS-1. One key function of the PS proteins is to contribute to the γ-secretase enzyme, which cleaves the amyloid precursor protein (APP) in its transmembrane region to generate the C-terminus of the Aβ peptide, the major component of the amyloid deposits in the Alzheimer brain. Most FAD mutations in PS-1 or 2 change the specificity of the γ-secretase enzyme such that there is increase the production of a highly amyloidegenic class of Aβ peptide, Aβx-42, compared to the more common Aβ x-40 [5,11,12,27,47]
Several lines of evidence indicate that abnormalities in one or more aspects of the cell cycle may contribute to AD pathogenesis [1,9,14,26,28,30,31,32,42,49,50]. For example, many sporadic and familial AD patients, including those carrying presenilin mutations, exhibit a defect in chromosome segregation that leads to aneuploidy, including trisomy 21, in many cells throughout the body [10,26,30,31,49,50]. This finding is intriguing because individuals with full trisomy 21 (Down syndrome) all develop AD pathology at a very early age [7,21,32].
To determine whether defects in chromosome segregation and trisomy 21 aneuploidy seen in AD patients might contribute to their disease rather than being merely a correlative manifestation of the disease process, we used transgenic mice and transfected cells in culture to test directly the involvement of PS-1 in chromosome segregation. All assays, tissues, and cells showed comparable results and allowed the novel conclusion that overexpression or FAD mutation of PS-1 leads to chromosome missegregation and aneuploidy, including trisomy 21. Immunocytochemical staining of the PS-1 transfected cells indicated that the presenilins cause chromosome missegregation by altering the structure/function of the mitotic spindle.
2. Materials and Methods
2.1 Mice
Transgenic mice expressing the human FAD mutant presenilins M146L or M146V, or a human wild-type presenilin under control of the PDGF promoter, and their non-transgenic littermates, 14–19 months of age, were gifts of Dr. Karen Duff, Nathan Klein Institute and New York University School of Medicine [5]. The background strain was Swiss WebsterXC57BL6DBA. Homozygous knock-in mice carrying a human mutant PS-1 (M146V) coding region in place of the mouse sequence, and WT nontransgenic mice of the same genetic background (129/SvXC57BL/6), 12–15 months of age were a gift of Dr. Mark Mattson and Dr. Steven Chan of the National Institute on Aging [10].
2.2 Primary Cells
Mouse spleens were harvested and immediately triturated in RPMI 1640 medium using the ground glass ends of two Superfrost Plus slides (Fisher). The cell suspension was cultured in RPMI 1640 medium supplemented with 10% FBS, 5% PSA, 2mM L-Glutamine, 50μM β-mercapthoethanol, and 5μg/mL Concanavilin-A (Sigma) for 42–44 hours at 37°C in a 5% CO2 humidified atmosphere. Prior to harvesting, the cells were treated with 100ng/mL of colcemid (Sigma) for 40–45 minutes.
Primary neuronal cultures from whole brains of similarly aged adult mice were prepared by a modification of the method of Liesi et al. [23]. After removal of the meninges, each brain was triturated in serum-free Modified Eagle Medium (MEM) approximately 20 times, and the solid tissue was allowed to settle and thereby separate from the partially dissociated cells in the supernatant. The cells in suspension were plated on single well chamber slides (Lab-Tek) previously coated with poly-d-lysine (50μg/mL) and incubated in Neural Basal Medium with B27 supplement (Gibco) at 37°C in a 5% CO2 humidified atmosphere for one hour. The medium together with the unattached glia was removed and replaced with fresh NBM-B27 and the cultures were further incubated at 37°C for 12 – 16 hours and then prepared for in situ hybridization.
2.3 Cell Line
The hTERT-HME1 cell line (Clontech) was maintained in Mammary Epithelium Basal Medium (MEBM, Cambrex-Clonetech) supplemented with 52 μg/ml BPE, 0.5 μg/ml hydrocortisone, 10 ng/ml hEGF, 5 μg/ml insulin, 50 μg/ml gentamicin, and 50 ng/ml amphotericin-B.
2.4 Plasmids
Plasmids were constructed by inserting WT PS-1, FAD mutant PS-1(M146L), and FAD mutant PS-1(M146V) cDNA into the pcDNA3 expression vector (Clontech).
2.5 Transient Transfections
One day prior to transfection, hTERT cells (1–3 × 105cells/2mL) were plated in a 6-well plate (9.4cm2/well) with supplemented MEBM. A FuGene6-DNA complex was prepared according to the manufacturer’s protocol using a ratio of Fugene 6: DNA of 3μL:0.5μg–1μg and applied to the cells. Twenty-four hours after transfection, the cells were harvested and transferred to 100mm dishes, single well chamber slides, or 22×22mm coverslips. At 48 hours post-transfection cells were either harvested immediately for in situ hybridization or treated with 33 ng/ml colcemid for 10 hours, harvested and scored for aneuploidy in metaphase chromosome spreads or by in situ hybridization. Parallel cultures were harvested for RNA analysis by Northern Blot to determine relative expression levels. All constructs expressed at the same level, except pcDNA3-wt, which expressed at about half the level of the others.
2.6 Metaphase Chromosome Analysis
Following colcemid treatment, cells were harvested according to standard cytogenetic methods. Briefly, hypotonic treatment in 0.075M KCl for 15 minutes in a 37°C water bath followed by 3:1 methanol: acetic acid fixative for a minimum of 30 minutes on ice. Cells were dropped onto glass slides (Fisherbrand Frosted Microscope Slides) and allowed to age. Metaphase spreads were stained with Giemsa (Fisher) and the chromosomes counted.
2.7 In Situ Hybridization
A bacterial artificial chromosome (BAC) containing a mouse chromosome 16-specific sequence was provided by Dr. Bruce Lamb at Case Western Reserve University [20]. The BAC probe was labeled by nick translation (Roche) with spectrum green dUTP (Vysis). For each assay, 60ng of labeled BAC probe was precipitated with 0.16mg/mL Mouse COT-1 DNA (Invitrogen), 1/10th volume of 3M Sodium Acetate, and 2.5 volumes cold 100% Ethanol. The precipitated BAC probe was re-suspended in 10μL Hybrisol I (Invitrogen) and incubated in a 37°C water bath overnight. The pre-incubated BAC probe was used for fluorescence in situ hybridization (FISH) of mouse primary cultures. Interphase and metaphase FISH of hTERT-HME1 cells was performed using either the LSI 21 SpectrumOrange probe or the LSI TEL/AML1 ES Dual Color Translocation Probe (Vysis). The LSI 21 probe detects the complementary DNA sequence on chromosome 21 while the LSI TEL/AML1 ES Dual Color Translocation Probe is a mixture of the LSI TEL probe labeled with SpectrumGreen and the LSI AML1 probe labeled with SpectrumOrange, and detects both chromosome 12 and 21. Hybridizations were done according to manufacturer’s instructions (Vysis) using the Hybrite hybridization chamber (HYBrite, Vysis) followed by counter-staining with Vectashield Mounting Medium with DAPI (4,6-dimidino-2-phenylidone)/antifade (Vector).
2.8 Image Acquisition and Analysis
To assure that only optimal slides were used for FISH, all slides were evaluated by phase-contrast microscopy prior to hybridization. Hybridization signals were scored and enumerated according to Vysis guidelines using a Nikon Eclipse E1000 microscope with a 4912 CCIR high performance COHU CCD Camera. Genus 2.81® software was used to process images (Applied Imaging). Only single non-overlapped cells that contained no visible cytoplasm surrounding the interphase nucleus were scored. Interphase spot counting was performed separately for each probe and only bright compact signals were counted using DAPI, FITC, and TRITC Nikon filter cubes with a Nikon Eclipse E1000 fluorescence microscope and Genus 2.81 software.
2.9 Immunocytochemistry
Plated primary neuronal cultures were fixed with cold 3:1 methanol: acetic acid for 30 minutes, then permeabilized in 4XSSC at 37°C for 45 minutes. Neurons were stained with NeuN antibody (Chemicon International), 1:100, followed by AlexaFluor 568-conjugated anti-mouse secondary antibody (Molecular Probes), 1:1000. After final washes, coverslips were mounted onto slides with Vectashield mounting medium containing DAPI (Vector Laboratories, Inc.).
To analyze mitotic spindles, post-transfection cells plated on 22×22 mm coverslips were fixed with 4% paraformaldehyde at 37°C for 30 minutes. Following fixation, cells were permeabilized with 0.2% Triton X-100 in PBS for 30 minutes. The microtubules were stained with anti-α-tubulin antibody (Sigma, clone B-5-1-2), 1:500, followed by AlexaFluor 488-conjugated anti-mouse secondary antibody (Molecular Probes, A11029), 1:1000. After final washes, coverslips were mounted onto slides with Vectashield mounting medium containing DAPI (Vector).
2.10 Statistical Analysis
The statistical significance of the differences in aneuploidy was determined using Student’s T test. A paired T test was used to compare the results of transfecting different plasmids into the hTERT–HME cells in multiple experiments after background aneuploidy from mock transfected cells was subtracted. When multiple experiments with different levels of background aneuploidy were combined as in the analysis of the mitotic spindles, the data are presented as fold increase over background, which best allows the inclusion of appropriate error bars. Five-ten mice of each group and three-seven transfections of each plasmid were analyzed.
3. Results
To investigate the potential role of the presenilins in the cell cycle and chromosome segregation, we used metaphase chromosome analysis and DNA in situ hybridization to count chromosomes in cells expressing either the normal human PS-1 gene or one of two FAD mutant PS-1 genes (M146V or M146L).
3.1 Aneuploidy in spleen cells from PS-1-FAD mice
We first examined the effect of PS-1 on chromosome segregation in transgenic mice in which the PDGF promoter induces high PS expression in CNS neurons and lower expression in other cells [5]. First, classical cytogenetics such as metaphase chromosome analysis was used to analyze primary mouse splenocytes from transgenic PS-1 (WT, M146L and M146V) and non-transgenic mice. Splenocytes were chosen for this analysis because they can be induced to divide by addition of Concanavilin A, a requirement for metaphase analysis, and because the PDGF promoter is active in the spleen. The dividing cells were arrested at metaphase by colcemid treatment and the chromosomes stained and counted. Blinded metaphase chromosome analysis of 3500 splenocytes showed twice the level of aneuploidy (ie 30–35% of cells with < or > the normal mouse complement of 40 chromosomes) in the PS-1-FAD animals in comparison to PS-1 WT transgenic or the non-transgenic animals (Fig. 1A).
Figure 1.

Aneuploidy induced in mouse spleen cells by a FAD mutant presenilin transgene. (A) Karyotype analysis of spleen cells of transgenic (WT: normal human PS-1; M146L: FAD mutant M146L PS-1; M146V: FAD mutant M146V PS-1) and non-transgenic mice (NON) revealed significantly higher levels of aneuploidy in the mutant PS-1 transgenic spleen cells. Mice were 15–17 months of age. (B) Fluorescence in situ hybridization (FISH) with a BAC probe (labeled with spectrum green) was used to score chromosome 16 in both interphase and metaphase spleen cells from mice transgenic for human PS-1 compared to nontransgenic mice. DAPI was used as a counter-stain. Diploid chromosome 16 is seen as two signals in the interphase cell on the left and seen as 4 signals, or 2 pairs of signals as in the metaphase cell on the right. (C) Quantitative in situ hybridization of spleen cells from transgenic and non-transgenic mice revealed significantly higher levels of trisomy 16 in the mutant PS-1 spleen cells. Mice were 14–19 months of age.
In contrast to metaphase chromosome analysis, fluorescence in situ DNA hybridization (FISH) allows one to assess aneuploidy for particular chromosomes in cells independent of their cell cycle phase. A hybridization probe was generated from a bacterial artificial chromosome (BAC) carrying a 300 kb fragment of mouse chromosome 16 [20] (the mouse chromosome most similar to human chromosome 21) and the DNA was labeled with spectrum green by nick translation. A total of 3000 splenocytes were hybridized and blindly scored for the number of fluorescent spots to measure both hybridization efficiency and aneuploidy for chromosome 16. Depending on the cell cycle phase in which the individual cells were harvested, metaphase and interphase cells from non-transgenic mice were expected to harbor an even number of chromosomes and thus show usually two and sometimes four signals for cells in the G-2-M phase of the cell cycle (Fig. 1B). To assure the accuracy of our data and to avoid any wrongful counting due to incomplete hybridization, we scored only triple signals (trisomy) as aneuploid. Cells with only one signal were scored as incomplete hybridizations. A comparison of the number of cells with one and two hybridization spots showed that the labeled BAC hybridized with greater than 90% efficiency. This finding, together with the count of the tetrasomic (ie G2-M phase) cells, assured us that the majority of cells with three spots were indeed trisomic and not the result of incomplete hybridization of the four chromosomes in G2-M phase cells. When trisomy 16 cells were scored, splenocytes from the nontransgenic and PS-1 WT transgenic mice showed 6.4% and 7.7% trisomy 16 respectively. In contrast, the PS-1 (M146L) mice showed 15% trisomy 16 and the PS-1 (M146V) mice showed 19% trisomy 16, more than two fold higher than the mice expressing the WT human PS-1 gene (Fig. 1C). Like the metaphase chromosome analysis, FISH revealed more aneuploidy in cells carrying the M146V mutation compared to cells carrying the M146L mutation. Overall, both chromosome counting methods showed that mutant PS-1 genes induce aneuploidy in the splenocytes of PS-1 transgenic mice.
3.2 Aneuploidy in neurons from PS-1 transgenic mice
FISH can also be used to measure aneuploidy in non-dividing cells such as neurons. Whole brains from PS-1 transgenic and non-transgenic mice were processed to yield primary neuronal cultures in which over 90% of the cells were positive for NeuN (data not shown). The isolated neurons were then hybridized with the mouse chromosome 16 BAC probe. The hybridization efficiency was approximately 80%, with most neurons being disomic, i.e. exhibiting two signals (Fig. 2A, left). In addition, we found approximately 3% trisomy 16; (Fig. 2A, right) in neurons from the PS-1-FAD (M146L) transgenic mice and 4% trisomy 16 in neurons from the PS-1-FAD (M146V) transgenic mice (Fig. 2B). Neurons from the nontransgenic mice showed almost no trisomy 16 aneuploidy and the PS-1 WT neurons showed less than 1%. Approximately 1500 neurons were blindly scored from each type of mouse.
Figure 2.

Aneuploidy induced in primary mouse brain neurons by a FAD PS-1 transgene. (A) FISH with a BAC probe (labeled with spectrum green) was used to score chromosome 16 in cultured neurons from PS-1 transgenic and non-transgenic mice. DAPI was used as a counter-stain. Chromosome 16 trisomy (right) reveals aneuploidy in neurons of PS-1 transgenic mice. Only 2 signals (disomy) were usually observed in neurons from non-transgenic mice (left). (B) Quantitative FISH of neurons from transgenic (WT: normal human PS-1; M146L: FAD mutant M146L PS-1; M146V: FAD mutant M146V PS-1) and non-transgenic mice (NON) revealed significantly higher levels of trisomy 16 only in the neurons from mice transgenic for FAD mutant PS-1. Mice were 14–19 months of age. (C) Quantitative FISH of cultured neurons from mutant (M146V) PS-1 Knock In (PS KI) and non-transgenic mice revealed significantly higher levels of trisomy 16 in the (M146V) PS-1 KI neurons. PS KI mice were 10–15 months of age and the non-transgenics were 17 months of age.
3.3 Aneuploidy in neurons from PS-1 knock-in mice
Because transgenic mice can exhibit features that are due in part to the location of the transgene and/or an abnormal level of transgene expression, we also investigated chromosome aneuploidy in the neurons of PS-1 knock-in mice in which an FAD-PS-1 (M146V) gene had been placed under the control of the normal mouse PS-1 promoter [10]. As shown in Figure 2C, trisomy 16 aneuploid neurons were detected at the same, greatly increased frequency in the brain neurons of the PS-1 (M146V) knock-in mice as they were detected in neurons from the PS-1 (M146V) transgenic mice. This result clearly shows that human FAD mutant PS-1 expressed at physiologically correct levels in the physiologically correct tissues leads to chromosome missegregation and aneuploidy.
3.4 Aneuploidy in PS-1 transfected cells
To determine whether the aneuploidy observed in FAD-PS-1 transgenic and knock-in mice was indeed caused directly by the PS-1 gene expression, we asked whether PS-1 genes could also induce aneuploidy after transient expression in mammalian cells in culture. A preliminary obstacle to this analysis was the karyotype instability that is characteristic of oncogene-immortalized cell lines. For example, our preliminary results showed that the aneugenic effect of transfected presenilin genes was possible but difficult to measure in human EBV-transformed lymphocytes [31]. To improve the signal/noise ratio in the assay, we turned to the hTERT-HME1 cell line, a primary human mammary epithelial cell line that expresses the telomerase reverse transcriptase from a permanently-transfected hTERT plasmid (Clontech) and is thus both immortal and karyotypically stable.
Parallel cultures of hTERT cells were transiently transfected with WT PS-1, mutant PS-1 (M146L), and control empty vector (pcDNA3). The effect of transient PS-1 expression was determined first by counting the number of copies of chromosome 21 by FISH using probes that detect either chromosome 21 alone or both chromosomes 21 and 12 as different colors. Overexpression of both WT PS-1 and FAD mutant PS-1 induced chromosome missegregation and the development of trisomy 21 cells (Fig. 3).
Figure 3.
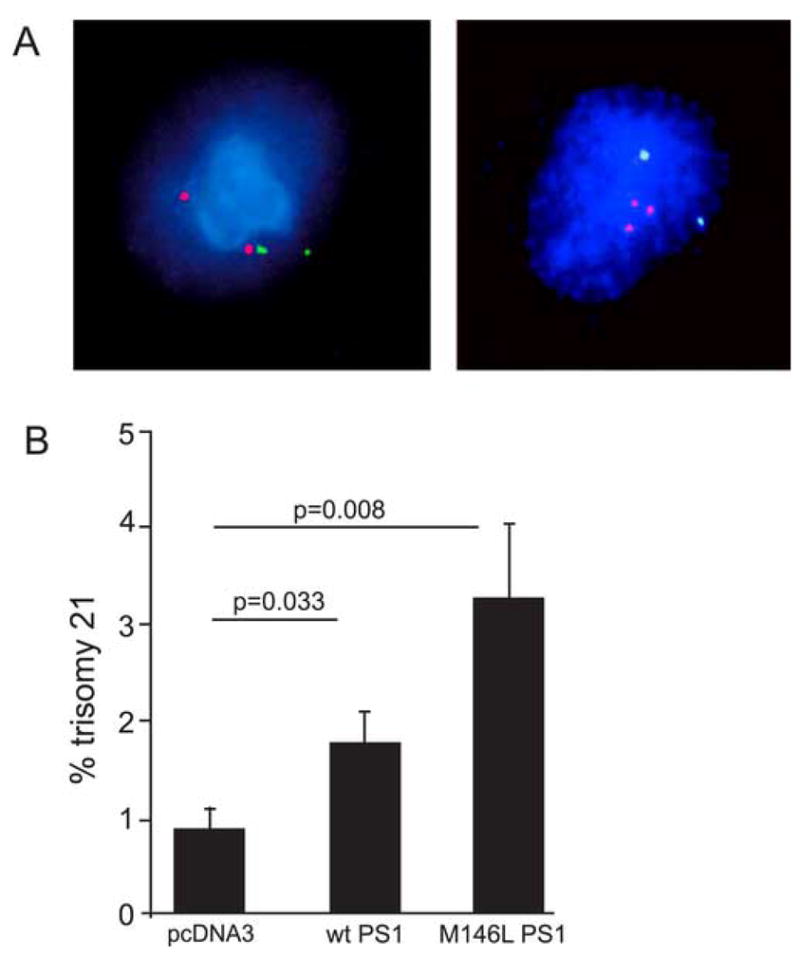
Increase in trisomy 21 aneuploidy in human cells induced by transfected normal and FAD mutant PS-1. (A) Examples of FISH with the LSI TEL/AML1 ES dual color probe used to detect both chromosome 12 (Spectrum Green) and 21 (Spectrum Orange) in PS-1 transfected hTERT cells. DAPI was used as a counter-stain. Chromosome 21 trisomy (right) reveals aneuploidy in a PS-1 transfected hTERT cell which is normal (disomic) for chromosome 12. A typical non-transfected hTERT cell (left) reveals normal (disomy) karyotype as evidenced by 2 red signals and 2 green signals. (B) Quantitative FISH of hTERT cells transfected with WT or mutant (M146L) PS-1 plasmids revealed significantly higher aneuploidy (trisomy 21) in both when compared to cells transfected with the empty vector.
Further analysis of hTERT cells over-expressing either WT or FAD mutant presenilin showed that trisomy 21 is not the only outcome of the PS-induced cell cycle defect. Specifically, cells trisomic for chromosome 12 were also observed (Fig. 4A), suggesting that presenilin affects the process of chromosome segregation generally. Indeed, by counting all chromosomes in the transfected cells in metaphase, we found that up to 30% of cells expressing wt or M146V PS-1 became aneuploid within 48 hours by either gaining or losing chromosomes during mitosis (Fig. 4B). Together these results show that the aneugenic activity of PS-1 expression likely affects all chromosomes and therefore probably alters some aspect of normal mitosis.
Figure 4.
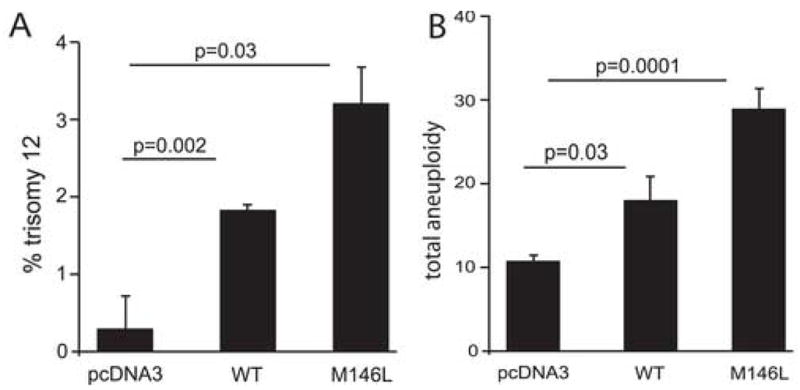
PS-1-transfected human cells develop aneuploidy for many chromosomes. (A) Quantitative FISH on hTERT cells transiently transfected with WT or mutant (M146L) PS-1 revealed significant trisomy 12 aneuploidy when compared to empty vector control. (B) Karyotype analysis of hTERT cells transiently transfected with WT or mutant (M146L) PS-1 revealed significant aneuploidy when compared to cells transiently transfected with the empty vector. Up to 30% of cells became aneuploid 48 hours after transfection, and all size classes of chromosomes were affected.
3.5 Overexpression of either WT or FAD-PS-1 causes abnormal mitotic spindles
For overexpression or FAD mutation in PS-1/γ-secretase to cause chromosome missegregation, an essential aspect of mitosis must be affected. We therefore examined the mitotic spindles of the cells transfected with PS-1 plasmids and found numerous abnormalities (Fig. 5A). The most common defect observed was a disruption or malformation of the microtubule array such that there was no classic spindle structure with two clear microtubule organizing centers/centrosomes connected by microtubules to the DNA. Cells with lagging chromosomes were also observed. Quantification of the results in Fig. 5B shows that transient over-expression of either WT or mutant PS-1 leads to a significant increase in the number of abnormal mitotic spindles. These data strongly suggest that the mechanism by which PS-1 over-expression/mutation causes chromosome missegregation and aneuploidy, including trisomy 21, is by interfering with the normal structure and function of the microtubules and/or mitotic spindle.
Figure 5.

Mitotic spindle abnormalities in PS-1 transfected cells. (A) hTERT cells were transfected for 48 hours with either the WT or M146L PS-1 plasmid or the empty vector and then stained with anti-α-tubulin monoclonal antibody and DAPI as a co-stain. Typical examples of cells in metaphase and anaphase are shown. Note the well-formed spindles, the correctly-positioned DNA and the clearly-defined microtubule organizing centers/centrosomes in the vector-transfected cells (a,b). In contrast, cultures transfected with PS-1-expressing plasmids developed numerous cells with abnormal mitotic spindles by 48 hours (c,d). For example, the first cell (c) appears to be in metaphase, however, there is no evidence that any centrosome or microtubule organizing center (MOC) has begun to develop, although the DNA is tightly arrayed along a metaphase plate. The next cell (d) appears to be in anaphase, but again the microtubule array and centrosomes are not well defined. Note also the apparent presence of lagging chromosomes left at the metaphase plate as the rest of the DNA has begun to move to opposite poles of the cell. (B) Quantification of the abnormal mitotic figures in cells transiently transfected with WT or mutant (M146L) PS-1 show more mitotic abnormalities (abnormal spindle appearance and lagging chromosomes) than cells transfected with the empty vector.
4. Discussion
Down syndrome patients carry three copies of chromosome 21 in all of their cells due to chromosome missegregation during meiosis, and invariably develop AD pathology at an early age [7,21]. The data of this paper reinforce and complement the connections between trisomy 21/Down syndrome and Alzheimer’s disease by providing evidence that chromosome missegregation during mitosis may also contribute to AD. Specifically, we have found that the expression of FAD mutant PS-1 in transgenic and knock-in mice and the expression of both wt and mutant PS-1 in transfected cells leads to the malformation/disruption of the mitotic spindle, the missegregation of chromosomes during mitosis, and the consequent development of aneuploid cells. In human cells, the presenilin-induced aneuploidy includes, but is not limited to trisomy 21. We found little evidence that overexpression or mutation of presenilin caused splenocytes, neurons, or hTERT cells to duplicate their chromosomes without dividing and thus become tetrasomic, another cell cycle abnormality that has been proposed and reported to develop in certain regions of the human AD brain, where it may contribute to neuronal cell death [14,28,42,49,50].
Together, our results suggest that mutations in the presenilin genes that change the level, structure or function of the protein may contribute to AD not only by increasing the production of the Aβ X-42 peptide and thus of amyloid deposits, but also by inducing chromosome missegregation and the development of aneuploid cells.
This conclusion is consistent with and may explain several previous findings about the presenilins. For example, as mentioned in the introduction, significant numbers of trisomy 21 and other aneuploid cells have been found in both sporadic and familial AD patients, including individuals carrying a mutation in PS-1 or PS-2 [9,26,31,49,50]. Similarly, two polymorphisms in PS-1, one in the coding sequence and one in the promoter have been found to be associated with both an increased risk of AD and an increased incidence of Down syndrome offspring due to chromosome missegregation during meiosis [24,29]. The finding of endogenous presenilin proteins in structures related to mitosis—centrosomes, kinetochores, and the nuclear envelope - also suggests that PS-1 and 2 play a role in the cell cycle and chromosome segregation [13,18,22]. Significantly, the microtubule-associated protein CLIP 170, which is also present in the centrosome and is required for centrosome function, binds to presenilin and is essential for the γ-secretase processing of APP into Aβ [17,40]. Presenilin also affects β-catenin signalling which then helps regulate the cell cycle [2], a process that could be disrupted by PS over expression or mutation. Finally, under or over-expressed or mutant presenilins inhibit the cell cycle [15,16], increase cells’ sensitivity to apoptosis [6,10,43,44,48] and, in transgenic mice, results in age-related neurodegeneration [3,36] and reduced neurogenesis [9,45,46,51]. All of these effects would be a natural consequence of aneuploidy activating the mitotic cell cycle checkpoint.
The analysis of chromosomes in PS-1 transgenic and knock-in mice indicates that, at low, physiological, levels of expression, FAD mutant PS-1 genes causes chromosome missegregation and aneuploidy while the WT PS-1 gene does not. The finding that exposure of cultured hTERT cells to over-expression of either WT or FAD mutant PS-1 both cause chromosome abnormalities is interesting and, at first sight, perhaps a little unexpected. However, it is consistent with the finding that FAD mutations in the presenilin genes lead to a dominant gain-of-function that might be mimicked by overexpression of the WT PS protein [11,12,47]. Evidently, lower levels of transfected PS-1 expression comparable to that present in the transgenic and knockin mice, are required to reveal the difference between WT and FAD mutant PS-1 on chromosome segregation. A more sensitive cell assay might more easily reveal the difference between aneugenic effects of WT and FAD-PS-1.
In as much as mutations in presenilin lead to both AD and chromosome missegregation, it will be of interest to discover how these two presenilin effects are related. Several possibilities can be envisioned and are the subject of current investigations. For example, the process of mitotic spindle malformation/disruption and consequent chromosome missegregation in neural stem cells could trigger a cell cycle checkpoint that leads to apoptosis and/or reduced neurogenesis, although recent work in mice suggests that aneuploid neurons survive and function well [19]. Also, the evident PS-induced disruption of the microtubule network indicates that PS overexpression or mutation likely interferes with microtubule formation or function, possibly through an effect on microtubule associated proteins such as Tau. In AD, the result could be tangle pathology, neuronal malfunction or death.
Alternatively or in addition, the aneuploid cells observed in AD patients, mice, and PS-1-transfected cells suggest that the product of chromosome missegregation, especially trisomy 21 cells in the brain, may also play an important role in AD [32]. Such trisomy 21 cells could trigger a cascade of events in classical AD that leads to neurodegeneration in a manner similar to what occurs in Down syndrome, but more slowly due to the modulating effect of the majority population of normal diploid cells in the same organ. Pathogenic trisomy 21 cells in the brain could include both the neurons, which produce the majority of APP and Aβ, and, perhaps equally importantly, the glial cells, which are activated in Down syndrome and AD brain and are essential for the development of AD pathology through their production of inflammatory cytokines and amyloid-promoting proteins, particularly apoE and antichymotrypsin [33].
Finally, it is relevant that early onset FAD has been reported in six families carrying a duplication of one of their APP genes and that APP promoter mutations that increase expression are linked to increased AD risk [35,42,44]. Furthermore, women who have a Downs syndrome/trisomy 21 child before the age of 35 are usually aneuploid for trisomy 21 themselves and have a five-fold increased risk of developing AD later in life [26,37,38]. These findings indicate that overexpression or an extra copy of normal APP, either in Down syndrome, in the duplicated APP families, or because of spontaneous or PS-induced trisomy 21 mosaicism can lead to AD.
The data indicate that increased/altered presnilin function interferes with microtubule function, leading to chromosome missegregation. Whether this novel function of presenilin involves γ-secretase cleavage and which cleavage products are responsible for the effect of presenilin on microtubules and the cell cycle is unknown, but the connections to AD reviewed above suggest that APP or one of its cleavage products such as AICD or Aβ is likely to be involved.
In sum, the data of this paper and previous results indicate that both sporadic and familial Alzheimer’s disease exhibit defects in mitosis and chromosome segregation, which can lead to trisomy 21 and other mosaicism, particularly through the action of the presenilin. Interestingly, age itself is the greatest risk factor for developing AD, and low levels of aneuploidy have also been shown to develop in mature brain neurons because of mitotic defects in neural precursor cells [19,34,51]. The fact that chromosome segregation defects underlie another disease of aging—cancer [4]—suggests that perhaps microtubule disfunction leading to different degrees and types of aneuploidy underlie many manifestations of “normal” aging in addition to predisposing to Alzheimer’s disease when chromosome 21 is affected [32].
Acknowledgments
We thank Dr. Karen Duff for providing PS-1 transgenic mice, Mark Mattson and Steven Chan for PS-1 knock-in mice, Dr. Todd Golde for the M146L PS-1 plasmid, and Dr. Bruce Lamb for the mouse chromosome 16 BAC. We also thank Ann Thomas and Danielle Conforto for some of the spleen counts reported and Dr. Maxine Sutcliffe for advice on cytogenetics and in situ techniques. The research was supported by the Alzheimer’s Association grant #IIRG-2 96-038, the Eric Pfeiffer Chair for Research in Alzheimer’s Disease at the Suncoast Gerontology Center at USF, the Johnnie B. Byrd Sr. Alzheimer’s Center and Research Institute, and private donors. Additional funding provided by NIA grant #AG09665. The majority of these data were presented at the 2004 Society for Neuroscience meeting.
Footnotes
5.1 Disclosure Statement: No author has a conflict of interest. All Animal studies have been approved by the USF IACUC.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arendt T, Rodel L, Gartner U, Holzer M. Expression of the cyclin-dependent kinase inhibitor p16 in alzheimer’s disease. Neuroreport. 1996;7(18):3047–3049. doi: 10.1097/00001756-199611250-00050. [DOI] [PubMed] [Google Scholar]
- 2.Chevallier NL, Soriano S, Kang DE, Masliah E, Hu G, Koo EH. Perturbed neurogenesis in the adult hippocampus associated with presenilin-1 A246E mutation. Am J Pathol. 2005;167(1):151–159. doi: 10.1016/S0002-9440(10)62962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, Inoue H, Shirotani K, Takahashi K, Gallyas F, Tabira T. Transgenic mice with alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999;5(5):560–564. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- 4.Duesberg P. Are centrosomes or aneuploidy the key to cancer? Science. 1999;284(5423):2091–2092. doi: 10.1126/science.284.5423.2089f. [DOI] [PubMed] [Google Scholar]
- 5.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383(6602):710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 6.Eckert A, Marques CA, Keil U, Schussel K, Muller WE. Increased apoptotic cell death in sporadic and genetic alzheimer’s disease. Ann NY Acad Sci. 2003;1010:604–609. doi: 10.1196/annals.1299.113. [DOI] [PubMed] [Google Scholar]
- 7.Epstein CJ. The consequences of chromosome imbalance. Am J Med Genet Suppl. 1990;7:31–37. doi: 10.1002/ajmg.1320370706. [DOI] [PubMed] [Google Scholar]
- 8.Feng R, Wang H, Wang J, Shrom D, Zeng X, Tsien JZ. Forebrain degeneration and ventricle enlargement caused by double knockout of alzheimer’s presenilin-1 and presenilin-2. Proc Natl Acad Sci USA. 2004;101(21):8162–8167. doi: 10.1073/pnas.0402733101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geller LN, Potter H. Chromosome missegregation and trisomy 21 mosaicism in alzheimer’s disease. Neurobiol Dis. 1999;6(3):167–179. doi: 10.1006/nbdi.1999.0236. [DOI] [PubMed] [Google Scholar]
- 10.Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5(1):101–106. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- 11.Haass C, De Strooper B. The presenilins in alzheimer’s disease--proteolysis holds the key. Science. 1999;286(5441):916–919. doi: 10.1126/science.286.5441.916. [DOI] [PubMed] [Google Scholar]
- 12.Hardy J, Selkoe DJ. The amyloid hypothesis of alzheimer’s disease: Progress and problems on the road to therapeutics. Science. 2002 Jul;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 13.Honda T, Nihonmatsu N, Yasutake K, Ohtake A, Sato K, Tanaka S, Murayama O, Murayama M, Takashima A. Familial alzheimer’s disease-associated mutations block translocation of full-length presenilin 1 to the nuclear envelope. Neurosci Res. 2000;37(2):101–111. doi: 10.1016/s0168-0102(00)00106-1. [DOI] [PubMed] [Google Scholar]
- 14.Husseman JW, Nochlin D, Vincent I. Mitotic activation: A convergent mechanism for a cohort of neurodegenerative diseases. Neurobiol Aging. 2000;21(6):815–828. doi: 10.1016/s0197-4580(00)00221-9. [DOI] [PubMed] [Google Scholar]
- 15.Janicki SM, Monteiro MJ. Presenilin overexpression arrests cells in the G1 phase of the cell cycle. arrest potentiated by the alzheimer’s disease PS2(N141I)mutant. Am J Pathol. 1999;155(1):135–144. doi: 10.1016/S0002-9440(10)65108-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janicki SM, Stabler SM, Monteiro MJ. Familial alzheimer’s disease presenilin-1 mutants potentiate cell cycle arrest. Neurobiol Aging. 2000;21(6):829–836. doi: 10.1016/s0197-4580(00)00222-0. [DOI] [PubMed] [Google Scholar]
- 17.Johnsingh AA, Johnston JM, Merz G, Xu J, Kotula L, Jacobsen JS, Tezapsidis N. Altered binding of mutated presenilin with cytoskeleton-interacting proteins. FEBS Lett. 2000;465(1):53–58. doi: 10.1016/s0014-5793(99)01664-6. [DOI] [PubMed] [Google Scholar]
- 18.Kimura N, Nakamura SI, Honda T, Takashima A, Nakayama H, Ono F, Sakakibara I, Doi K, Kawamura S, Yoshikawa Y. Age-related changes in the localization of presenilin-1 in cynomolgus monkey brain. Brain Res. 2001;922(1):30–41. doi: 10.1016/s0006-8993(01)03146-8. [DOI] [PubMed] [Google Scholar]
- 19.Kingsbury MA, Friedman B, McConnell MJ, Rehen SK, Yang AH, Kaushal D, Chun J. Aneuploid neurons are functionally active and integrated into brain circuitry. Proc Natl Acad Sci USA. 2005;102(17):6143–6147. doi: 10.1073/pnas.0408171102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kulnane LS, Lehman EJ, Hock BJ, Tsuchiya KD, Lamb BT. Rapid and efficient detection of transgene homozygosity by FISH of mouse fibroblasts. Mamm Genome. 2002;13(4):223–226. doi: 10.1007/s00335-001-2128-5. [DOI] [PubMed] [Google Scholar]
- 21.Lemere CA, Lopera F, Kosik KS, Lendon CL, Ossa J, Saido TC, Yamaguchi H, Ruiz A, Martinez A, Madrigal L, Hincapie L, Arango JC, Anthony DC, Koo EH, Goate AM, Selkoe DJ, Arango JC. The E280A presenilin 1 alzheimer mutation produces increased A beta 42 deposition and severe cerebellar pathology. Nat Med. 1996;2(10):1146–1150. doi: 10.1038/nm1096-1146. [DOI] [PubMed] [Google Scholar]
- 22.Li J, Xu M, Zhou H, Ma J, Potter H. Alzheimer presenilins in the nuclear membrane, interphase kinetochores, and centrosomes suggest a role in chromosome segregation. Cell. 1997;90(5):917–927. doi: 10.1016/s0092-8674(00)80356-6. [DOI] [PubMed] [Google Scholar]
- 23.Liesi P, Fried G, Stewart RR. Neurons and glial cells of the embryonic human brain and spinal cord express multiple and distinct isoforms of laminin. J Neurosci Res. 2001;64(2):144–167. doi: 10.1002/jnr.1061. [DOI] [PubMed] [Google Scholar]
- 24.Lucarelli P, Piciullo A, Palmarino M, Verdecchia M, Saccucci P, Arpino C, Curatolo P. Association between presenilin-1 -48C/T polymorphism and down’s syndrome. Neurosci Lett. 2004;367(1):88–91. doi: 10.1016/j.neulet.2004.05.086. [DOI] [PubMed] [Google Scholar]
- 25.Migliore L, Boni G, Bernardini R, Trippi F, Colognato R, Fontana I, Coppede F, Sbrana I. Susceptibility to chromosome malsegregation in lymphocytes of women who had a down syndrome child in young age. Neurobiol Aging. 2006;27(5):710–716. doi: 10.1016/j.neurobiolaging.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 26.Migliore L, Botto N, Scarpato R, Petrozzi L, Cipriani G, Bonuccelli U. Preferential occurrence of chromosome 21 malsegregation in peripheral blood lymphocytes of alzheimer disease patients. Cytogenet Cell Genet. 1999;87(1–2):41–46. doi: 10.1159/000015389. [DOI] [PubMed] [Google Scholar]
- 27.Murphy MP, Uljon SN, Fraser PE, Fauq A, Lookingbill HA, Findlay KA, Smith TE, Lewis PA, McLendon DC, Wang R, Golde TE. Presenilin 1 regulates pharmacologically distinct gamma -secretase activities. implications for the role of presenilin in gamma -secretase cleavage. J Biol Chem. 2000;275(34):26277–26284. doi: 10.1074/jbc.M002812200. [DOI] [PubMed] [Google Scholar]
- 28.Obrenovich ME, Raina AK, Ogawa O, Atwood CS, Morelli L, Smith MA. In: Alzheimer disease-A new beginning, or a final exit? Nicoletti, Ferdinando, Agata Copani., editors. eurekah.com: Landes Bioscience; 2003. [Google Scholar]
- 29.Petersen MB, Karadima G, Samaritaki M, Avramopoulos D, Vassilopoulos D, Mikkelsen M. Association between presenilin-1 polymorphism and maternal meiosis II errors in down syndrome. Am J Med Genet. 2000;93(5):366–372. doi: 10.1002/1096-8628(20000828)93:5<366::aid-ajmg5>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 30.Potter H, Ma J, Das S, Geller LN, Benjamin M, Kayyali SS, Dressler D. Beyond b-protein: New steps in the pathogenic pathway to Alzheimer’s disease. In: Iqbal K, Mortimer JA, Winblad B, Wisniewski WH, editors. In research advances in Alzheimer’s disease and related disorders. New York: John Wiley and Sons; 1995. pp. 643–654. [Google Scholar]
- 31.Potter H. Cell cycle and chromosome segregation defects in alzheimer’s disease. In: Nicoletti, Ferdinando, Agata Copani., editors. Cell-cycle mechanisms in neuronal death. Eurekah.com: Landes Bioscience; 2003. [Google Scholar]
- 32.Potter H. Review and hypothesis: Alzheimer disease and down syndrome--chromosome 21 nondisjunction may underlie both disorders. American Journal of Human Genetics. 1991;48(6):1192–1200. [PMC free article] [PubMed] [Google Scholar]
- 33.Potter H, Wefes IM, Nilsson LN. The inflammation-induced pathological chaperones ACT and apo-E are necessary catalysts of alzheimer amyloid formation. Neurobiol Aging. 2001;22(6):923–930. doi: 10.1016/s0197-4580(01)00308-6. [DOI] [PubMed] [Google Scholar]
- 34.Rehen SK, McConnell MJ, Kaushal D, Kingsbury MA, Yang AH, Chun J. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc Natl Acad Sci USA. 2001;98(23):13361–13366. doi: 10.1073/pnas.231487398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere, Vital A, Dumanchin C, Feuillet S, Brice A, Vercilletto M, Dubas F, Frebourg T, Campion D. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Geneti. 2006;38(1):11–12. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 36.Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, 3rd, Kandel ER, Duff K, Kirkwood A, Shen J. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron. 2004;42(1):23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- 37.Schupf N, Kapell D, Lee JH, Ottman R, Mayeux R. Increased risk of alzheimer’s disease in mothers of adults with down’s syndrome. Lancet. 1994;344(8919):353–356. doi: 10.1016/s0140-6736(94)91398-6. [DOI] [PubMed] [Google Scholar]
- 38.Schupf N, Kapell D, Nightingale B, Lee JH, Mohlenhoff J, Bewley S, Ottman R, Mayeux R. Specificity of the fivefold increase in AD in mothers of adults with down syndrome. Neurology. 2001;57(6):979–984. doi: 10.1212/wnl.57.6.979. [DOI] [PubMed] [Google Scholar]
- 39.Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, Del-Favero J, Cruts M, van Duijn CM, Van Broeckhoven C. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain. 2006 Aug 18; doi: 10.1093/brain/awl203. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 40.Tezapsidis N, Merz PA, Merz G, Hong H. Microtubular interactions of presenilin direct kinesis of Abeta peptide and its precursors. FASEB J. 2003;17(10):1322–1324. doi: 10.1096/fj.02-0980fje. [DOI] [PubMed] [Google Scholar]
- 41.Theuns J, Brouwers N, Engelborghs S, Sleegers K, Bogaerts V, Corsmit E, De Pooter T, van Duijn CM, De Deyn PP, Van Broeckhoven C. Promoter mutations that increase amyloid precursor-protein expression are associated with Alzheimer disease. Am J Hum Genet. 2006 Jun;78(6):936–46. doi: 10.1086/504044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vincent I, Rosado M, Davies P. Mitotic mechanisms in alzheimer’s disease? J Cell Biol. 1996;132(3):413–425. doi: 10.1083/jcb.132.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vito P, Lacana E, D’Adamio L. Interfering with apoptosis: Ca(2+)-binding protein ALG-2 and alzheimer’s disease gene ALG-3. Science. 1996;271(5248):521–525. doi: 10.1126/science.271.5248.521. [DOI] [PubMed] [Google Scholar]
- 44.Vito P, Wolozin B, Ganjei JK, Iwasaki K, Lacana E, D’Adamio L. Requirement of the familial alzheimer’s disease gene PS2 for apoptosis. opposing effect of ALG-3. J Biol Chem. 1996;271(49):31025–31028. doi: 10.1074/jbc.271.49.31025. [DOI] [PubMed] [Google Scholar]
- 45.Wang R, Dineley KT, Sweatt JD, Zheng H. Presenilin 1 familial alzheimer’s disease mutation leads to defective associative learning and impaired adult neurogenesis. Neuroscience. 2004;126(2):305–312. doi: 10.1016/j.neuroscience.2004.03.048. [DOI] [PubMed] [Google Scholar]
- 46.Wen PH, Hof PR, Chen X, Gluck K, Austin G, Younkin SG, Younkin LH, DeGasperi R, Gama Sosa MA, Robakis NK, Haroutunian V, Elder GA. The presenilin-1 familial alzheimer disease mutant P117L impairs neurogenesis in the hippocampus of adult mice. Exp Neurol. 2004;188(2):224–237. doi: 10.1016/j.expneurol.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 47.Wolfe MS. The secretases of alzheimer’s disease. Curr Top Dev Biol. 2003;54:233–261. doi: 10.1016/s0070-2153(03)54011-x. [DOI] [PubMed] [Google Scholar]
- 48.Wolozin B, Iwasaki K, Vito P, Ganjei JK, Lacana E, Sunderland T, Zhao B, Kusiak JW, Wasco W, D’Adamio L. Participation of presenilin 2 in apoptosis: Enhanced basal activity conferred by an alzheimer mutation. Science. 1996;274(5293):1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- 49.Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in alzheimer’s disease. J Neurosci. 2001;21(8):2661–2668. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of alzheimer’s disease. J Neurosci. 2003;23(7):2557–2563. doi: 10.1523/JNEUROSCI.23-07-02557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuasa S, Nakajima M, Aizawa H, Sahara N, Koizumi K, Sakai T, Usami M, Kobayashi S, Kuroyanagi H, Mori H, Koseki H, Shirasawa T. Impaired cell cycle control of neuronal precursor cells in the neocortical primordium of presenilin-1-deficient mice. J Neurosci Res. 2002;70(3):501–513. doi: 10.1002/jnr.10430. [DOI] [PubMed] [Google Scholar]